
The Immunobiology of Nipah Virus

 , , , , and
, , , , and 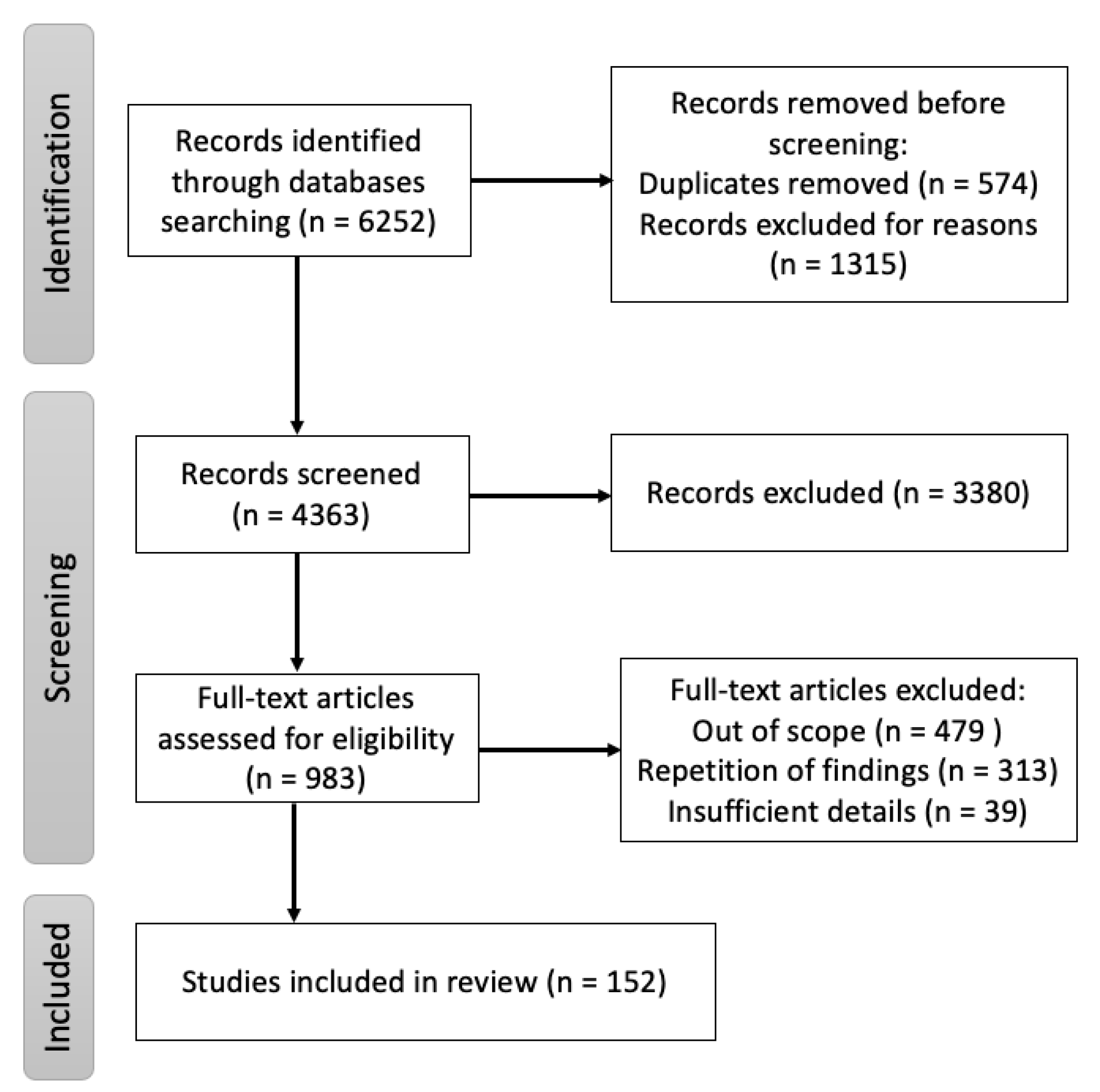{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
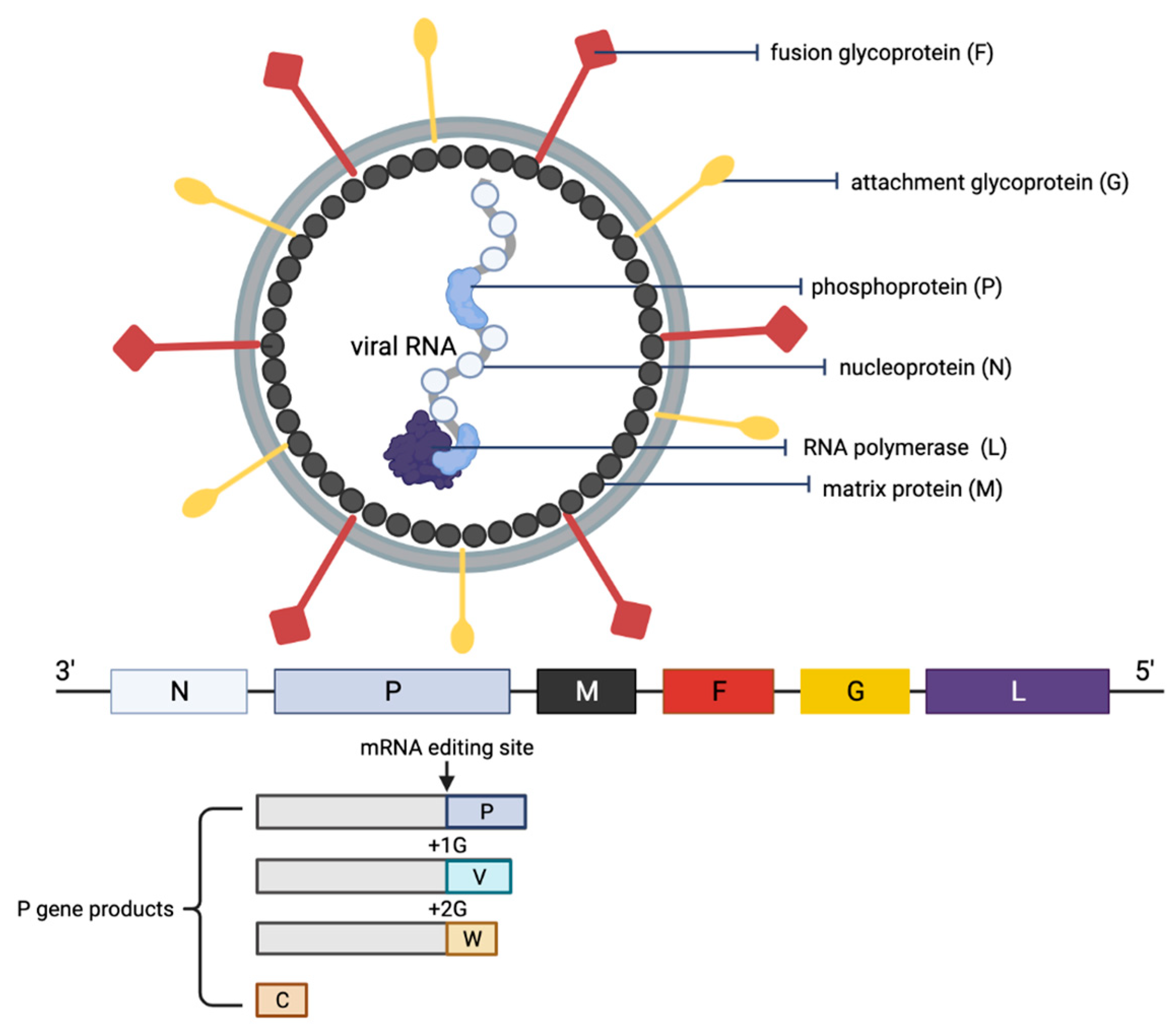
3. Replication Cycle of NiV
4. Pathogenesis of NV
5. NiV F and G Glycoproteins
NiV F and G Glycoproteins as Therapeutic Targets
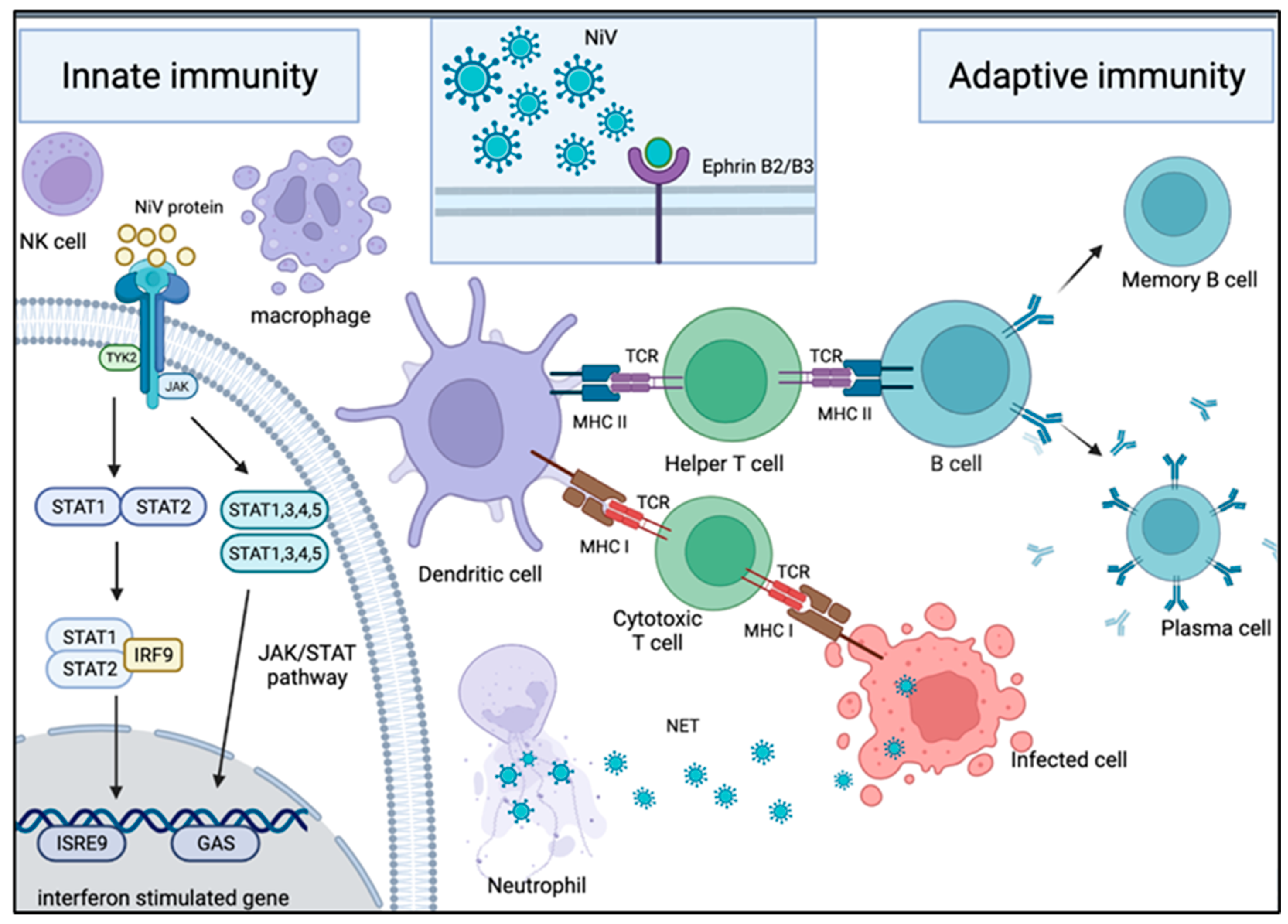
6. Innate Immunity
7. Adaptive Immunity
7.1. Humoral Immunity—B Lymphocytes
7.2. Cellular Immunity—T Lymphocytes
8. Immunomodulatory Impact Targets—Cytokines
9. Therapeutics and Vaccines—Host and Immune Responses
10. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chua, K.B.; Goh, K.J.; Wong, K.T.; Kamarulzaman, A.; Seow, P.; Tan, K.; Ksiazek, T.G.; Zaki, S.R.; Paul, G.; Lam, S.K.; et al. Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet 1999, 354, 1257–1259. [Google Scholar] [CrossRef]
- World Health Organization. WHO R&D Nipah Baseline Situation Analysis; WHO: Geneva, Switzerland, 2018; p. 41.
- CEPI. Priority Diseases. Available online: https://cepi.net/research_dev/priority-diseases/ (accessed on 31 March 2022).
- UK Vaccine Network. Available online: https://www.gov.uk/government/groups/uk-vaccines-network (accessed on 31 March 2022).
- Chua, K.; Bellini, W.; Rota, P.; Harcourt, B.; Tamin, A.; Lam, S.K.; Ksiazek, T.G.; Rollin, P.E.; Zaki, S.R.; Shieh, W.J.; et al. Nipah virus: A recently emergent deadly paramyxovirus. Science 2000, 288, 1432–1436. [Google Scholar] [CrossRef]
- Harcourt, B.H.; Tamin, A.; Ksiazek, T.G.; Rollin, P.E.; Anderson, L.J.; Bellini, W.J.; Rota, P.A. Molecular characterization of Nipah virus, a newly emergent paramyxovirus. Virology 2000, 271, 334–349. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, B.H.; Lowe, L.; Tamin, A.; Liu, X.; Bankamp, B.; Bowden, N.; Rollin, P.E.; Comer, J.A.; Ksiazek, T.G.; Hossain, M.J.; et al. Genetic characterization of Nipah virus, Bangladesh, 2004. Emerg. Infect. Dis. 2005, 11, 1594–1597. [Google Scholar] [CrossRef]
- Chan, Y.P.; Chua, K.B.; Koh, C.L.; Lim, M.E.; Lam, S.K. Complete nucleotide sequences of Nipah virus isolates from Malaysia. J. Gen. Virol. 2001, 82, 2151–2155. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Harcourt, B.H.; Yu, M.; Tamin, A.; Rota, P.A.; Bellini, W.J.; Eaton, B.T. Molecular biology of Hendra and Nipah viruses. Microbes Infect. 2001, 3, 279–287. [Google Scholar] [CrossRef]
- Yadav, P.D.; Shete, A.M.; Kumar, G.A.; Sarkale, P.; Sahay, R.R.; Radhakrishnan, C.; Lakra, R.; Pardeshi, P.; Gupta, N.; Gangakhedkar, R.R.; et al. Nipah virus sequences from humans and bats during Nipah outbreak, Kerala, India, 2018. Emerg. Infect. Dis. 2019, 25, 1003–1006. [Google Scholar] [CrossRef]
- AbuBakar, S.; Chang, L.Y.; Ali, A.R.; Sharifah, S.H.; Yusoff, K.; Zamrod, Z. Isolation and molecular identification of Nipah virus from pigs. Emerg. Infect. Dis. 2004, 10, 2228–2230. [Google Scholar] [CrossRef] [PubMed]
- Clayton, B.A.; Middleton, D.; Arkinstall, R.; Frazer, L.; Wang, L.F.; Marsh, G.A. The nature of exposure drives transmission of Nipah viruses from Malaysia and Bangladesh in ferrets. PLoS Negl. Trop. Dis. 2016, 10, e0004775. [Google Scholar] [CrossRef]
- Clayton, B.A.; Middleton, D.; Bergfeld, J.; Haining, J.; Arkinstall, R.; Wang, L.; Marsh, G.A. Transmission routes for Nipah virus from Malaysia and Bangladesh. Emerg. Infect. Dis. 2012, 18, 12–18. [Google Scholar] [CrossRef]
- Mire, C.E.; Satterfield, B.A.; Geisbert, J.B.; Agans, K.N.; Borisevich, V.; Yan, L.; Chan, Y.P.; Cross, R.W.; Fenton, K.A.; Broder, C.C.; et al. Pathogenic differences between Nipah virus Bangladesh and Malaysia strains in primates: Implications for antibody therapy. Sci. Rep. 2016, 6, 30916. [Google Scholar] [CrossRef] [PubMed]
- Pro-Med. Nipah Virus—Bangladesh, India; WHO: Geneva, Switzerland, 2021; p. 20210910.28660529. [Google Scholar]
- Chong, H.T.; Hossain, M.J.; Tan, C.T. Differences in epidemiologic and clinical features of Nipah virus encephalitis between the Malaysian and Bangladesh outbreaks. Neurol. Asia 2008, 13, 23–26. [Google Scholar]
- Chandni, R.; Renjith, T.P.; Fazal, A.; Yoosef, N.; Ashhar, C.; Thulaseedharan, N.K.; Suraj, K.P.; Sreejith, M.K.; Sajeeth Kumar, K.G.; Rajendran, V.R.; et al. Clinical manifestations of Nipah virus-infected patients who presented to the Emergency Department during an outbreak in Kerala state in India, May 2018. Clin. Infect. Dis. 2020, 71, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Rahmat, K.; Goh, K.J. Late-onset Nipah virus encephalitis 11 years after the initial outbreak: A case report. Neurol. Asia 2012, 17, 71–74. [Google Scholar]
- Tan, C.T.; Goh, K.J.; Wong, K.T.; Sarji, S.A.; Chua, K.B.; Chew, N.K.; Murugasu, P.; Loh, Y.L.; Chong, H.T.; Tan, K.S.; et al. Relapsed and late-onset Nipah encephalitis. Ann. Neurol. 2002, 51, 703–708. [Google Scholar] [CrossRef]
- Chua, K.B.; Lek Koh, C.; Hooi, P.S.; Wee, K.F.; Khong, J.H.; Chua, B.H.; Chan, Y.P.; Lim, M.E.; Lam, S.K. Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect. 2002, 4, 145–151. [Google Scholar] [CrossRef]
- Halpin, K.; Hyatt, A.D.; Fogarty, R.; Middleton, D.; Bingham, J.; Epstein, J.H.; Rahman, S.A.; Hughes, T.; Smith, C.; Field, H.E.; et al. Pteropid bats are confirmed as the reservoir hosts of henipaviruses: A comprehensive experimental study of virus transmission. Am. J. Trop. Med. Hyg. 2011, 85, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.A.; Hassan, S.S.; Olival, K.J.; Mohamed, M.; Chang, L.Y.; Hassan, L.; Saad, N.M.; Shohaimi, S.A.; Mamat, Z.C.; Naim, M.S.; et al. Characterization of Nipah virus from naturally infected Pteropus vampyrus bats, Malaysia. Emerg. Infect. Dis. 2010, 16, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Sazzad, H.M.S.; Satter, S.M.; Sultana, S.; Hossain, M.J.; Hasan, M.; Rahman, M.; Campbell, S.; Cannon, D.L.; Ströher, U.; et al. Nipah virus transmission from bats to humans associated with drinking traditional liquor made from date palm sap, Bangladesh, 2011–2014. Emerg. Infect. Dis. 2016, 22, 664–670. [Google Scholar] [CrossRef]
- Homaira, N.; Rahman, M.; Hossain, M.J.; Epstein, J.H.; Sultana, R.; Khan, M.S.U.; Podder, G.; Nahar, K.; Ahmed, B.; Gurley, E.S.; et al. Nipah virus outbreak with person-to-person transmission in a district of Bangladesh, 2007. Epidemiol. Infect. 2010, 138, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Salah Uddin Khan, M.; Hossain, J.; Gurley, E.S.; Nahar, N.; Sultana, R.; Luby, S.P. Use of infrared camera to understand bats’ access to date palm sap: Implications for preventing Nipah virus transmission. Ecohealth 2010, 7, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Gurley, E.; Montgomery, J.; Hossain, M.; Bell, M.; Azad, A.; Islam, M.; Molla, M.; Carroll, D.; Ksiazek, T.; Rota, P.; et al. Person-to-person transmission of Nipah virus in a Bangladeshi community. Emerg. Infect. Dis. 2007, 13, 1031–1037. [Google Scholar] [CrossRef]
- Luby, S.P.; Hossain, M.J.; Gurley, E.S.; Ahmed, B.N.; Banu, S.; Khan, S.U.; Homaira, N.; Rota, P.A.; Rollin, P.E.; Comer, J.A.; et al. Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001–2007. Emerg. Infect. Dis. 2009, 15, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Devnath, P.; Masud, H.M.A.A. Nipah virus: A potential pandemic agent in the context of the current severe acute respiratory syndrome coronavirus 2 pandemic. New Microbes New Infect. 2021, 41, 100873. [Google Scholar] [CrossRef]
- Luby, S.P. The pandemic potential of Nipah virus. Antivir. Res. 2013, 100, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Gomez Roman, R.; Tornieporth, N.; Cherian, N.G.; Shurtleff, A.C.; L’Azou Jackson, M.; Yeskey, D.; Hacker, A.; Mungai, E.; Le, T.T. Medical countermeasures against henipaviruses: A review and public health perspective. Lancet Infect. Dis. 2022, 22, e13–e27. [Google Scholar] [CrossRef]
- Bonaparte, M.I.; Dimitrov, A.S.; Bossart, K.N.; Crameri, G.; Mungall, B.A.; Bishop, K.A.; Choudhry, V.; Dimitrov, D.S.; Wang, L.F.; Eaton, B.T.; et al. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc. Natl. Acad. Sci. USA 2005, 102, 10652–10657. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Rajashankar, K.R.; Chan, Y.-P.; Himanen, J.P.; Broder, C.C.; Nikolov, D.B. Host cell recognition by the henipaviruses: Crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc. Natl. Acad. Sci. USA 2008, 105, 9953–9958. [Google Scholar] [CrossRef] [PubMed]
- Negrete, O.A.; Wolf, M.C.; Aguilar, H.C.; Enterlain, S.; Wang, W.; Mühlberger, E.; Su, S.V.; Bertolotti-Ciarlet, A.; Flick, R.; Lee, B. Two key residues in ephrinB3 are critical for its use as an alternative receptor for Nipah virus. PLoS Pathog. 2006, 2, e7. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Hickey, A.C.; Smith, M.A.; Chan, Y.-P.; Wang, L.-F.; Mattapallil, J.J.; Geisbert, J.B.; Bossart, K.N.; Broder, C.C. Development of an acute and highly pathogenic nonhuman primate model of Nipah virus infection. PLoS ONE 2010, 5, e10690. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, V.; Wong, K.T.; Looi, R.; Georges-Courbot, M.-C.; Barrot, L.; Buckland, R.; Wild, T.F.; Horvat, B. Acute Hendra virus infection: Analysis of the pathogenesis and passive antibody protection in the hamster model. Virology 2009, 387, 459–465. [Google Scholar] [CrossRef]
- Bossart, K.N.; Tachedjian, M.; McEachern, J.A.; Crameri, G.; Zhu, Z.; Dimitrov, D.S.; Broder, C.C.; Wang, L.-F. Functional studies of host-specific ephrin-B ligands as Henipavirus receptors. Virology 2008, 372, 357–371. [Google Scholar] [CrossRef] [PubMed]
- DeBuysscher, B.L.; Scott, D.P.; Rosenke, R.; Wahl, V.; Feldmann, H.; Prescott, J. Nipah virus efficiently replicates in human smooth muscle cells without cytopathic effect. Cells 2021, 10, 1319. [Google Scholar] [CrossRef] [PubMed]
- Tiong, V.; Shu, M.-H.; Wong, W.F.; AbuBakar, S.; Chang, L.-Y. Nipah virus infection of immature dendritic cells increases its transendothelial migration across human brain microvascular endothelial cells. Front. Microbiol. 2018, 9, 2747. [Google Scholar] [CrossRef]
- Liu, J.; Coffin, K.M.; Johnston, S.C.; Babka, A.M.; Bell, T.M.; Long, S.Y.; Honko, A.N.; Kuhn, J.H.; Zeng, X. Nipah virus persists in the brains of nonhuman primate survivors. JCI Insight 2019, 4, e129629. [Google Scholar] [CrossRef]
- Wong, K.; Robertson, T.; Ong, B.; Chong, J.; Yaiw, K.; Wang, L.; Ansford, A.; Tannenberg, A. Human Hendra virus infection causes acute and relapsing encephalitis. Neuropathol. Appl. Neurobiol. 2009, 35, 296–305. [Google Scholar] [CrossRef]
- Liu, Q.; Stone, J.A.; Bradel-Tretheway, B.; Dabundo, J.; Benavides Montano, J.A.; Santos-Montanez, J.; Biering, S.B.; Nicola, A.V.; Iorio, R.M.; Lu, X. Unraveling a three-step spatiotemporal mechanism of triggering of receptor-induced Nipah virus fusion and cell entry. PLoS Pathog. 2013, 9, e1003770. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Bradel-Tretheway, B.; Monreal, A.I.; Saludes, J.P.; Lu, X.; Nicola, A.V.; Aguilar, H.C. Nipah virus attachment glycoprotein stalk C-terminal region links receptor binding to fusion triggering. J. Virol. 2015, 89, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.-P.; Lu, M.; Dutta, S.; Yan, L.; Barr, J.; Flora, M.; Feng, Y.-R.; Xu, K.; Nikolov, D.B.; Wang, L.-F. Biochemical, conformational, and immunogenic analysis of soluble trimeric forms of henipavirus fusion glycoproteins. J. Virol. 2012, 86, 11457–11471. [Google Scholar] [CrossRef]
- Wong, J.J.; Young, T.A.; Zhang, J.; Liu, S.; Leser, G.P.; Komives, E.A.; Lamb, R.A.; Zhou, Z.H.; Salafsky, J.; Jardetzky, T.S. Monomeric ephrinB2 binding induces allosteric changes in Nipah virus G that precede its full activation. Nat. Commun. 2017, 8, 781. [Google Scholar] [CrossRef]
- Wang, Z.; Amaya, M.; Addetia, A.; Dang, H.V.; Reggiano, G.; Yan, L.; Hickey, A.C.; DiMaio, F.; Broder, C.C.; Veesler, D. Architecture and antigenicity of the Nipah virus attachment glycoprotein. Science 2022, 375, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Chan, Y.-P.; Bradel-Tretheway, B.; Akyol-Ataman, Z.; Zhu, Y.; Dutta, S.; Yan, L.; Feng, Y.; Wang, L.-F.; Skiniotis, G. Crystal structure of the pre-fusion Nipah virus fusion glycoprotein reveals a novel hexamer-of-trimers assembly. PLoS Pathog. 2015, 11, e1005322. [Google Scholar] [CrossRef]
- Stone, J.A.; Vemulapati, B.M.; Bradel-Tretheway, B.; Aguilar, H.C. Multiple strategies reveal a bidentate interaction between the Nipah virus attachment and fusion glycoproteins. J. Virol. 2016, 90, 10762–10773. [Google Scholar] [CrossRef]
- Wong, J.J.; Chen, Z.; Chung, J.K.; Groves, J.T.; Jardetzky, T.S. EphrinB2 clustering by Nipah virus G is required to activate and trap F intermediates at supported lipid bilayer-cell interfaces. Sci. Adv. 2021, 7, eabe1235. [Google Scholar] [CrossRef]
- Foster, S.L.; Woolsey, C.; Borisevich, V.; Agans, K.N.; Prasad, A.N.; Deer, D.J.; Geisbert, J.B.; Dobias, N.S.; Fenton, K.A.; Cross, R.W. A recombinant VSV-vectored vaccine rapidly protects nonhuman primates against lethal Nipah virus disease. Proc. Natl. Acad. Sci. USA 2022, 119, e2200065119. [Google Scholar] [CrossRef] [PubMed]
- Loomis, R.J.; DiPiazza, A.T.; Falcone, S.; Ruckwardt, T.J.; Morabito, K.M.; Abiona, O.M.; Chang, L.A.; Caringal, R.T.; Presnyak, V.; Narayanan, E. Chimeric fusion (F) and attachment (G) glycoprotein antigen delivery by mRNA as a candidate Nipah vaccine. Front. Immunol. 2021, 12, 772864. [Google Scholar] [CrossRef] [PubMed]
- Loomis, R.J.; Stewart-Jones, G.B.; Tsybovsky, Y.; Caringal, R.T.; Morabito, K.M.; McLellan, J.S.; Chamberlain, A.L.; Nugent, S.T.; Hutchinson, G.B.; Kueltzo, L.A. Structure-based design of Nipah virus vaccines: A generalizable approach to paramyxovirus immunogen development. Front. Immunol. 2020, 11, 842. [Google Scholar] [CrossRef]
- Dong, J.; Cross, R.W.; Doyle, M.P.; Kose, N.; Mousa, J.J.; Annand, E.J.; Borisevich, V.; Agans, K.N.; Sutton, R.; Nargi, R. Potent henipavirus neutralization by antibodies recognizing diverse sites on Hendra and Nipah virus receptor binding protein. Cell 2020, 183, 1536–1550.e17. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Hemmers, S.; Teijaro, J.R.; Arandjelovic, S.; Mowen, K.A. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS ONE 2011, 6, e22043. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Zychlinsky, A. NETs: A new strategy for using old weapons. Trends Immunol. 2009, 30, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Hayes, G. Neutrophil extracellular traps. Curr. Biol. 2011, 21, R297–R298. [Google Scholar] [CrossRef] [PubMed]
- Drescher, B.; Bai, F. Neutrophil in viral infections, friend or foe? Virus Res. 2013, 171, 1–7. [Google Scholar] [CrossRef]
- Arcanjo, A.; Logullo, J.; Menezes, C.C.B.; de Souza Carvalho Giangiarulo, T.C.; Dos Reis, M.C.; de Castro, G.M.M.; da Silva Fontes, Y.; Todeschini, A.R.; Freire-de-Lima, L.; Decote-Ricardo, D.; et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci. Rep. 2020, 10, 19630. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Q.; Yin, Y.; Zhang, Y.; Cao, Y.; Lin, X.; Huang, L.; Hoffmann, D.; Lu, M.; Qiu, Y. Excessive neutrophils and neutrophil extracellular traps in COVID-19. Front. Immunol. 2020, 11, 2063. [Google Scholar] [CrossRef]
- Rawat, S.; Vrati, S.; Banerjee, A. Neutrophils at the crossroads of acute viral infections and severity. Mol. Aspects Med. 2021, 81, 100996. [Google Scholar] [CrossRef] [PubMed]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, L.; Zhang, Y.; Pu, L.; Liu, J.; Li, X.; Chen, Z.; Hao, Y.; Wang, B.; Han, J.; et al. High level of neutrophil extracellular traps correlates with poor prognosis of severe influenza A infection. J. Infect. Dis. 2018, 217, 428–437. [Google Scholar] [CrossRef]
- Funchal, G.A.; Jaeger, N.; Czepielewski, R.S.; Machado, M.S.; Muraro, S.P.; Stein, R.T.; Bonorino, C.B.; Porto, B.N. Respiratory syncytial virus fusion protein promotes TLR-4-dependent neutrophil extracellular trap formation by human neutrophils. PLoS ONE 2015, 10, e0124082. [Google Scholar] [CrossRef]
- Muraro, S.P.; De Souza, G.F.; Gallo, S.W.; Da Silva, B.K.; De Oliveira, S.D.; Vinolo, M.A.R.; Saraiva, E.M.; Porto, B.N. Respiratory syncytial virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci. Rep. 2018, 8, 14166. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiedzka-Rystwej, P.; Repka, W.; Tokarz-Deptula, B.; Deptula, W. “In sickness and in health”—How neutrophil extracellular trap (NET) works in infections, selected diseases and pregnancy. J. Inflamm. 2019, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J.; Coombs, R.W. Viricidal effect of Lactobacillus acidophilus on human immunodeficiency virus type 1: Possible role in heterosexual transmission. J. Exp. Med. 1991, 174, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J.; Coombs, R.W. Viricidal effect of polymorphonuclear leukocytes on human immunodeficiency virus-1. Role of the myeloperoxidase system. J. Clin. Investig. 1992, 89, 2014–2017. [Google Scholar] [CrossRef]
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral mechanisms of human defensins. J. Mol. Biol. 2013, 425, 4965–4980. [Google Scholar] [CrossRef]
- Tumpey, T.M.; Garcia-Sastre, A.; Taubenberger, J.K.; Palese, P.; Swayne, D.E.; Pantin-Jackwood, M.J.; Schultz-Cherry, S.; Solorzano, A.; Van Rooijen, N.; Katz, J.M.; et al. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: Functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J. Virol. 2005, 79, 14933–14944. [Google Scholar] [CrossRef] [PubMed]
- Marcos, V.; Zhou, Z.; Yildirim, A.O.; Bohla, A.; Hector, A.; Vitkov, L.; Wiedenbauer, E.M.; Krautgartner, W.D.; Stoiber, W.; Belohradsky, B.H.; et al. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat. Med. 2010, 16, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Kohno, S.; Newstead, M.W.; Zeng, X.; Bhan, U.; Lukacs, N.W.; Kunkel, S.L.; Standiford, T.J. Critical role of IL-1 receptor-associated kinase-M in regulating chemokine-dependent deleterious inflammation in murine influenza pneumonia. J. Immunol. 2010, 184, 1410–1418. [Google Scholar] [CrossRef]
- Tate, M.D.; Brooks, A.G.; Reading, P.C. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir. Res. 2008, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Cortjens, B.; de Boer, O.J.; de Jong, R.; Antonis, A.F.; Sabogal Pineros, Y.S.; Lutter, R.; van Woensel, J.B.; Bem, R.A. Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease. J. Pathol. 2016, 238, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.K.; Miller, D.; Aljofan, M.; Mungall, B.A.; Rollin, P.E.; Bellini, W.J.; Rota, P.A. Characterization of the antiviral and inflammatory responses against Nipah virus in endothelial cells and neurons. Virology 2010, 404, 78–88. [Google Scholar] [CrossRef]
- Habjan, M.; Andersson, I.; Klingstrom, J.; Schumann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Muhlberger, E.; et al. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE 2008, 3, e2032. [Google Scholar] [CrossRef] [PubMed]
- Leon, A.J.; Borisevich, V.; Boroumand, N.; Seymour, R.; Nusbaum, R.; Escaffre, O.; Xu, L.; Kelvin, D.J.; Rockx, B. Host gene expression profiles in ferrets infected with genetically distinct henipavirus strains. PLoS Negl. Trop. Dis. 2018, 12, e0006343. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Tachedjian, M.; Wynne, J.W.; Boyd, V.; Cui, J.; Smith, I.; Cowled, C.; Ng, J.H.; Mok, L.; Michalski, W.P.; et al. Contraction of the type I IFN locus and unusual constitutive expression of IFN-alpha in bats. Proc. Natl. Acad. Sci. USA 2016, 113, 2696–2701. [Google Scholar] [CrossRef]
- Zhou, P.; Cowled, C.; Marsh, G.A.; Shi, Z.; Wang, L.F.; Baker, M.L. Type III IFN receptor expression and functional characterisation in the pteropid bat, Pteropus alecto. PLoS ONE 2011, 6, e25385. [Google Scholar] [CrossRef]
- Zhou, P.; Cowled, C.; Todd, S.; Crameri, G.; Virtue, E.R.; Marsh, G.A.; Klein, R.; Shi, Z.; Wang, L.F.; Baker, M.L. Type III IFNs in pteropid bats: Differential expression patterns provide evidence for distinct roles in antiviral immunity. J. Immunol. 2011, 186, 3138–3147. [Google Scholar] [CrossRef] [PubMed]
- Keiffer, T.R.; Ciancanelli, M.J.; Edwards, M.R.; Basler, C.F. Interactions of the Nipah virus P, V, and W proteins across the STAT family of transcription factors. mSphere 2020, 5, e00449-20. [Google Scholar] [CrossRef]
- Sugai, A.; Sato, H.; Takayama, I.; Yoneda, M.; Kai, C. Nipah and Hendra virus nucleoproteins inhibit nuclear accumulation of signal transducer and activator of transcription 1 (STAT1) and STAT2 by interfering with their complex formation. J. Virol. 2017, 91, e01136-17. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Wang, L.F.; Horvath, C.M. Hendra virus V protein inhibits interferon signaling by preventing STAT1 and STAT2 nuclear accumulation. J. Virol. 2003, 77, 11842–11845. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Taniguchi, T. Toll-like receptor signaling and IRF transcription factors. IUBMB Life 2006, 58, 290–295. [Google Scholar] [CrossRef]
- Davis, M.E.; Wang, M.K.; Rennick, L.J.; Full, F.; Gableske, S.; Mesman, A.W.; Gringhuis, S.I.; Geijtenbeek, T.B.; Duprex, W.P.; Gack, M.U. Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host Microbe 2014, 16, 19–30. [Google Scholar] [CrossRef]
- Rodriguez, K.R.; Horvath, C.M. Paramyxovirus V protein interaction with the antiviral sensor LGP2 disrupts MDA5 signaling enhancement but is not relevant to LGP2-mediated RLR signaling inhibition. J. Virol. 2014, 88, 8180–8188. [Google Scholar] [CrossRef] [PubMed]
- Parisien, J.P.; Bamming, D.; Komuro, A.; Ramachandran, A.; Rodriguez, J.J.; Barber, G.; Wojahn, R.D.; Horvath, C.M. A shared interface mediates paramyxovirus interference with antiviral RNA helicases MDA5 and LGP2. J. Virol. 2009, 83, 7252–7260. [Google Scholar] [CrossRef] [PubMed]
- Seto, J.; Qiao, L.; Guenzel, C.A.; Xiao, S.; Shaw, M.L.; Hayot, F.; Sealfon, S.C. Novel Nipah virus immune-antagonism strategy revealed by experimental and computational study. J. Virol. 2010, 84, 10965–10973. [Google Scholar] [CrossRef]
- Rajsbaum, R.; Versteeg, G.A.; Schmid, S.; Maestre, A.M.; Belicha-Villanueva, A.; Martinez-Romero, C.; Patel, J.R.; Morrison, J.; Pisanelli, G.; Miorin, L.; et al. Unanchored K48-linked polyubiquitin synthesized by the E3-ubiquitin ligase TRIM6 stimulates the interferon-IKKepsilon kinase-mediated antiviral response. Immunity 2014, 40, 880–895. [Google Scholar] [CrossRef]
- Bharaj, P.; Wang, Y.E.; Dawes, B.E.; Yun, T.E.; Park, A.; Yen, B.; Basler, C.F.; Freiberg, A.N.; Lee, B.; Rajsbaum, R. The matrix protein of Nipah virus targets the E3-ubiquitin ligase TRIM6 to inhibit the IKKepsilon kinase-mediated type-I IFN antiviral response. PLoS Pathog. 2016, 12, e1005880. [Google Scholar] [CrossRef]
- Rockx, B.; Brining, D.; Kramer, J.; Callison, J.; Ebihara, H.; Mansfield, K.; Feldmann, H. Clinical outcome of henipavirus infection in hamsters is determined by the route and dose of infection. J. Virol. 2011, 85, 7658–7671. [Google Scholar] [CrossRef] [PubMed]
- Dhondt, K.P.; Mathieu, C.; Chalons, M.; Reynaud, J.M.; Vallve, A.; Raoul, H.; Horvat, B. Type I interferon signaling protects mice from lethal henipavirus infection. J. Infect. Dis. 2013, 207, 142–151. [Google Scholar] [CrossRef]
- Georges-Courbot, M.C.; Contamin, H.; Faure, C.; Loth, P.; Baize, S.; Leyssen, P.; Neyts, J.; Deubel, V. Poly(I)-poly(C12U) but not ribavirin prevents death in a hamster model of Nipah virus infection. Antimicrob. Agents Chemother. 2006, 50, 1768–1772. [Google Scholar] [CrossRef]
- Darif, D.; Hammi, I.; Kihel, A.; El Idrissi Saik, I.; Guessous, F.; Akarid, K. The pro-inflammatory cytokines in COVID-19 pathogenesis: What goes wrong? Microb. Pathog. 2021, 153, 104799. [Google Scholar] [CrossRef]
- Ang, B.S.P.; Lim, T.C.C.; Wang, L. Nipah Virus Infection. J. Clin. Microbiol. 2018, 56, e01875-17. [Google Scholar] [CrossRef] [PubMed]
- Sejvar, J.J.; Hossain, J.; Saha, S.K.; Gurley, E.S.; Banu, S.; Hamadani, J.D.; Faiz, M.A.; Siddiqui, F.M.; Mohammad, Q.D.; Mollah, A.H.; et al. Long-term neurological and functional outcome in Nipah virus infection. Ann. Neurol. 2007, 62, 235–242. [Google Scholar] [CrossRef]
- Ademokun, A.A.; Dunn-Walters, D. Immune Responses: Primary and Secondary. Encycl. Life Sci. 2010. [Google Scholar] [CrossRef]
- Gray, D. A role for antigen in the maintenance of immunological memory. Nat. Rev. Immunol. 2002, 2, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Taub, D.D.; Ershler, W.B.; Janowski, M.; Artz, A.; Key, M.L.; McKelvey, J.; Muller, D.; Moss, B.; Ferrucci, L.; Duffey, P.L.; et al. Immunity from smallpox vaccine persists for decades: A longitudinal study. Am. J. Med. 2008, 121, 1058–1064. [Google Scholar] [CrossRef]
- Koepke, R.; Eickhoff, J.C.; Ayele, R.A.; Petit, A.B.; Schauer, S.L.; Hopfensperger, D.J.; Conway, J.H.; Davis, J.P. Estimating the effectiveness of tetanus-diphtheria-acellular pertussis vaccine (Tdap) for preventing pertussis: Evidence of rapidly waning immunity and difference in effectiveness by Tdap brand. J. Infect. Dis. 2014, 210, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Viana, P.O.; Ono, E.; Miyamoto, M.; Salomao, R.; Costa-Carvalho, B.T.; Weckx, L.Y.; de Moraes-Pinto, M.I. Humoral and cellular immune responses to measles and tetanus: The importance of elapsed time since last exposure and the nature of the antigen. J. Clin. Immunol. 2010, 30, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wang, G.; Wang, Y.; Zhang, Q.; Ren, L.; Gu, X.; Huang, T.; Zhong, J.; Wang, Y.; Wang, X.; et al. SARS-CoV-2-specific antibody and T-cell responses 1 year after infection in people recovered from COVID-19: A longitudinal cohort study. Lancet Microbe 2022, 3, e348–e356. [Google Scholar] [CrossRef]
- Arunkumar, G.; Devadiga, S.; McElroy, A.K.; Prabhu, S.; Sheik, S.; Abdulmajeed, J.; Robin, S.; Sushama, A.; Jayaram, A.; Nittur, S.; et al. Adaptive immune responses in humans during Nipah virus acute and convalescent phases of infection. Clin. Infect. Dis. 2019, 69, 1752–1756. [Google Scholar] [CrossRef]
- Schountz, T.; Baker, M.L.; Butler, J.; Munster, V. Immunological control of viral infections in bats and the emergence of viruses highly pathogenic to humans. Front. Immunol. 2017, 8, 1098. [Google Scholar] [CrossRef]
- Berhane, Y.; Weingartl, H.M.; Lopez, J.; Neufeld, J.; Czub, S.; Embury-Hyatt, C.; Goolia, M.; Copps, J.; Czub, M. Bacterial infections in pigs experimentally infected with Nipah virus. Transbound. Emerg. Dis. 2008, 55, 165–174. [Google Scholar] [CrossRef]
- Lara, A.; Cong, Y.; Jahrling, P.B.; Mednikov, M.; Postnikova, E.; Yu, S.; Munster, V.; Holbrook, M.R. Peripheral immune response in the African green monkey model following Nipah-Malaysia virus exposure by intermediate-size particle aerosol. PLoS Negl. Trop. Dis. 2019, 13, e0007454. [Google Scholar] [CrossRef] [PubMed]
- Riddell, M.A.; Moss, W.J.; Hauer, D.; Monze, M.; Griffin, D.E. Slow clearance of measles virus RNA after acute infection. J. Clin. Virol. 2007, 39, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding roles for CD4+ T cells in immunity to viruses. Nat. Rev. Immunol. 2012, 12, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Lentz, M.R.; Lara, A.; Alexander, I.; Bartos, C.; Bohannon, J.K.; Hammoud, D.; Huzella, L.; Jahrling, P.B.; Janosko, K.; et al. Loss in lung volume and changes in the immune response demonstrate disease progression in African green monkeys infected by small-particle aerosol and intratracheal exposure to Nipah virus. PLoS Negl. Trop. Dis. 2017, 11, e0005532. [Google Scholar] [CrossRef] [PubMed]
- Pickering, B.S.; Hardham, J.M.; Smith, G.; Weingartl, E.T.; Dominowski, P.J.; Foss, D.L.; Mwangi, D.; Broder, C.C.; Roth, J.A.; Weingartl, H.M. Protection against henipaviruses in swine requires both, cell-mediated and humoral immune response. Vaccine 2016, 34, 4777–4786. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Wen, Z.; Su, H.; Ge, J.; Chen, W.; Wang, X.; Wu, C.; Yang, C.; Chen, H.; Bu, Z. Newcastle disease virus-vectored Nipah encephalitis vaccines induce B and T cell responses in mice and long-lasting neutralizing antibodies in pigs. Virology 2012, 432, 327–335. [Google Scholar] [CrossRef]
- Kalodimou, G.; Veit, S.; Jany, S.; Kalinke, U.; Broder, C.C.; Sutter, G.; Volz, A. A soluble version of nipah virus glycoprotein G delivered by vaccinia virus MVA activates specific CD8 and CD4 T cells in mice. Viruses 2020, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- de Vries, R.D.; de Jong, A.; Verburgh, R.J.; Sauerhering, L.; van Nierop, G.P.; van Binnendijk, R.S.; Osterhaus, A.D.M.E.; Maisner, A.; Koopmans, M.P.G.; de Swart, R.L. Human paramyxovirus infections induce T cells that cross-react with zoonotic henipaviruses. mBio 2020, 11, e00972-20. [Google Scholar] [CrossRef]
- Dinarello, C.A. Historical insights into cytokines. Eur. J. Immunol. 2007, 37, S34–S45. [Google Scholar] [CrossRef] [PubMed]
- Schountz, T. Immunology of bats and their viruses: Challenges and opportunities. Viruses 2014, 6, 4880–4901. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Guillaume, V.; Sabine, A.; Ong, K.C.; Wong, K.T.; Legras-Lachuer, C.; Horvat, B. Lethal Nipah virus infection induces rapid overexpression of CXCL10. PLoS ONE 2012, 7, e32157. [Google Scholar] [CrossRef]
- Sui, Y.; Potula, R.; Dhillon, N.; Pinson, D.; Li, S.; Nath, A.; Anderson, C.; Turchan, J.; Kolson, D.; Narayan, O. Neuronal apoptosis is mediated by CXCL10 overexpression in simian human immunodeficiency virus encephalitis. Am. J. Pathol. 2004, 164, 1557–1566. [Google Scholar] [CrossRef]
- Sui, Y.; Stehno-Bittel, L.; Li, S.; Loganathan, R.; Dhillon, N.K.; Pinson, D.; Nath, A.; Kolson, D.; Narayan, O.; Buch, S. CXCL10-induced cell death in neurons: Role of calcium dysregulation. Eur. J. Neurosci. 2006, 23, 957–964. [Google Scholar] [CrossRef]
- Woon, A.P.; Boyd, V.; Todd, S.; Smith, I.; Klein, R.; Woodhouse, I.B.; Riddell, S.; Crameri, G.; Bingham, J.; Wang, L.-F. Acute experimental infection of bats and ferrets with Hendra virus: Insights into the early host response of the reservoir host and susceptible model species. PLoS Pathog. 2020, 16, e1008412. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Lo, M.K.; Spiropoulou, C.F. Activation and cell death in human dendritic cells infected with Nipah virus. Virology 2013, 441, 49–56. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Prasad, A.N.; Woolsey, C.; Geisbert, J.B.; Agans, K.N.; Borisevich, V.; Deer, D.J.; Mire, C.E.; Cross, R.W.; Fenton, K.A.; Broder, C.C. Resistance of Cynomolgus monkeys to Nipah and Hendra virus disease is associated with cell-mediated and humoral immunity. J. Infect. Dis. 2020, 221, S436–S447. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Shakui, P.; Keep, R.F.; Moore, B.B.; Kunkel, S.L.; Van Rooijen, N.; Andjelkovic, A.V. Monocyte chemoattractant protein-1 regulation of blood–brain barrier permeability. J. Cereb. Blood Flow Metab. 2005, 25, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Escaffre, O.; Borisevich, V.; Vergara, L.A.; Wen, J.W.; Long, D.; Rockx, B. Characterization of Nipah virus infection in a model of human airway epithelial cells cultured at an air–liquid interface. J. Gen. Virol. 2016, 97, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Valbuena, G.; Halliday, H.; Borisevich, V.; Goez, Y.; Rockx, B. A human lung xenograft mouse model of Nipah virus infection. PLoS Pathog. 2014, 10, e1004063. [Google Scholar] [CrossRef]
- Levroney, E.L.; Aguilar, H.C.; Fulcher, J.A.; Kohatsu, L.; Pace, K.E.; Pang, M.; Gurney, K.B.; Baum, L.G.; Lee, B. Novel innate immune functions for galectin-1: Galectin-1 inhibits cell fusion by Nipah virus envelope glycoproteins and augments dendritic cell secretion of proinflammatory cytokines. J. Immunol. 2005, 175, 413–420. [Google Scholar] [CrossRef]
- Aditi; Shariff, M. Nipah virus infection: A review. Epidemiol. Infect. 2019, 147, e95. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Bossart, K.N.; Bishop, K.A.; Crameri, G.; Dimitrov, A.S.; McEachern, J.A.; Feng, Y.; Middleton, D.; Wang, L.F.; Broder, C.C.; et al. Exceptionally potent cross-reactive neutralization of Nipah and Hendra viruses by a human monoclonal antibody. J. Infect. Dis. 2008, 197, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Bossart, K.N.; Zhu, Z.; Middleton, D.; Klippel, J.; Crameri, G.; Bingham, J.; McEachern, J.A.; Green, D.; Hancock, T.J.; Chan, Y.P.; et al. A neutralizing human monoclonal antibody protects against lethal disease in a new ferret model of acute nipah virus infection. PLoS Pathog. 2009, 5, e1000642. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Mire, C.E.; Geisbert, J.B.; Chan, Y.P.; Agans, K.N.; Feldmann, F.; Fenton, K.A.; Zhu, Z.; Dimitrov, D.S.; Scott, D.P.; et al. Therapeutic treatment of Nipah virus infection in nonhuman primates with a neutralizing human monoclonal antibody. Sci. Transl. Med. 2014, 6, 242ra82. [Google Scholar] [CrossRef] [PubMed]
- Playford, E.G.; Munro, T.; Mahler, S.M.; Elliott, S.; Gerometta, M.; Hoger, K.L.; Jones, M.L.; Griffin, P.; Lynch, K.D.; Carroll, H.; et al. Safety, tolerability, pharmacokinetics, and immunogenicity of a human monoclonal antibody targeting the G glycoprotein of henipaviruses in healthy adults: A first-in-human, randomised, controlled, phase 1 study. Lancet Infect. Dis. 2020, 20, 445–454. [Google Scholar] [CrossRef]
- Mire, C.E.; Chan, Y.P.; Borisevich, V.; Cross, R.W.; Yan, L.; Agans, K.N.; Dang, H.V.; Veesler, D.; Fenton, K.A.; Geisbert, T.W.; et al. A cross-reactive humanized monoclonal antibody targeting fusion glycoprotein function protects ferrets against lethal Nipah virus and Hendra virus infection. J. Infect. Dis. 2020, 221, S471–S479. [Google Scholar] [CrossRef]
- Dang, H.V.; Chan, Y.P.; Park, Y.J.; Snijder, J.; Da Silva, S.C.; Vu, B.; Yan, L.; Feng, Y.R.; Rockx, B.; Geisbert, T.W.; et al. An antibody against the F glycoprotein inhibits Nipah and Hendra virus infections. Nat. Struct. Mol. Biol. 2019, 26, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, V.; Contamin, H.; Loth, P.; Georges-Courbot, M.C.; Lefeuvre, A.; Marianneau, P.; Chua, K.B.; Lam, S.K.; Buckland, R.; Deubel, V.; et al. Nipah virus: Vaccination and passive protection studies in a hamster model. J. Virol. 2004, 78, 834. [Google Scholar] [CrossRef]
- Mungall, B.A.; Middleton, D.; Crameri, G.; Bingham, J.; Halpin, K.; Russell, G.; Green, D.; McEachern, J.; Pritchard, L.I.; Eaton, B.T.; et al. Feline model of acute Nipah virus infection and protection with a soluble glycoprotein-based subunit vaccine. J. Virol. 2006, 80, 12293–12302. [Google Scholar] [CrossRef]
- Pallister, J.A.; Klein, R.; Arkinstall, R.; Haining, J.; Long, F.; White, J.R.; Payne, J.; Feng, Y.R.; Wang, L.F.; Broder, C.C.; et al. Vaccination of ferrets with a recombinant G glycoprotein subunit vaccine provides protection against Nipah virus disease for over 12 months. Virol. J. 2013, 10, 237. [Google Scholar] [CrossRef] [PubMed]
- Khetawat, D.; Broder, C.C. A functional henipavirus envelope glycoprotein pseudotyped lentivirus assay system. Virol. J. 2010, 7, 312. [Google Scholar] [CrossRef]
- Middleton, D.; Pallister, J.; Klein, R.; Feng, Y.R.; Haining, J.; Arkinstall, R.; Frazer, L.; Huang, J.A.; Edwards, N.; Wareing, M.; et al. Hendra virus vaccine, a one health approach to protecting horse, human, and environmental health. Emerg. Infect. Dis. 2014, 20, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Bobb, K.; Borisevich, V.; Geisbert, J.B.; Agans, K.N.; Cross, R.W.; Prasad, A.N.; Fenton, K.A.; Yu, H.; Fouts, T.R.; et al. A single dose investigational subunit vaccine for human use against Nipah virus and Hendra virus. NPJ Vaccines 2021, 6, 23. [Google Scholar] [CrossRef]
- Weingartl, H.M.; Berhane, Y.; Caswell, J.L.; Loosmore, S.; Audonnet, J.-C.; Roth, J.A.; Czub, M. Recombinant Nipah virus vaccines protect pigs against challenge. J. Virol. 2006, 80, 7929. [Google Scholar] [CrossRef]
- Broder, C.C.; Geisbert, T.W.; Xu, K.; Nikolov, D.B.; Wang, L.-F.; Middleton, D.; Pallister, J.; Bossart, K.N. Immunization strategies against henipaviruses. Curr. Top. Microbiol. Immunol. 2012, 359, 197–223. [Google Scholar] [PubMed]
- Yoneda, M.; Georges-Courbot, M.C.; Ikeda, F.; Ishii, M.; Nagata, N.; Jacquot, F.; Raoul, H.; Sato, H.; Kai, C. Recombinant measles virus vaccine expressing the Nipah virus glycoprotein protects against lethal Nipah virus challenge. PLoS ONE 2013, 8, e58414. [Google Scholar] [CrossRef]
- Prescott, J.; DeBuysscher, B.L.; Feldmann, F.; Gardner, D.J.; Haddock, E.; Martellaro, C.; Scott, D.; Feldmann, H. Single-dose live-attenuated vesicular stomatitis virus-based vaccine protects African green monkeys from Nipah virus disease. Vaccine 2015, 33, 2823–2829. [Google Scholar] [CrossRef]
- Keshwara, R.; Shiels, T.; Postnikova, E.; Kurup, D.; Wirblich, C.; Johnson, R.F.; Schnell, M.J. Rabies-based vaccine induces potent immune responses against Nipah virus. NPJ Vaccines 2019, 4, 15. [Google Scholar] [CrossRef]
- Lo, M.K.; Spengler, J.R.; Welch, S.R.; Harmon, J.R.; Coleman-Mccray, J.D.; Scholte, F.E.M.; Shrivastava-Ranjan, P.; Montgomery, J.M.; Nichol, S.T.; Weissman, D.; et al. Evaluation of a single-dose nucleoside-modified messenger RNA vaccine encoding Hendra virus-soluble glycoprotein against lethal Nipah virus challenge in Syrian hamsters. J. Infect. Dis. 2020, 221, S493–S498. [Google Scholar] [CrossRef]
- Goh, K.J.; Tan, C.T.; Chew, N.K.; Tan, P.S.K.; Kamarulzaman, A.; Sarji, S.A.; Wong, K.T.; Abdullah, B.J.J.; Chua, K.B.; Lam, S.K. Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. N. Engl. J. Med. 2000, 342, 1229–1235. [Google Scholar] [PubMed]
- Freiberg, A.N.; Worthy, M.N.; Lee, B.; Holbrook, M.R. Combined chloroquine and ribavirin treatment does not prevent death in a hamster model of Nipah and Hendra virus infection. J. Gen. Virol. 2010, 91, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Niyas, V.K.M.; Soneja, M.; Shibeesh, A.P.; Basheer, M.; Sadanandan, R.; Wig, N.; Biswas, A. First experience of ribavirin postexposure prophylaxis for Nipah virus, tried during the 2018 outbreak in Kerala, India. J. Infect. 2019, 78, 491–503. [Google Scholar]
- Lo, M.K.; Feldmann, F.; Gary, J.M.; Jordan, R.; Bannister, R.; Cronin, J.; Patel, N.R.; Klena, J.D.; Nichol, S.T.; Cihlar, T.; et al. Remdesivir (GS-5734) protects African green monkeys from Nipah virus challenge. Sci. Transl. Med. 2019, 11, 9242. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Paton, N.I.; Leo, Y.S.; Zaki, S.R.; Auchus, A.P.; Lee, K.E.; Ling, A.E.; Chew, S.K.; Ang, B.; Rollin, P.E.; Umapathi, T.; et al. Outbreak of Nipah-virus infection among abattoir workers in Singapore. Lancet 1999, 354, 1253–1256. [Google Scholar] [PubMed]
- Srinivasan, K.; Rao, M. Understanding the clinical utility of favipiravir (T-705) in coronavirus disease of 2019: A review. Ther. Adv. Infect. Dis. 2021, 8, 204993612110630. [Google Scholar] [CrossRef] [PubMed]
- Dawes, B.E.; Kalveram, B.; Ikegami, T.; Juelich, T.; Smith, J.K.; Zhang, L.; Park, A.; Lee, B.; Komeno, T.; Furuta, Y.; et al. Favipiravir (T-705) protects against Nipah virus infection in the hamster model. Sci. Rep. 2018, 8, 7604. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liew, Y.J.M.; Ibrahim, P.A.S.; Ong, H.M.; Chong, C.N.; Tan, C.T.; Schee, J.P.; Gómez Román, R.; Cherian, N.G.; Wong, W.F.; Chang, L.-Y. The Immunobiology of Nipah Virus. Microorganisms 2022, 10, 1162. https://doi.org/10.3390/microorganisms10061162
Liew YJM, Ibrahim PAS, Ong HM, Chong CN, Tan CT, Schee JP, Gómez Román R, Cherian NG, Wong WF, Chang L-Y. The Immunobiology of Nipah Virus. Microorganisms. 2022; 10(6):1162. https://doi.org/10.3390/microorganisms10061162
Chicago/Turabian StyleLiew, Yvonne Jing Mei, Puteri Ainaa S. Ibrahim, Hui Ming Ong, Chee Ning Chong, Chong Tin Tan, Jie Ping Schee, Raúl Gómez Román, Neil George Cherian, Won Fen Wong, and Li-Yen Chang. 2022. "The Immunobiology of Nipah Virus" Microorganisms 10, no. 6: 1162. https://doi.org/10.3390/microorganisms10061162
APA StyleLiew, Y. J. M., Ibrahim, P. A. S., Ong, H. M., Chong, C. N., Tan, C. T., Schee, J. P., Gómez Román, R., Cherian, N. G., Wong, W. F., & Chang, L.-Y. (2022). The Immunobiology of Nipah Virus. Microorganisms, 10(6), 1162. https://doi.org/10.3390/microorganisms10061162