
Seasonal and Zonal Succession of Bacterial Communities in North Sea Salt Marsh Sediments

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Site Description
2.2. Nucleotide Extraction and 16S rRNA Amplicon Sequencing
2.3. Processing of 16S rRNA Amplicon Data
2.4. Statistical Analyses
2.5. Functional Gene Prediction (tax4fun2)
3. Results
3.1. Bacterial Alpha-Diversity Varies along the Transect
3.2. Distance–Decay Analysis Shows Higher Dissimilarity with Distance
3.3. Zonation Outweighs Seasonality Effect on Bacterial Diversity
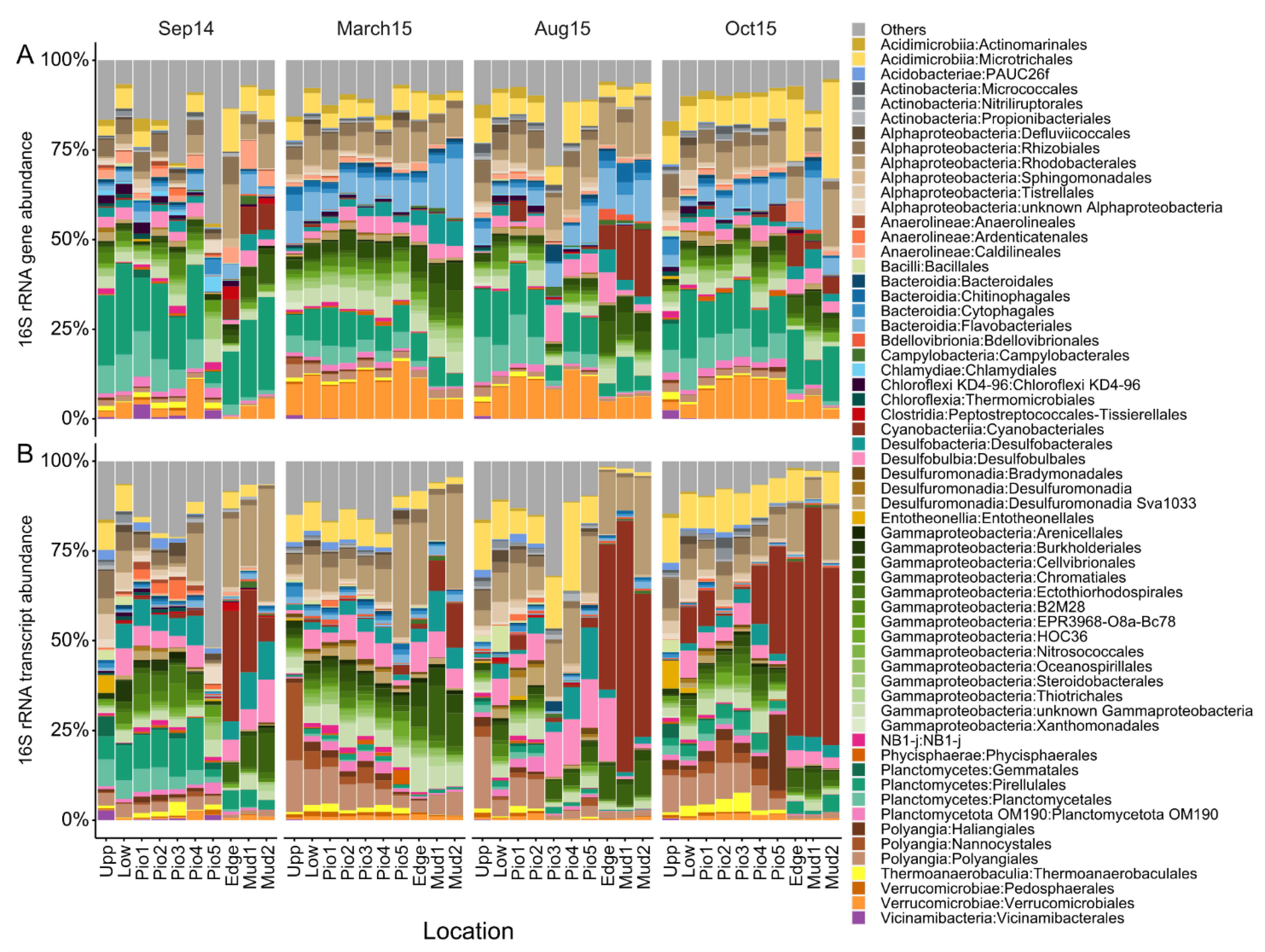
3.4. Bacterial Community Composition Shifts Are Driven by Specific Taxa
3.5. Functional Gene Prediction Resolves Zonal Variations
4. Discussion
4.1. Composition of Salt Marsh Mircrobial Communities Changes on Spatial and Seasonal Scales
4.2. Cyanobacteria and Verrucomicrobiae Exhibit Different Response Strategies to Changing Environmental Conditions
4.3. Inter- and Intrazonal Successions of the Bacterial Communities along the Salt Marsh Transect
4.4. Predicted Gene Abundances Show Gradual Changes in Nitrogen and Sulfur Cycling Potentials
4.5. Ecological Considerations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alongi, D.M. Carbon Balance in Salt Marsh and Mangrove Ecosystems: A Global Synthesis. J. Mar. Sci. Eng. 2020, 8, 767. [Google Scholar] [CrossRef]
- Gedan, K.B.; Silliman, B.R.; Bertness, M.D. Centuries of human-driven change in salt marsh ecosystems. Ann. Rev. Mar. Sci. 2009, 1, 117–141. [Google Scholar] [CrossRef] [PubMed]
- Deegan, L.A.; Johnson, D.S.; Warren, R.S.; Peterson, B.J.; Fleeger, J.W.; Fagherazzi, S.; Wollheim, W.M. Coastal eutrophication as a driver of salt marsh loss. Nature 2012, 490, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Silliman, B.R.; Bertness, M.D. A trophic cascade regulates salt marsh primary production. Proc. Natl. Acad. Sci. USA 2002, 99, 10500–10505. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.T.; Sundberg, K.; Hopkinson, C.S. Salt Marsh Primary Production and Its Responses to Relative Sea Level and Nutrients in Estuaries at Plum Island, Massachusetts, and North Inlet, South Carolina, USA. Oceanography 2013, 26, 78–84. [Google Scholar] [CrossRef]
- Koretsky, C.M.; Van Cappellen, P.; DiChristina, T.J.; Kostka, J.E.; Lowe, K.L.; Moore, C.M.; Roychoudhury, A.N.; Viollier, E. Salt marsh pore water geochemistry does not correlate with microbial community structure. Estuar. Coast. Shelf Sci. 2005, 62, 233–251. [Google Scholar] [CrossRef]
- Lu, Q.Q.; Bai, J.H.; Zhang, G.L.; Zhao, Q.Q.; Wu, J.J. Spatial and seasonal distribution of carbon, nitrogen, phosphorus, and sulfur and their ecological stoichiometry in wetland soils along a water and salt gradient in the Yellow River Delta, China. Phys. Chem. Earth 2018, 104, 9–17. [Google Scholar] [CrossRef]
- McKew, B.A.; Taylor, J.D.; McGenity, T.J.; Underwood, G.J. Resistance and resilience of benthic biofilm communities from a temperate saltmarsh to desiccation and rewetting. ISME J 2011, 5, 30–41. [Google Scholar] [CrossRef]
- Silvestri, S.; Defina, A.; Marani, M. Tidal regime, salinity and salt marsh plant zonation. Estuar. Coast. Shelf Sci. 2005, 62, 119–130. [Google Scholar] [CrossRef]
- Correll, M.D.; Hantson, W.; Hodgman, T.P.; Cline, B.B.; Elphick, C.S.; Gregory Shriver, W.; Tymkiw, E.L.; Olsen, B.J. Fine-Scale Mapping of Coastal Plant Communities in the Northeastern USA. Wetlands 2018, 39, 17–28. [Google Scholar] [CrossRef]
- Janousek, C.N.; Thorne, K.M.; Takekawa, J.Y. Vertical Zonation and Niche Breadth of Tidal Marsh Plants Along the Northeast Pacific Coast. Estuaries Coasts 2019, 42, 85–98. [Google Scholar] [CrossRef]
- Balke, T.; Lohmus, K.; Hillebrand, H.; Zielinski, O.; Haynert, K.; Meier, D.; Hodapp, D.; Minden, V.; Kleyer, M. Experimental salt marsh islands: A model system for novel metacommunity experiments. Estuar. Coast. Shelf Sci. 2017, 198, 288–298. [Google Scholar] [CrossRef]
- Balke, T.; Stock, M.; Jensen, K.; Bouma, T.J.; Kleyer, M. A global analysis of the seaward salt marsh extent: The importance of tidal range. Water Resour. Res. 2016, 52, 3775–3786. [Google Scholar] [CrossRef]
- Sala, N.M.; Bertness, M.D.; Silliman, B.R. The dynamics of bottom-up and top-down control in a New England salt marsh. Oikos 2008, 117, 1050–1056. [Google Scholar] [CrossRef]
- Altieri, A.H.; Bertness, M.D.; Coverdale, T.C.; Herrmann, N.C.; Angelini, C. A trophic cascade triggers collapse of a salt-marsh ecosystem with intensive recreational fishing. Ecology 2012, 93, 1402–1410. [Google Scholar] [CrossRef]
- Bouchard, V.; Lefeuvre, J.C. Primary production and macro-detritus dynamics in a European salt marsh: Carbon and nitrogen budgets. Aquat. Bot. 2000, 67, 23–42. [Google Scholar] [CrossRef]
- De Borger, E.; Braeckman, U.; Soetaert, K. Rapid organic matter cycling in North Sea sediments. Cont. Shelf Res. 2021, 214, 104327. [Google Scholar] [CrossRef]
- Luo, M.; Huang, J.F.; Zhu, W.F.; Tong, C. Impacts of increasing salinity and inundation on rates and pathways of organic carbon mineralization in tidal wetlands: A review. Hydrobiologia 2019, 827, 31–49. [Google Scholar] [CrossRef]
- Li, Z.; Tian, D.; Wang, B.; Wang, J.; Wang, S.; Chen, H.Y.H.; Xu, X.; Wang, C.; He, N.; Niu, S. Microbes drive global soil nitrogen mineralization and availability. Glob. Chang. Biol. 2019, 25, 1078–1088. [Google Scholar] [CrossRef]
- Tilman, D.; Isbell, F.; Cowles, J.M. Biodiversity and Ecosystem Functioning. Annu. Rev. Ecol. Evol. Syst. 2014, 45, 471–493. [Google Scholar] [CrossRef]
- Froelich, P.N.; Klinkhammer, G.P.; Bender, M.L.; Luedtke, N.A.; Heath, G.R.; Cullen, D.; Dauphin, P.; Hammond, D.; Hartman, B.; Maynard, V. Early Oxidation of Organic-Matter in Pelagic Sediments of the Eastern Equatorial Atlantic—Suboxic Diagenesis. Geochim. Cosmochim. Acta 1979, 43, 1075–1090. [Google Scholar] [CrossRef]
- Angermeyer, A.; Crosby, S.C.; Huber, J.A. Decoupled distance-decay patterns between dsrA and 16S rRNA genes among salt marsh sulfate-reducing bacteria. Environ. Microbiol. 2016, 18, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.S.; Liu, C.; Paterson, A.T.; Anderson, L.C.; Turner, R.E.; Overton, E.B. Salt Marsh Bacterial Communities before and after the Deepwater Horizon Oil Spill. Appl. Environ. Microbiol. 2017, 83, e00784-17. [Google Scholar] [CrossRef] [PubMed]
- Pennings, S.C.; Stanton, L.E.; Brewer, J.S. Nutrient effects on the composition of salt marsh plant communities along the southern Atlantic and Gulf coasts of the United States. Estuaries 2002, 25, 1164–1173. [Google Scholar] [CrossRef]
- Angermeyer, A.; Crosby, S.C.; Huber, J.A. Salt marsh sediment bacterial communities maintain original population structure after transplantation across a latitudinal gradient. PeerJ 2018, 6, e4735. [Google Scholar] [CrossRef]
- Hernandez, E.G.; Baraza, E.; Smit, C.; Berg, M.P.; Falcao Salles, J. Salt Marsh Elevation Drives Root Microbial Composition of the Native Invasive Grass Elytrigia atherica. Microorganisms 2020, 8, 1619. [Google Scholar] [CrossRef]
- Garcia Hernandez, E.; Berg, M.P.; Van Oosten, A.R.; Smit, C.; Falcao Salles, J. Linking Bacterial Communities Associated with the Environment and the Ecosystem Engineer Orchestia gammarellus at Contrasting Salt Marsh Elevations. Microb. Ecol. 2021, 82, 537–548. [Google Scholar] [CrossRef]
- Dinter, T.; Geihser, S.; Gube, M.; Daniel, R.; Kuzyakov, Y. Impact of sea level change on coastal soil organic matter, priming effects and prokaryotic community assembly. FEMS Microbiol. Ecol. 2019, 95, fiz129. [Google Scholar] [CrossRef]
- Yao, Z.; Du, S.; Liang, C.; Zhao, Y.; Dini-Andreote, F.; Wang, K.; Zhang, D. Bacterial Community Assembly in a Typical Estuarine Marsh with Multiple Environmental Gradients. Appl. Environ. Microbiol. 2019, 85. [Google Scholar] [CrossRef]
- Neubauer, S.C.; Piehler, M.F.; Smyth, A.R.; Franklin, R.B. Saltwater Intrusion Modifies Microbial Community Structure and Decreases Denitrification in Tidal Freshwater Marshes. Ecosystems 2018, 22, 912–928. [Google Scholar] [CrossRef]
- Dang, C.; Morrissey, E.M.; Neubauer, S.C.; Franklin, R.B. Novel microbial community composition and carbon biogeochemistry emerge over time following saltwater intrusion in wetlands. Glob. Chang. Biol. 2019, 25, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Dini-Andreote, F.; de Cassia Pereira e Silva, M.; Triado-Margarit, X.; Casamayor, E.O.; van Elsas, J.D.; Salles, J.F. Dynamics of bacterial community succession in a salt marsh chronosequence: Evidences for temporal niche partitioning. ISME J. 2014, 8, 1989–2001. [Google Scholar] [CrossRef] [PubMed]
- Graul, C. LeafletR: Interactive Web-Maps Based on the Leaflet JavaScript Library; R Package Version 0.4-0; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Esri, D. GeoEye, i-cubed, USDA FSA, USGS, AEX, Getmapping, Aerogrid, IGN, IGP, swisstopo, GIS-Anwender-Community. Available online: https://www.arcgis.com/home/item.html?id=10df2279f9684e4a9f6a7f08febac2a9 (accessed on 6 October 2021).
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic. Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Herlemann, D.P.; Labrenz, M.; Jurgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Yeh, Y.-C.; McNichol, J.C.; Needham, D.M.; Fichot, E.B.; Fuhrman, J.A. Comprehensive single-PCR 16S and 18S rRNA community analysis validated with 2 mock communities and denoising algorithms. bioRxiv 2019. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic. Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- R-Core-Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.; Simpson, G.; Solymos, P.; et al. Vegan: Community Ecology Package; R Package Version 2.5-6. 2019; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Jost, L. Partitioning diversity into independent alpha and beta components. Ecology 2007, 88, 2427–2439. [Google Scholar] [CrossRef]
- Martinez Arbizu, P. Pairwise Adonis: Pairwise Multilevel Comparison Using Adonis; R Package Version 0.4; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T.; Wemheuer, B. Tax4Fun2: Prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ. Microbiome 2020, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Chuvochina, M.; Adame, M.F.; Guyot, A.; Lovelock, C.; Lockington, D.; Gamboa-Cutz, J.N.; Dennis, P.G. Drivers of bacterial diversity along a natural transect from freshwater to saline subtropical wetlands. Sci. Total Environ. 2021, 759, 143455. [Google Scholar] [CrossRef] [PubMed]
- Martiny, J.B.; Eisen, J.A.; Penn, K.; Allison, S.D.; Horner-Devine, M.C. Drivers of bacterial beta-diversity depend on spatial scale. Proc. Natl. Acad. Sci. USA 2011, 108, 7850–7854. [Google Scholar] [CrossRef]
- Barranguet, C.; Kromkamp, J.; Peene, J. Factors controlling primary production and photosynthetic characteristics of intertidal microphytobenthos. Mar. Ecol. Prog. Ser. 1998, 173, 117–126. [Google Scholar] [CrossRef]
- Watermann, F.; Hillebrand, H.; Gerdes, G.; Krumbein, W.E.; Sommer, U. Competition between benthic cyanobacteria and diatoms as influenced by different grain sizes and temperatures. Mar. Ecol. Prog. Ser. 1999, 187, 77–87. [Google Scholar] [CrossRef]
- Binder, B.J.; Liu, Y.C. Growth rate regulation of rRNA content of a marine synechococcus (Cyanobacterium) strain. Appl. Environ. Microbiol. 1998, 64, 3346–3351. [Google Scholar] [CrossRef]
- Blazewicz, S.J.; Barnard, R.L.; Daly, R.A.; Firestone, M.K. Evaluating rRNA as an indicator of microbial activity in environmental communities: Limitations and uses. ISME J. 2013, 7, 2061–2068. [Google Scholar] [CrossRef]
- White, R.A., 3rd; Bottos, E.M.; Roy Chowdhury, T.; Zucker, J.D.; Brislawn, C.J.; Nicora, C.D.; Fansler, S.J.; Glaesemann, K.R.; Glass, K.; Jansson, J.K. Moleculo Long-Read Sequencing Facilitates Assembly and Genomic Binning from Complex Soil Metagenomes. mSystems 2016, 1, e00045-16. [Google Scholar] [CrossRef]
- Franklin, R.B.; Blum, L.K.; McComb, A.C.; Mills, A.L. A geostatistical analysis of small-scale spatial variability in bacterial abundance and community structure in salt marsh creek bank sediments. FEMS Microbiol. Ecol. 2002, 42, 71–80. [Google Scholar] [CrossRef][Green Version]
- Moffett, K.B.; Robinson, D.A.; Gorelick, S.M. Relationship of Salt Marsh Vegetation Zonation to Spatial Patterns in Soil Moisture, Salinity, and Topography. Ecosystems 2010, 13, 1287–1302. [Google Scholar] [CrossRef]
- van Regteren, M.; Amptmeijer, D.; de Groot, A.V.; Baptist, M.J.; Elschot, K. Where does the salt marsh start? Field-based evidence for the lack of a transitional area between a gradually sloping intertidal flat and salt marsh. Estuar. Coast. Shelf Sci. 2020, 243, 106909. [Google Scholar] [CrossRef]
- Kolton, M.; Rolando, J.L.; Kostka, J.E. Elucidation of the rhizosphere microbiome linked to Spartina alterniflora phenotype in a salt marsh on Skidaway Island, Georgia, USA. FEMS Microbiol. Ecol. 2020, 96, fiaa026. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Liu, W.; Zhang, M.; Lin, X.; Zhang, Y.; Tian, Y. Different Height Forms of Spartina alterniflora Might Select Their Own Rhizospheric Bacterial Communities in Southern Coast of China. Microb. Ecol. 2019, 77, 124–135. [Google Scholar] [CrossRef]
- Czapla, K.M.; Anderson, I.C.; Currin, C.A. The Effect of Fertilization on Biomass and Metabolism in North Carolina Salt Marshes: Modulated by Location-Specific Factors. J. Geophys. Res. Biogeosci. 2020, 125. [Google Scholar] [CrossRef]
- McFarlin, C.R.; Brewer, J.S.; Buck, T.L.; Pennings, S.C. Impact of fertilization on a salt marsh food web in Georgia. Estuaries Coasts 2008, 31, 313–325. [Google Scholar] [CrossRef]
- Anisfeld, S.C.; Hill, T.D. Fertilization Effects on Elevation Change and Belowground Carbon Balance in a Long Island Sound Tidal Marsh. Estuaries Coasts 2011, 35, 201–211. [Google Scholar] [CrossRef]
- Darby, F.A.; Turner, R.E. Effects of eutrophication on salt marsh root and rhizome biomass accumulation. Mar. Ecol. Prog. Ser. 2008, 363, 63–70. [Google Scholar] [CrossRef]
- Weller, D.E.; Jordan, T.E. Inexpensive spot sampling provides unexpectedly effective indicators of watershed nitrogen status. Ecosphere 2020, 11, e03224. [Google Scholar] [CrossRef]
- Johnson, D.S.; Warren, R.S.; Deegan, L.A.; Mozdzer, T.J. Saltmarsh plant responses to eutrophication. Ecol. Appl. 2016, 26, 2647–2659. [Google Scholar] [CrossRef]
- Bowen, J.L.; Giblin, A.E.; Murphy, A.E.; Bulseco, A.N.; Deegan, L.A.; Johnson, D.S.; Nelson, J.A.; Mozdzer, T.J.; Sullivan, H.L. Not All Nitrogen Is Created Equal: Differential Effects of Nitrate and Ammonium Enrichment in Coastal Wetlands. Bioscience 2020, 70, 1108–1119. [Google Scholar] [CrossRef]
- Murphy, A.E.; Bulseco, A.N.; Ackerman, R.; Vineis, J.H.; Bowen, J.L. Sulphide addition favours respiratory ammonification (DNRA) over complete denitrification and alters the active microbial community in salt marsh sediments. Environ. Microbiol. 2020, 22, 2124–2139. [Google Scholar] [CrossRef] [PubMed]
- Hey, D.L.; Kostel, J.A.; Crumpton, W.G.; Mitsch, W.J.; Scott, B. The roles and benefits of wetlands in managing reactive nitrogen. J. Soil Water Conserv. 2012, 67, 47a–53a. [Google Scholar] [CrossRef]
- Ledford, T.C.; Mortazavi, B.; Tatariw, C.; Starr, S.F.; Smyth, E.; Wood, A.G.; Simpson, L.T.; Cherry, J.A. Ecosystem carbon exchange and nitrogen removal rates in two 33-year-old constructed salt marshes are similar to those in a nearby natural marsh. Restor. Ecol. 2021, 29, e13439. [Google Scholar] [CrossRef]
- Bashagaluke, J.B.; Logah, V.; Opoku, A.; Sarkodie-Addo, J.; Quansah, C. Soil nutrient loss through erosion: Impact of different cropping systems and soil amendments in Ghana. PLoS ONE 2018, 13, e0208250. [Google Scholar] [CrossRef] [PubMed]
- Arkema, K.K.; Guannel, G.; Verutes, G.; Wood, S.A.; Guerry, A.; Ruckelshaus, M.; Kareiva, P.; Lacayo, M.; Silver, J.M. Coastal habitats shield people and property from sea-level rise and storms. Nat. Clim. Chang. 2013, 3, 913–918. [Google Scholar] [CrossRef]
- Howarth, R.W.; Marino, R. Nitrogen as the limiting nutrient for eutrophication in coastal marine ecosystems: Evolving views over three decades. Limnol. Oceanogr. 2006, 51, 364–376. [Google Scholar] [CrossRef]
- Teeling, H.; Fuchs, B.M.; Becher, D.; Klockow, C.; Gardebrecht, A.; Bennke, C.M.; Kassabgy, M.; Huang, S.; Mann, A.J.; Waldmann, J.; et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 2012, 336, 608–611. [Google Scholar] [CrossRef]
- Andersen, J.H.; Conley, D.J. Eutrophication in coastal marine ecosystems: Towards better understanding and management strategies. In Eutrophication in Coastal Ecosystems; Springer: Dordrecht, The Netherland, 2009; pp. 1–4. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tebbe, D.A.; Geihser, S.; Wemheuer, B.; Daniel, R.; Schäfer, H.; Engelen, B. Seasonal and Zonal Succession of Bacterial Communities in North Sea Salt Marsh Sediments. Microorganisms 2022, 10, 859. https://doi.org/10.3390/microorganisms10050859
Tebbe DA, Geihser S, Wemheuer B, Daniel R, Schäfer H, Engelen B. Seasonal and Zonal Succession of Bacterial Communities in North Sea Salt Marsh Sediments. Microorganisms. 2022; 10(5):859. https://doi.org/10.3390/microorganisms10050859
Chicago/Turabian StyleTebbe, Dennis Alexander, Simone Geihser, Bernd Wemheuer, Rolf Daniel, Hendrik Schäfer, and Bert Engelen. 2022. "Seasonal and Zonal Succession of Bacterial Communities in North Sea Salt Marsh Sediments" Microorganisms 10, no. 5: 859. https://doi.org/10.3390/microorganisms10050859
APA StyleTebbe, D. A., Geihser, S., Wemheuer, B., Daniel, R., Schäfer, H., & Engelen, B. (2022). Seasonal and Zonal Succession of Bacterial Communities in North Sea Salt Marsh Sediments. Microorganisms, 10(5), 859. https://doi.org/10.3390/microorganisms10050859