Abstract
The ongoing Coronavirus Disease 19 (COVID-19) pandemic has had a profound impact on the global healthcare system. As the SARS-CoV-2 virus, responsible for this pandemic, continues to spread and develop mutations in its genetic material, new variants of interest (VOIs) and variants of concern (VOCs) are emerging. These outbreaks lead to a decrease in the efficacy of existing treatments such as vaccines or drugs, highlighting the urgency of new therapies for COVID-19. Therefore, in this study, we aimed to identify potential SARS-CoV-2 antivirals using a virtual screening protocol and molecular dynamics simulations. These techniques allowed us to predict the binding affinity of a database of compounds with the virus Mpro protein. This in silico approach enabled us to identify twenty-two chemical structures from a public database (QSAR Toolbox Ver 4.5 ) and ten promising molecules from our in-house database. The latter molecules possess advantageous qualities, such as two-step synthesis, cost-effectiveness, and long-lasting physical and chemical stability. Consequently, these molecules can be considered as promising alternatives to combat emerging SARS-CoV-2 variants.
1. Introduction
At the end of 2019, numerous cases of a mysterious respiratory disease were reported in Wuhan, China [1]. As time passed, the number of cases and deaths increased, and soon enough, the causative agent was discovered to be a novel betacoronavirus, known today as Severe Acute Respiratory Syndrome coronavirus 2 (SARS-CoV-2) [2]. SARS-CoV-2 has spread throughout the world, leading to the global COVID-19 pandemic [3]. As a countermeasure to slow the spread of SARS-CoV-2, several countries decided to implement lockdown measures, which had significant repercussions on public health, the environment, human psychology, the global socioeconomic situation, and education [4]. Despite these measures, the virus continues to spread, and as of 23 June 2024, there are over 775 million confirmed COVID-19 cumulative cases of infection, of which more than 7 million have resulted in death [5].
Coronaviruses (CoVs) are enveloped positive-strand RNA viruses with the largest known RNA genomes of 30–32 kilobases (kb) [6]. Both animals and humans can be infected with these viruses, which raises the importance of keeping track of their zoonotic potential [7]. There are seven known human coronaviruses, four of which are endemic and generally cause mild to moderate upper respiratory tract symptoms [8]. These include 229-CoV, OC43-CoV, HKU1-CoV, and NL63-CoV [8]. The remaining three CoVs are betacoronaviruses known to cause pandemics in the 21st century, including SARS-CoV, MERS-CoV, and SARS-CoV-2 [9]. At the nucleotide level, SARS-CoV-2 shares approximately 79% sequence identity with SARS-CoV and approximately 50% with MERS-CoV, both of which were responsible for coronaviral pandemics in the 20th century [10]. Phylogenetically, since SARS-CoV-2 is a betacoronavirus, it belongs to the order Nidovirales, family Coronaviridae, subfamily Orthocoronavirinae, genus Betacoronavirinae, and subgenus Sarbecovirus [11].
The SARS-CoV-2 genome is composed of different open reading frames (ORFs), of which ORF 1a/b occupies two-thirds; the rest are occupied by other ORFs coding for accessory (i.e., ORF3a, ORF6, ORF7, ORF8, and ORF9) and structural (spike, nucleocapsid, membrane, and envelope) proteins [12]. Accessory proteins such as ORF3a, ORF6, ORF7, and ORF8 have various functions, including type IFN-I antagonism and suppression of viral responses [13]. Structural proteins include surface glycoprotein S, envelope protein E, membrane protein M, and nucleocapsid protein N, which are necessary for virus assembly and infectivity [14]. To invade host cells, SARS-CoV-2 uses its spike protein, which allows it to bind to the host cell receptor angiotensin converting enzyme 2 (ACE2); this binding is followed by a conformational change in the S protein, ultimately facilitating the fusion of the viral envelope with the cell membrane [15]. In this way, SARS-CoV-2 releases its genomic RNA into the host cell, which is directly converted to ppa1a and pp1ab, which, in turn, are proteolytically cleaved, giving rise to 10 and 16 proteins, respectively [16], which form the viral replicase transcriptase complex (RTC) [17]. This cleavage is achieved by the action of viral-encoded proteases known as papain-like proteases (PLpro, protease domain of Nsp3) and chymotrypsin-like (3CLpro) or main protease domains (Mpro) of Nsp5 [17]. Both the latter, along with the spike protein, are important therapeutic targets [18]. PLpro is the largest nonstructural protein encoded by SARS-CoV-2 and cleaves the viral polyprotein pp1ab to produce Nsp1-3 [19]. However, the homodimeric cysteine protease Mpro can proteolytically cleave the pp1ab polyprotein at 11 cleavage sites, which makes it important in the viral replication cycle [20,21]. In its monomeric form, Mpro has a molecular weight of 34.21 kDa and comprises three domains: domain I (residues 8–101), domain II (residues 102–184), and domain III (residues 201–303), where domains I and II have an antiparallel β-barrel structure and domain III contains five α-helices arranged into a largely antiparallel globular cluster [22,23]. The catalytic site of Mpro is located at the intersection of domains I and II, with a catalytic dyad formed by Cys145 and His41. It is important to note that Mpro is less enzymatically active than its dimeric form [22].
Currently, there are many ways to control the COVID-19 pandemic: vaccines [24,25], neutralizing monoclonal antibodies (mAbs) against the spike protein, and the antiviral drugs ritonavir-boosted nirmatrelvir (targeting the Mpro) [26], molnupiravir, and remdesivir (both targeting RNA-dependent RNA polymerase—RdRp) [27,28]. However, as SARS-CoV-2 continues to spread, novel mutations are detected in its genome, resulting in the emergence of variants that have the potential to evade immunity against SARS-CoV-2, thus making existing treatments no longer effective [29,30,31]. This highlights the importance of having an arsenal of antiviral drugs against SARS-CoV-2 that target the different stages of its replicative cycle.
A useful tool for discovering therapeutic targets and drug candidates is computational chemistry. It helps us to understand protein–ligand interactions, using software to explore and calculate molecular properties [32]. For instance, computer-aided drug design (CADD) identified HIV drugs saquinavir and indinavir, which are approved for use in patients. Similarly, computational methods have aided in COVID-19 drug discovery [33,34,35,36].
2. Methods
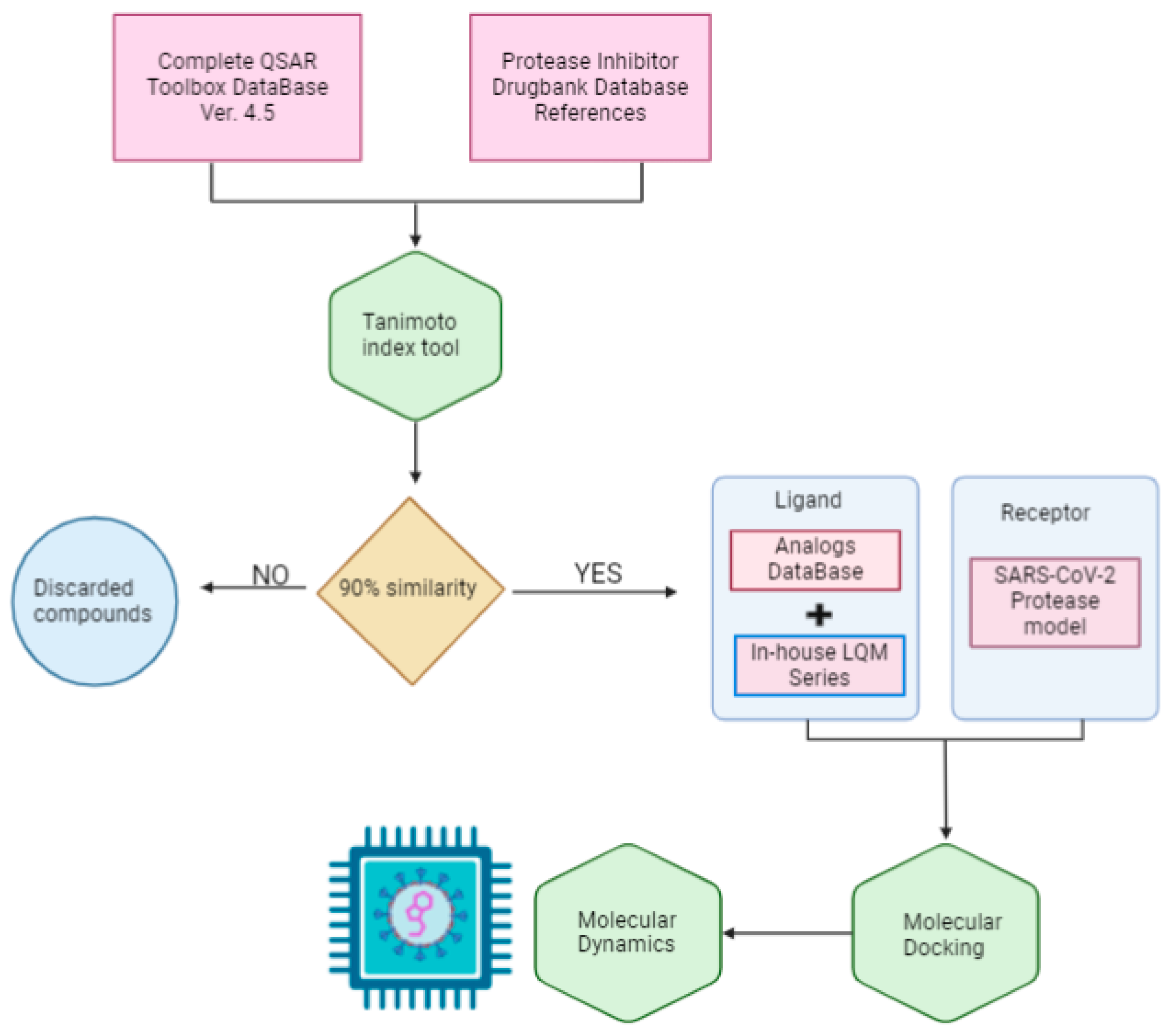
The scope of this research is to identify potential hit molecules and propose a therapeutic strategy aimed at inhibiting the major protease of SARS-CoV-2. Figure 1 shows the computational approach taken in this work to screen and evaluate two sources of molecular structure data: the public QSAR Toolbox database and our internal database. Our in-house collection of chemical compounds has been compiled over 30 years of research and currently contains 250 compounds spanning a wide range of structures, functional groups, and activities, including antiviral, antineoplastic, antihypertensive, antiparasitic, and other properties [37,38].
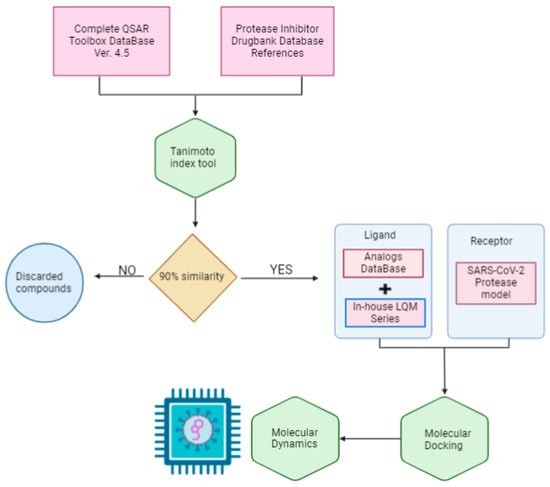
Figure 1.
General workflow diagram.
The QSAR Toolbox database provided access to a wide variety of compounds. From this database, molecules were selected if they had a similarity of more than 90% to previously reported protease inhibitors. The Tanimoto coefficient was used as the decision criterion [39].
To identify these reference molecules, a broad search for protease inhibitors of any type was conducted in the DrugBank database. A total of 25 molecules were obtained and used as reference compounds to calculate the respective similarity coefficients. The resulting compounds were added to the molecules in our internal database to form the joint database.
Although the compounds in the LQM series had been previously studied in different diseases, it is not possible to encompass this research within the traditional concept of drug repositioning, since the QSAR Toolbox database is not specifically made up of molecules tested as drugs. It is important to clarify that the current research serves primarily as a screening process to identify potential hit molecules within this joint database. This approach has the potential to significantly accelerate the discovery of promising candidates for the development of effective drugs against COVID-19.
3. Results and Analysis
3.1. Mpro Structure Selection
SARS-CoV-2 Mpro is an important biological and pharmacological target [40]. Its structural integrity remains remarkably conserved across various coronavirus proteins owing to the high degree of conservation in its amino acid composition. In other words, its protein structure shows minimal mutations, resulting in a consistent conformational behavior [41].
An initial search was conducted within the PDB (Protein Data Bank), where several structures of Mpro were found. These structures are identified by the following codes: 7TOB, 5R81, 6R7Y, 6R83, 5R7Z, 5R82, 5R84, 6LU7, 6M03, 6Y2F, 6Y2G, 6Y8E, and 6YB7. All structures were aligned and superposed and the resulting model is illustrated in Figure 2. This preliminary exploration serves as a focal point for our structure selection. This is based on its status as one of the most up-to-date structures available in the PDB database compared to other proteins at the time [42]. The selection criteria for docking, virtual screening, and molecular dynamics simulations are discussed below.
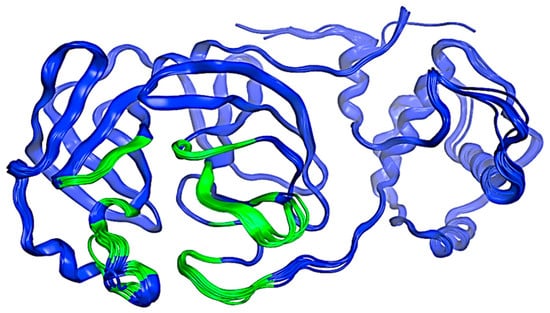
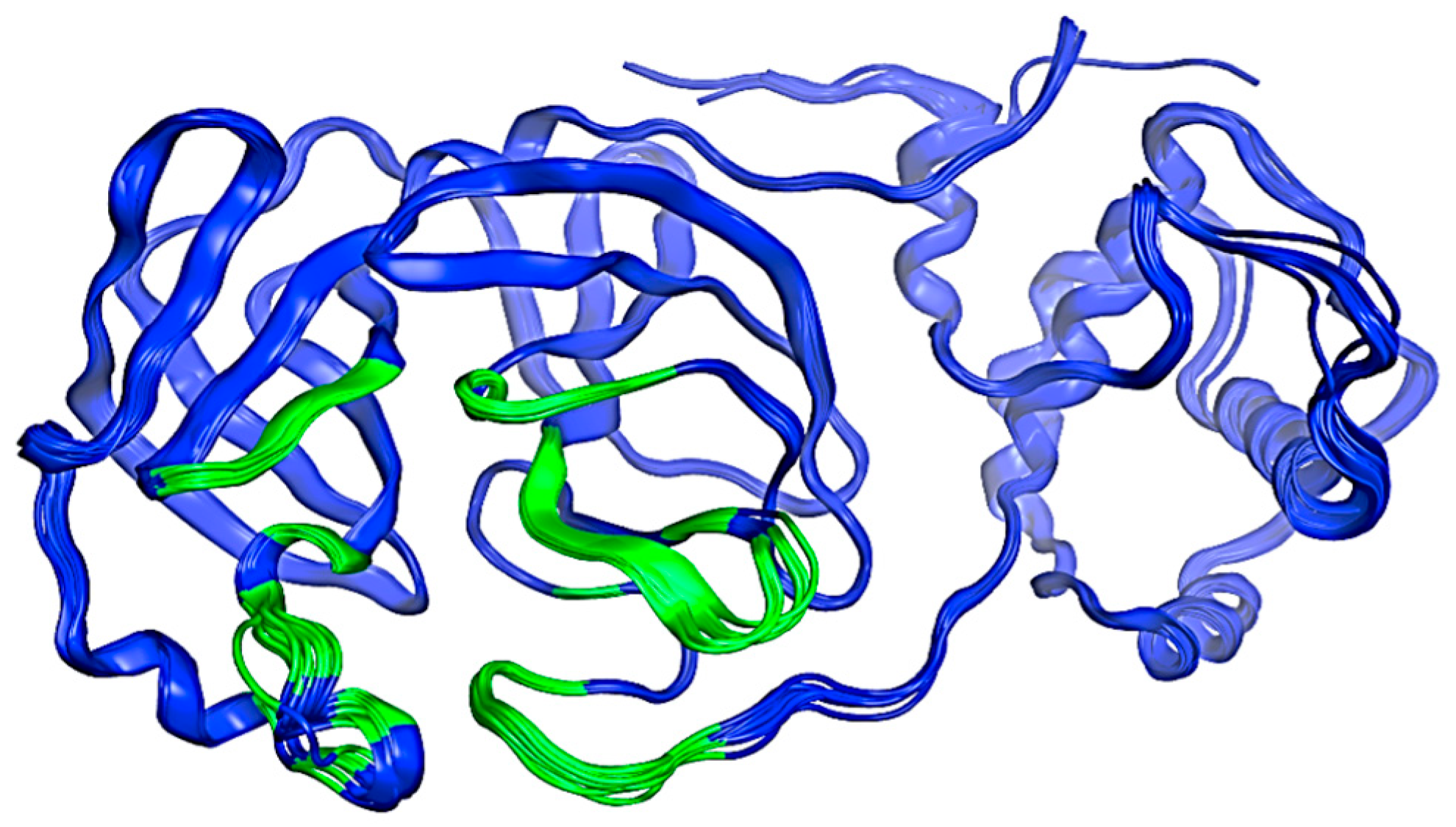
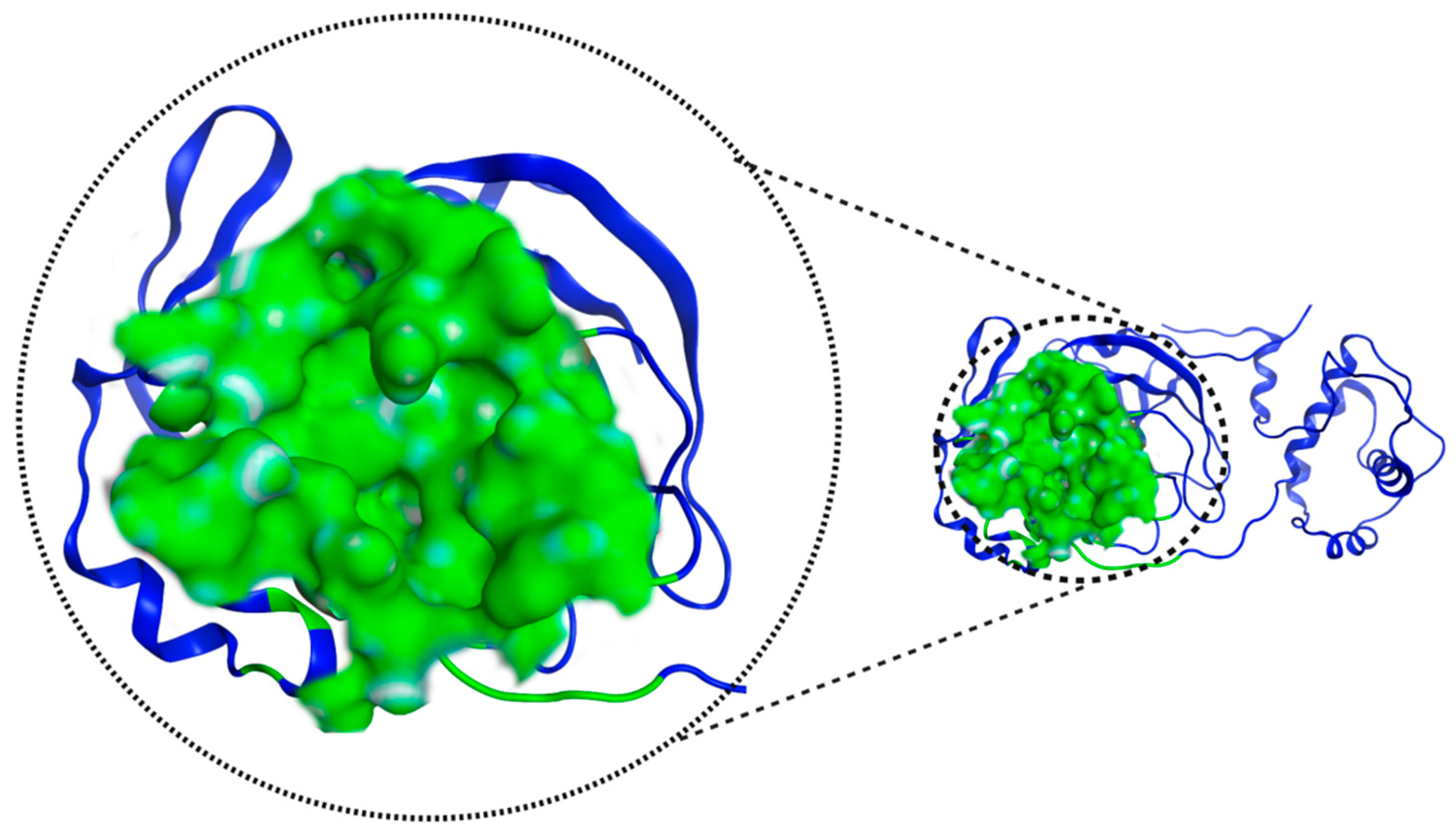
Figure 2.
Structure of the SARS-CoV-2 Mpro protein. The possible interaction sites are colored green, and the rest of the protein is blue. Image created in the Molecular Operating Environment, MOE 2022.02.
To identify potential active sites within the protein, the MOE SiteFinder tool was used [43]. This tool allows the detection of potential active sites in a receptor from its three-dimensional coordinates. It is a geometric method that considers the accessibility of the receptor atoms and a classification by chemical type. It excludes sites exposed to the solvent and classifies the different regions as hydrophobic or hydrophilic. Hydrophobic regions are represented as gray spheres, while hydrophilic regions are represented as red spheres. In Figure 3, the SARS-CoV-2 protein is presented along with the sites showing the highest interaction with the ligands. It is noteworthy that the amino acids His41 and Cys145 form a catalytic dyad [44], making them crucial elements for the subsequent experimental steps.
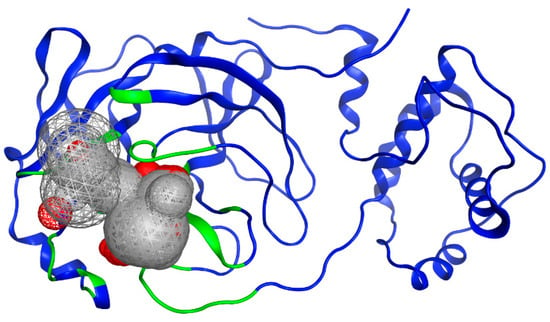
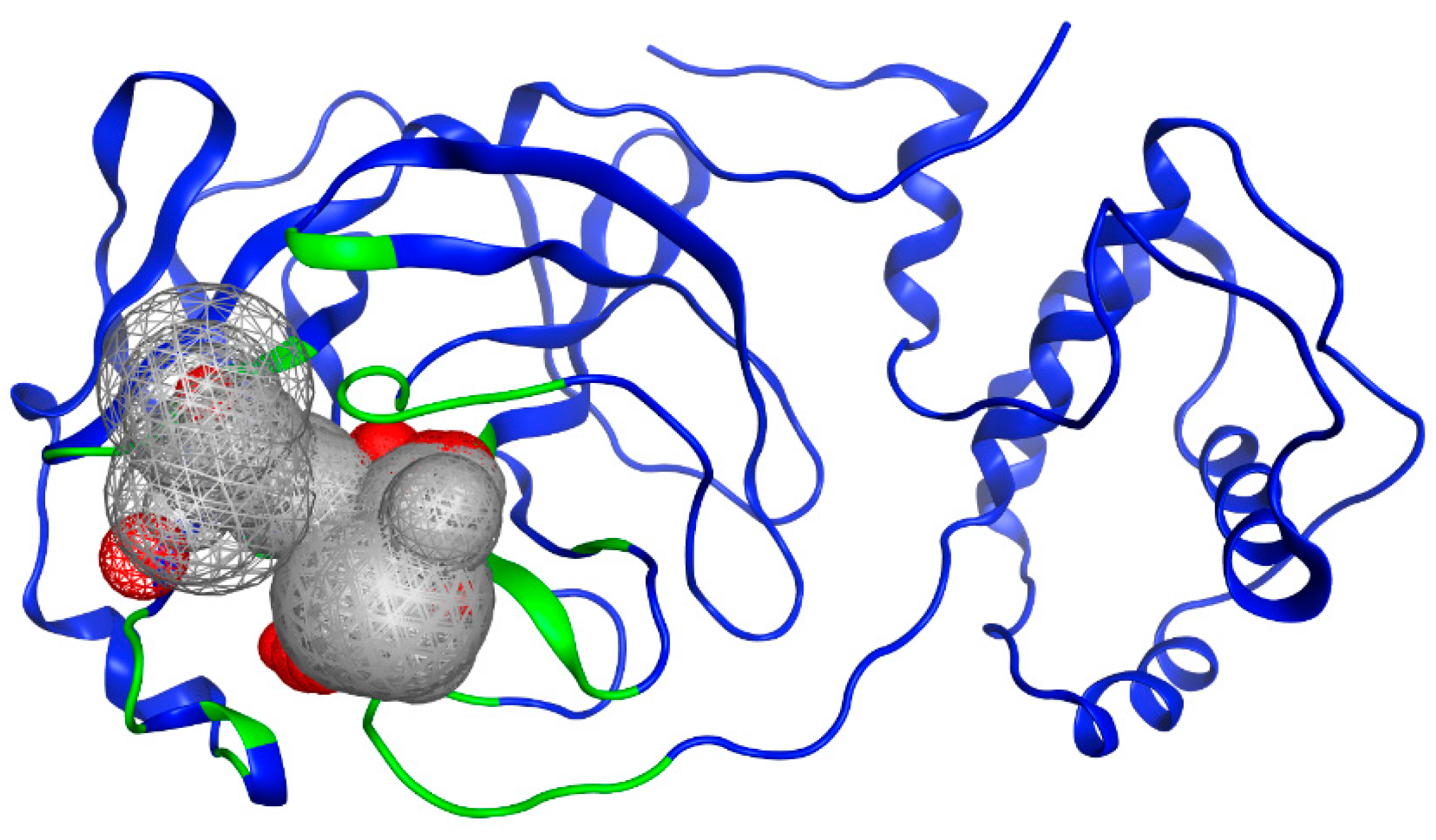
Figure 3.
Potential binding site evaluated by SiteFinder. The size of the sphere represents how tightly packed the atoms are in the receptor; the larger the volume, the more accessible the atoms become. Spheres are colored by hydrophobic (gray) and hydrophilic (red). Image generated in MOE, 2022.02.
To identify potential protein binding sites or targets, ProteinsPlus, an online platform developed by the University of Hamburg, was utilized [45]. This platform primarily focuses on structure-based modeling, with an emphasis on protein-ligand interactions. Its functionalities include predicting protonation and tautomerization, categorizing various interactions in protein-protein complexes, and predicting binding sites and assessing their druggability, among other tools [45].
The approach is based on the concept of “druggability”, and the result of this assessment is a score that evaluates the feasibility of modifying a target using a small-molecule drug. This evaluation is crucial in the progression of a drug discovery project, transitioning from the computational “hit to lead” phase [46]. The druggability is used in drug discovery to elucidate a biological target, and it can be predicted with an affinity for a drug.
We applied the DoGSiteScorer tool, which is specialized in detecting binding sites, relying solely on the three-dimensional structure of the protein to deduce the overall properties of the pocket, including size, shape, and key characteristics. This tool generates a druggability score for each pocket, which is calculated based on a combination of three descriptors: volume, surface, and DrugScore. A higher score on this last parameter indicates a pocket with greater potential and favorability to be the target of a drug. In this context, it is desirable to have values that meet an acceptable threshold. These values range from 0.5 to 1.0, and those closer to 1.0 indicate pockets with strong druggability potential [47].
Table 1 shows the volume, surface, and DrugScore values, allowing us to identify the Mpro structure that is the most suitable for drug targeting based on these three values, thus advancing us to the next phase of this study.

Table 1.
DoGSiteScorer values for different Mpro models.
For each SARS-CoV-2 main protease protein structure, between seven and nine potential binding pockets were identified. According to the results presented in Table 1, the 7TOB structure exhibits a druggability value of 0.81, positioning it as the candidate with the highest druggability potential. The 7TOB model corresponds to the Omicron variant 1.1.529, which is characterized by a highly conserved protein among the Mpro variants [41]. Therefore, we decided to select this protein to advance to the next phase of our project. Virtual screening (VS) studies have been conducted on this main protease structure to identify natural compounds as inhibitors, like theaflavin and ginkgetin [48].
Figure 4 shows the pocket where the catalytic dyad is located and presents an important feature of 7TOB: a volume of 673.22 Å3 and a surface area of 790.0 Å2.
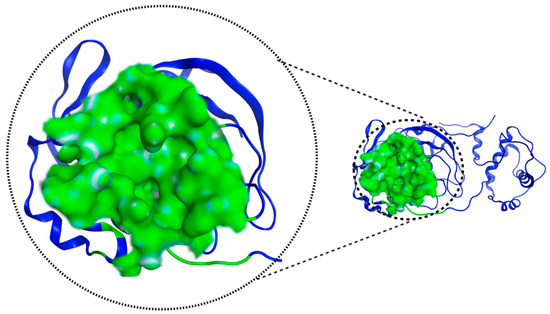
Figure 4.
Mpro 7TOB potential interaction site, in green, defined by “ProteinsPlus” online server.
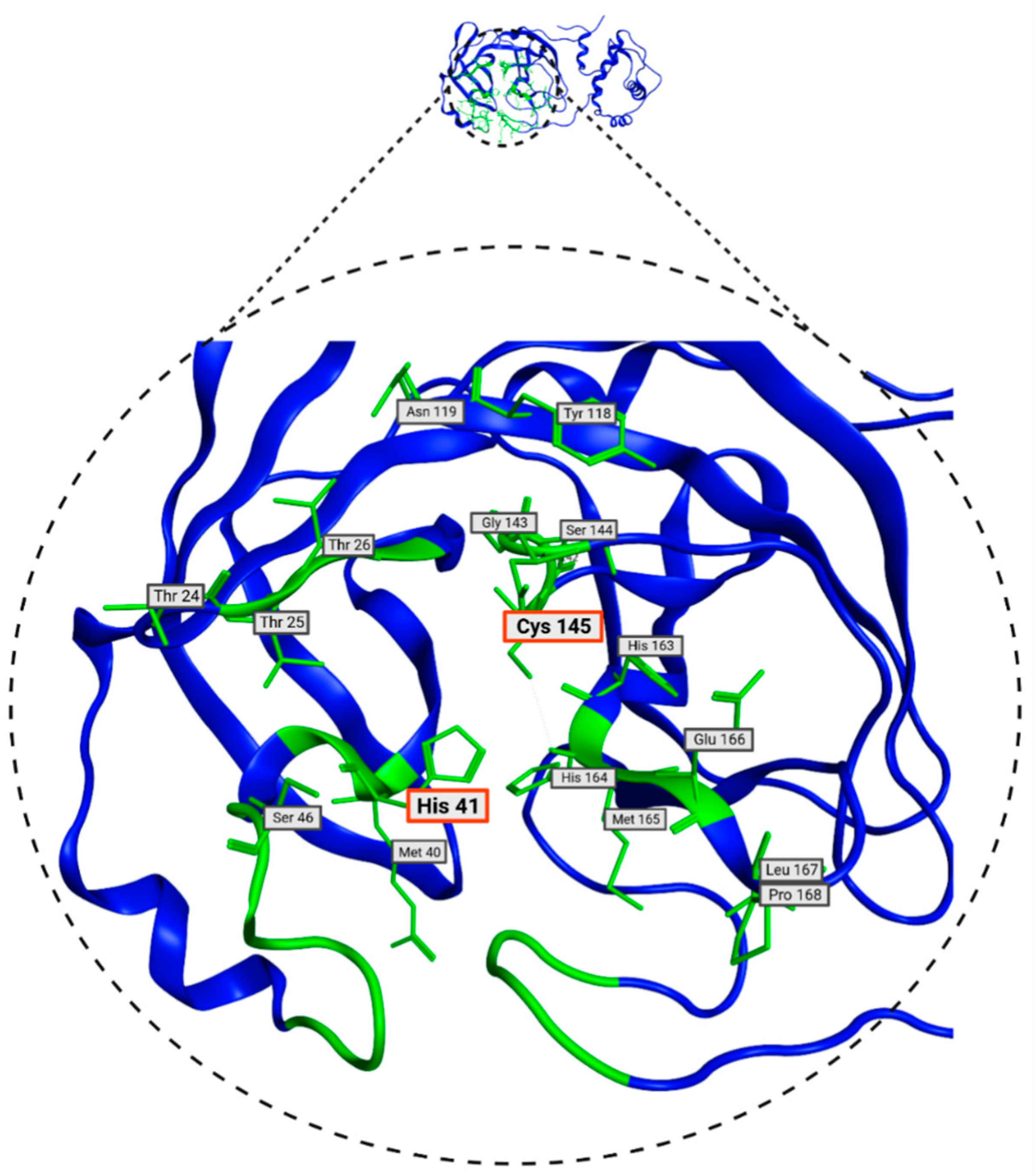
In Table 2, the residues defined by ProteinsPlus (DrugScore), which match with the SiteFinder process, are presented. These residues are the most likely to interact with molecules from the database and will be used in molecular docking. These residues are visualized in Figure 5 and play a crucial role in molecular docking processes. Some of the amino acids in the Mpro protein have already been identified as relevant in the literature. These are the His41 and Cys145 residues, which, as mentioned above, form a catalytic dyad and can inhibit protein activity [22]. Given the presence of these two residues, they prove to be a crucial part of inhibitor design [44].
Table 2.
List of amino acids that make up the pocket of SARS-CoV-2 Mpro (7TOB).
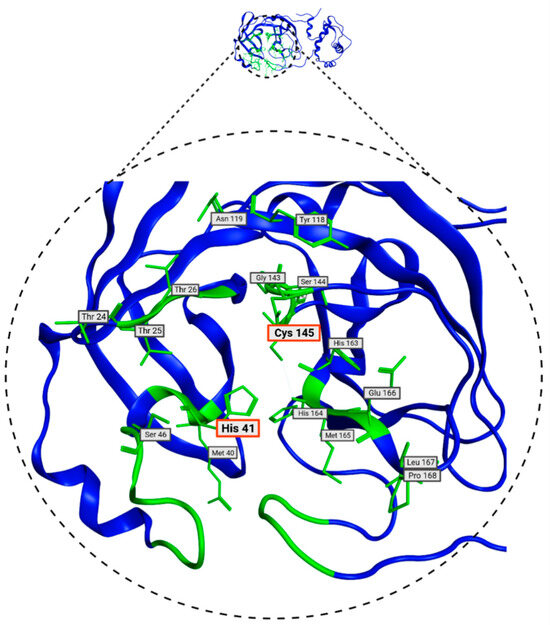
Figure 5.
Consensus amino acids belonging to Mpro 7TOB site identified with DoGSiteScorer and SiteFinder from MOE 2022 tools.
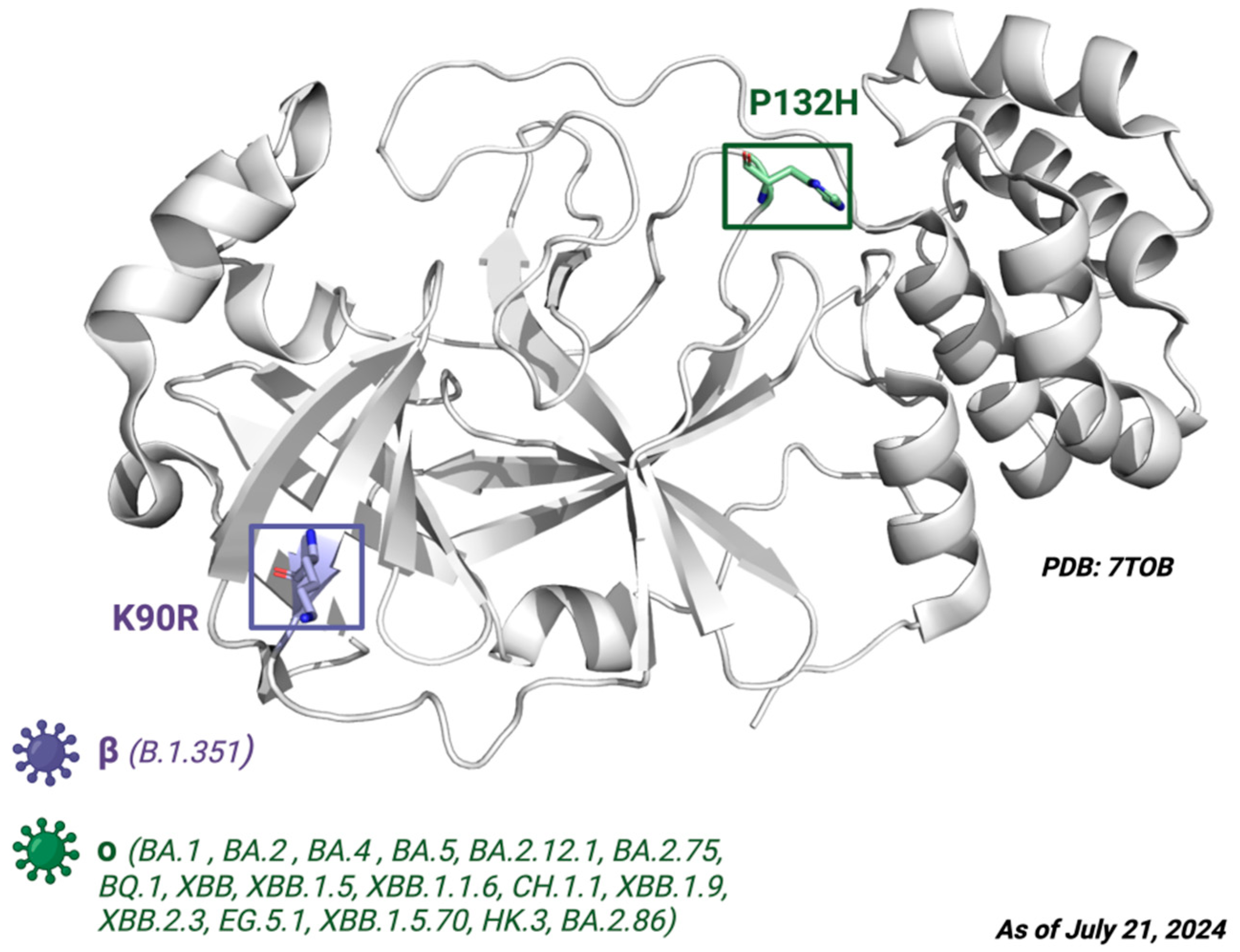
Next, the mutations defining the variants of interest (VOIs) such as B.1.1.7, B.1.351, P, B.1.617.2, BA.1, BA.2, BA.4, BA.5, BA.2.12.1, BA.2.75, BQ.1, XB, XBB.1.5, XBB.1.1.6, CH.1.1, XBB.1.9, XBB.2.3, EG.5.1, XBB.1.5.70, HK.3, and BA.2.86 were examined. ORF1ab substitutions shown in the CoVariants online platform [49] were converted to their respective amino acid sequence changes in Nsp5, then BLAST (Basic Local Alignment Search Tool, NIH) was used to align the polyprotein 1ab sequences. From this analysis, none of the VOIs had mutations in the target sites of the Mpro ligand. The only defining amino acid substitutions found were K90R for the beta variant B.1.351 and P132H for the Omicron variants BA.1, BA.2, BA.4, BA.5, BA.2.12.1, BA.2.75, BQ.1, XBB, XBB.1.5, XBB.1.1.6, CH.1.1, XBB.1.9, XBB.2.3, EG.5.1, XBB.1.5.70, HK.3, and BA.2.86 (Figure 6).
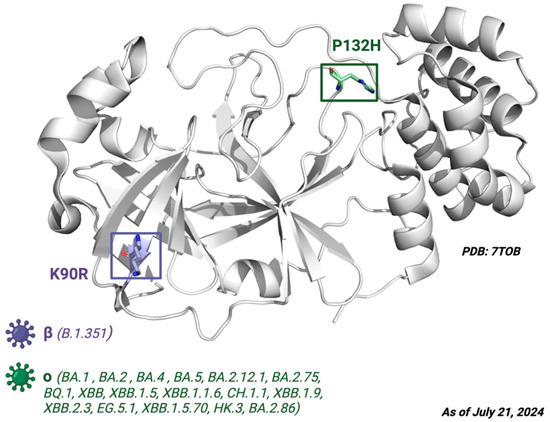
Figure 6.
The crystal structure of Mpro. Depiction of the Mpro amino acid substitutions in the SARS-CoV-2 variants of concern. The purple residue represents the K90R substitution present in the beta B.1.351 variant, whereas the green residue represents the P132H substitution present in the Omicron variants BA.1, BA.2, BA.4, BA.5, BA.2.12.1, BA.2.75, BQ.1, and XBB. Defining amino acid changes are those that appear at the phylogenetic root of a variant. Figure made with BioRender with a purchased license.
3.2. Selection of Potential Inhibitors of SARS-CoV-2 Mpro
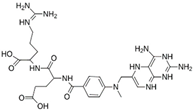
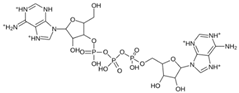
Once the Mpro structure and its corresponding binding site were defined, we proceeded to screen the quantitative structure–activity relationship (QSAR) Toolbox molecular database (QSAR Analytics S.A., Mexico), which contains 311,750 substances with various biological activities. We filtered the database using molecular similarity virtual screening using the Tanimoto coefficient as the decision criterion. We used twenty-five chemical structures of commercial antivirals previously identified as protease inhibitors, obtained from DrugBank, as references to determine compounds with high similarity with the QSAR Toolbox database, as shown in Table 3.
Table 3.
Count of compounds filtered by Tanimoto coefficient and their protease inhibitor reference.
Atom-centered fragments together with saturated and aromatic cycles and incident pi bond (π bond) similarity variants were employed due to the structural characteristics of the antiviral compounds. We exclusively selected chemical structures that exhibited a similarity equal to or greater than 90% with respect to each of the antivirals used as references, resulting in a total of 2565 identified molecules. The selected molecules, sourced from DrugBank, share the common feature of being protease inhibitors and were considered due to their potential impact on the SARS-CoV-2 Mpro protein. As a result, 2565 molecules were obtained, as shown in Table 3.
We then proceeded to filter the compounds from our joint database (public + in-house) by virtual screening. The three-dimensional model of the Mpro 7TOB protein of SARS-CoV-2 was used as the receptor and the total of 2565 (QSAR Toolbox) and 250 (LQM series) compounds as ligands.
Virtual screening was performed in duplicate using two docking engines (AutoDock Vina and MOE) as consensus to compare the trend of the calculated scores [43,74,75]. Ensitrelvir (S-217622) and Atazanavir (BMS-232632) molecules were also included in the screening as reference ligands to compare the binding affinity score with the molecules studied here as these ligands have shown in vitro and in vivo activity and are even in clinical trial phases [76].
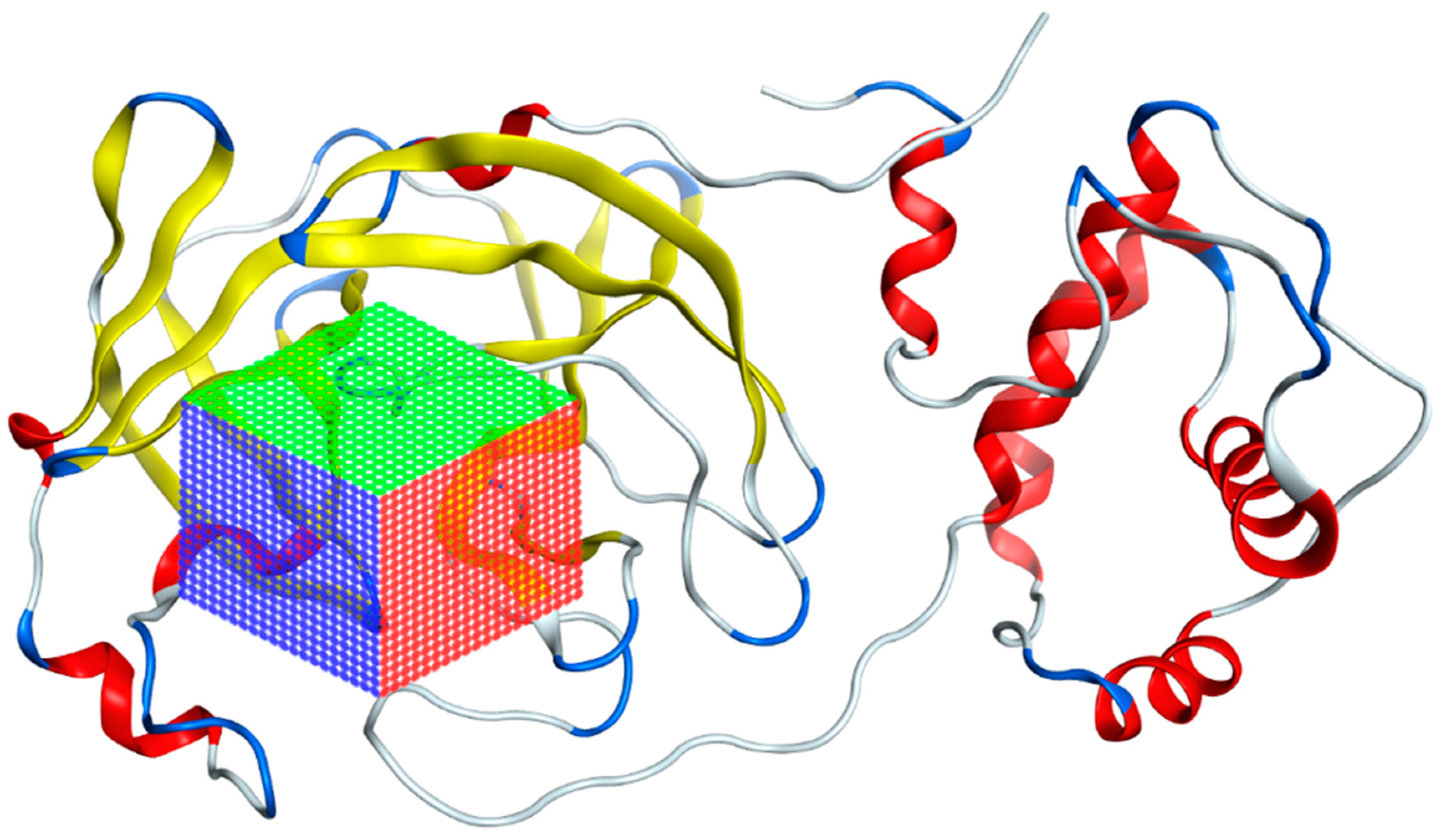
The preparation of AutoDock Vina input files was as follows: Using the AutoDockTools (ADT), the atomic coordinates of the SARS-CoV-2 main protease model were read from the PDB 7TOB file. The corresponding protein protonation was performed; then, the non-polar hydrogens were merged and the corresponding atomic partial Kollman charges were added. The receptor–ligand interaction zone was defined to include the amino acids identified above, generating a search box with the following dimensions: 25.50 Å × 28.05 Å × 25.50 Å (x, y, z) with the center located at an average distance from the atoms of the catalytic dyad (Cys145 and His41). Figure 7 illustrates the search box used for virtual screening. To prepare the ligand files, the PyRx tool was used to convert our compounds to the pdbqt format required for use in AutoDock Vina [77].
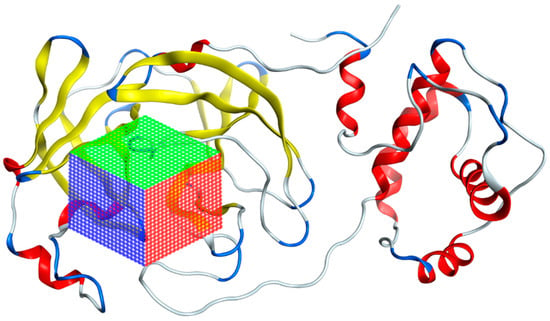
Figure 7.
Grid of the 7TOB protein of SARS-CoV-2 in which the area where interactions with ligands were calculated is presented.
The MOE protocol was as follows: the alpha triangle matching algorithm was used to calculate initial poses for each molecule and, using the London dG scoring function, the 100 poses were obtained. These poses were refined and optimized, keeping the receptor rigid and evaluating with the GBVI/WSA dG scoring function, resulting in a total of 50 pose results for each ligand.
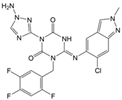
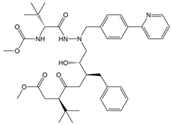
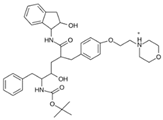
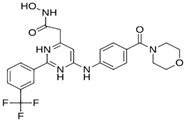
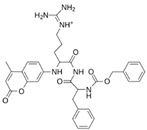
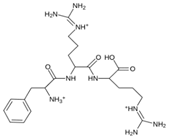
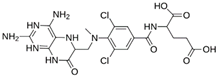
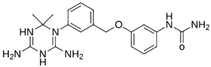
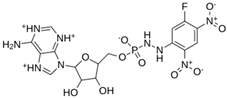
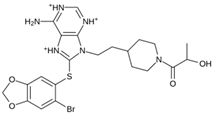
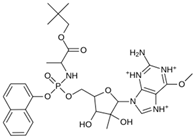
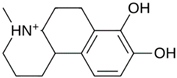
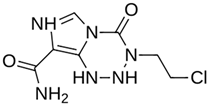
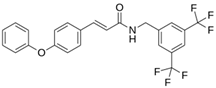
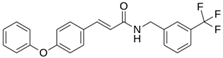
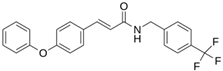
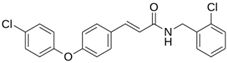
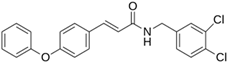
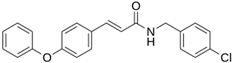
As a result of this filtering process, 22 compounds from the QSAR Toolbox database and 10 compounds from the LQM series were identified and are presented in the Table 4. The score values for each of the ligands obtained by the different docking engines are presented. The ligands with the most favorable interaction energy among the 32 compounds are highlighted: MproL6 and LQM 778.
Table 4.
Predicted score values by AutoDock Vina and MOE for 32 selected compounds.
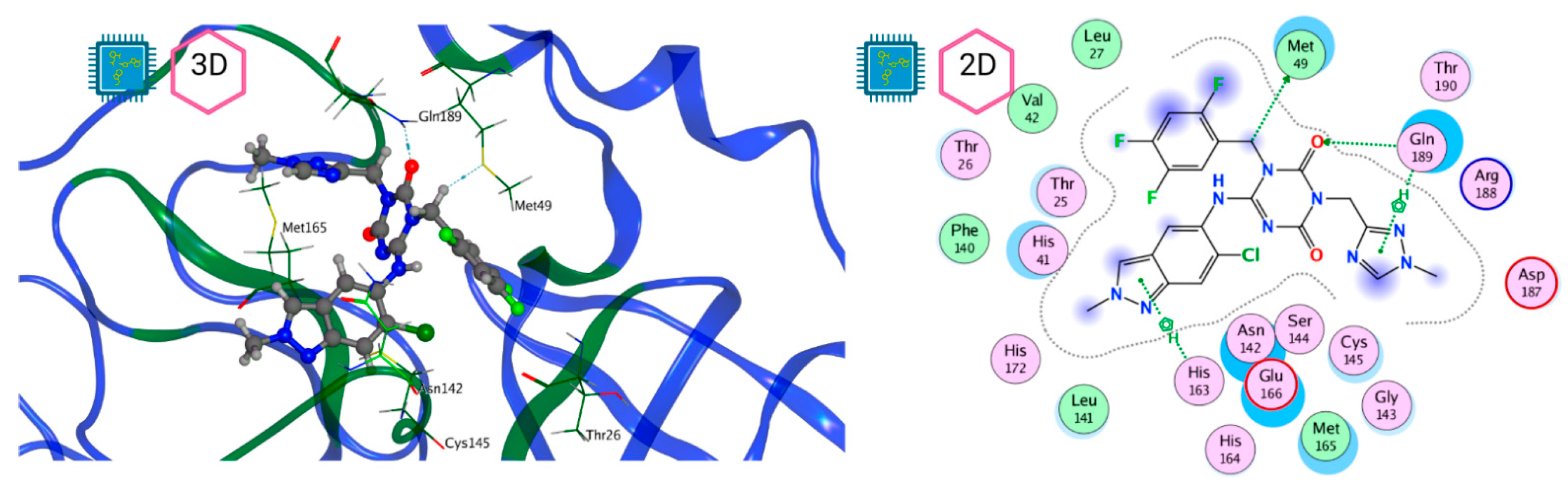
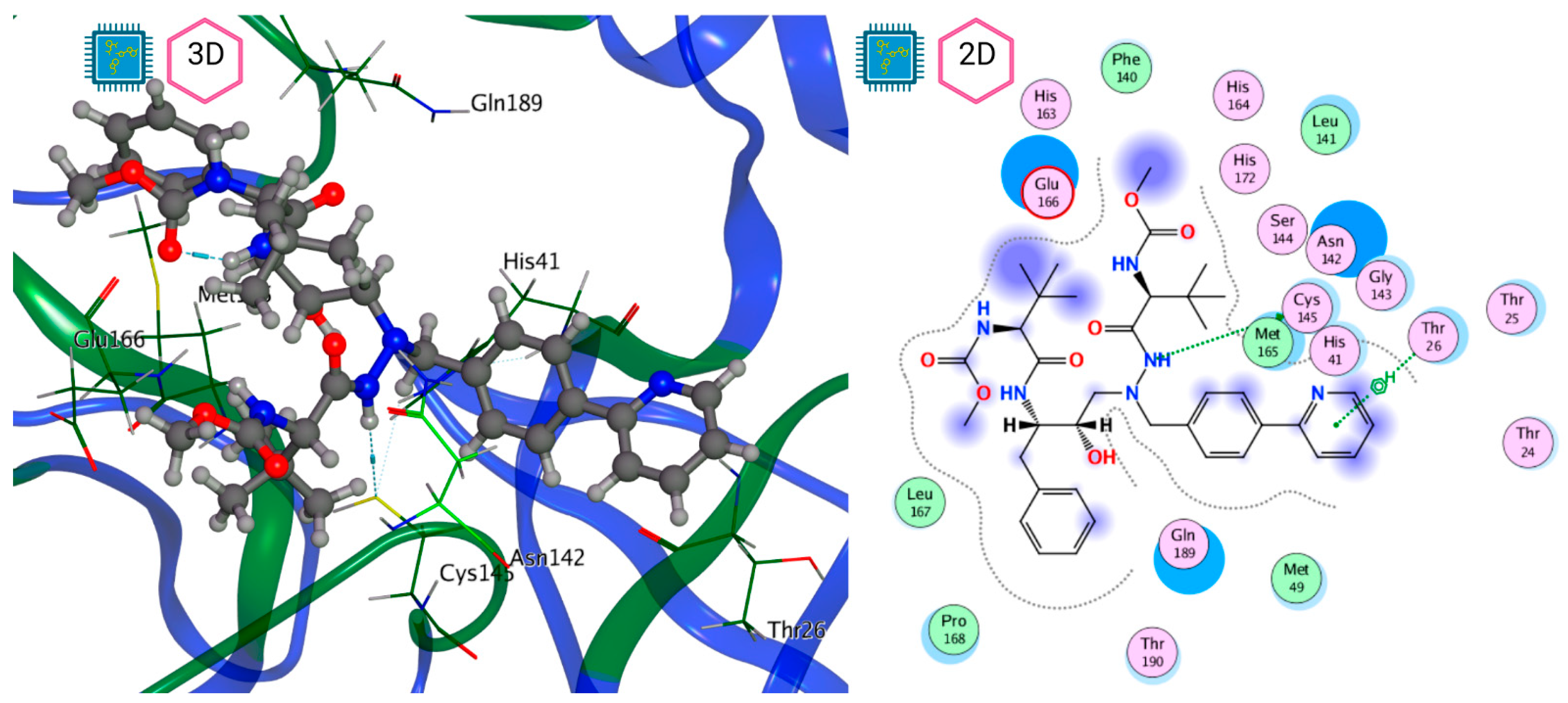
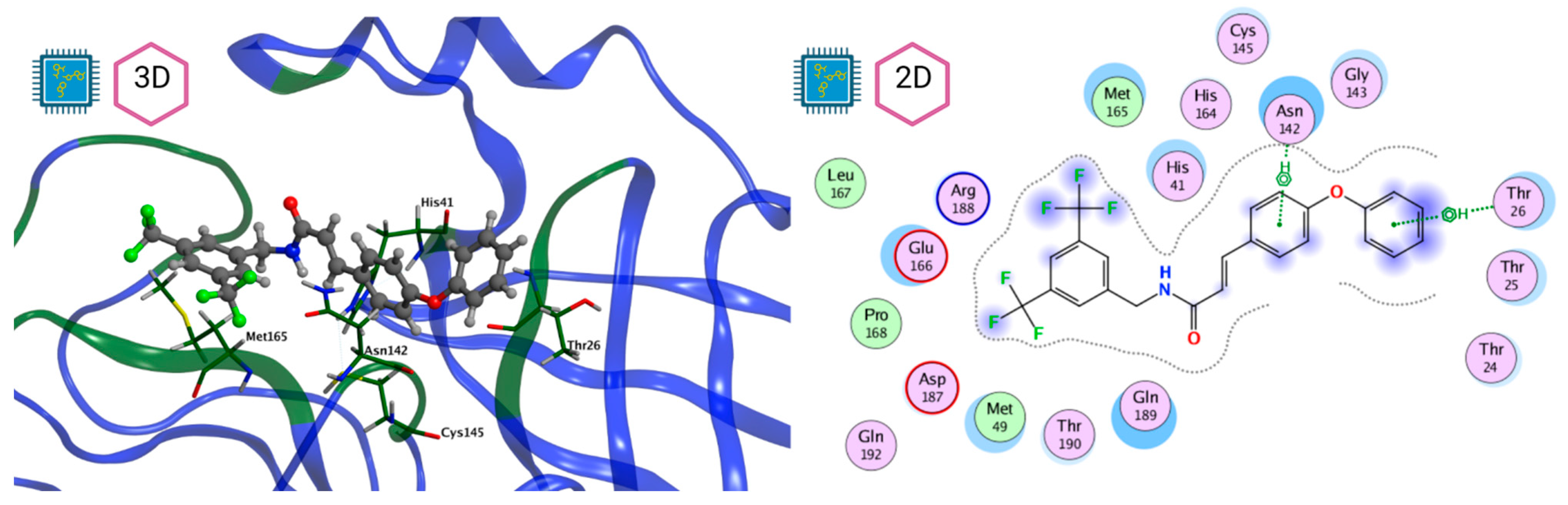
Figure 8 and Figure 9 show the active site of Mpro complexes with Ensitrelvir (S-217622) and Atazanavir (BMS-232632) respectively. Interactions as a hydrogen bond with Thr-26 phenoxy group (H–-π interaction) and Asn 142 amide sidechain also appear in the complex with the compound LQM 778. Figure 10 shows the active site of the complex formed with the ligand LQM 778. Although some nearby amino acids do not form direct interactions with the ligand, there is geometric and hydrophilic complementarity.
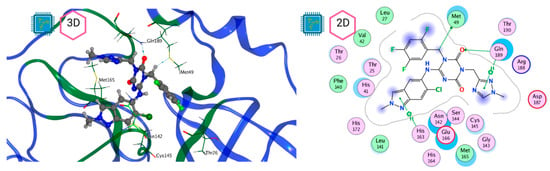
Figure 8.
Mpro–Ensitrelvir complex active site. The direct protein–ligand interactions are represented in the dotted line. Three-dimensional (left) and two-dimensional (right) representations.
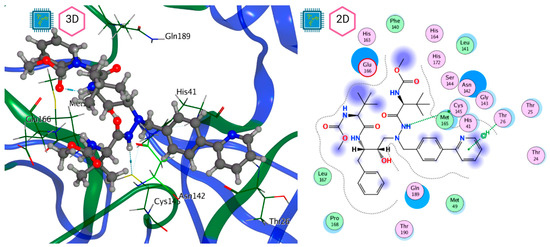
Figure 9.
Mpro–Atazanavir complex active site. The direct protein–ligand interactions are represented in the dotted line. Three-dimensional (left) and two-dimensional (right) representations.
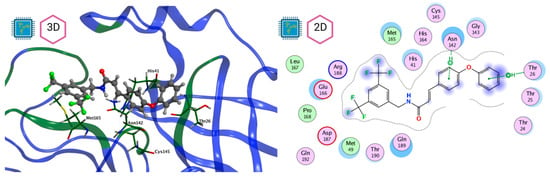
Figure 10.
Mpro–LQM 778 complex active site. The direct protein–ligand interactions are represented in the dotted line. Three-dimensional (left) and two-dimensional (right) representations.
Three molecular dynamics simulations of systems involving the Mpro protein were performed: one simulation of the protein without the ligand, called apo-form, and two simulations with Mpro complexes, one with the ligand MproL6 and the other with LQM 778.
The systems for molecular dynamics simulations were prepared using the MOE 2022.02. software. Starting from the representative pose for each system, the system was solvated in a cubic water box at periodic conditions (P1, 95 Å per side) and Na+ and Cl- counterions were added. Once solvated, a structural minimization of the whole system was performed. The input files were generated to run the molecular dynamics simulations in NAMD 2.13 software [78] using the Amber 14 force field, and the following protocol was established: first, a gradual heating stage from 0 to 300 K for 1 ns, followed by an equilibrium stage at 300 K and 1 atm pressure for 4 ns, to finally generate a 100 ns production stage where the pressure and temperature conditions were 300 K and 1 atm. For the molecular dynamic simulations, we used a cutoff of 12 and a time step of 0.002 fs.
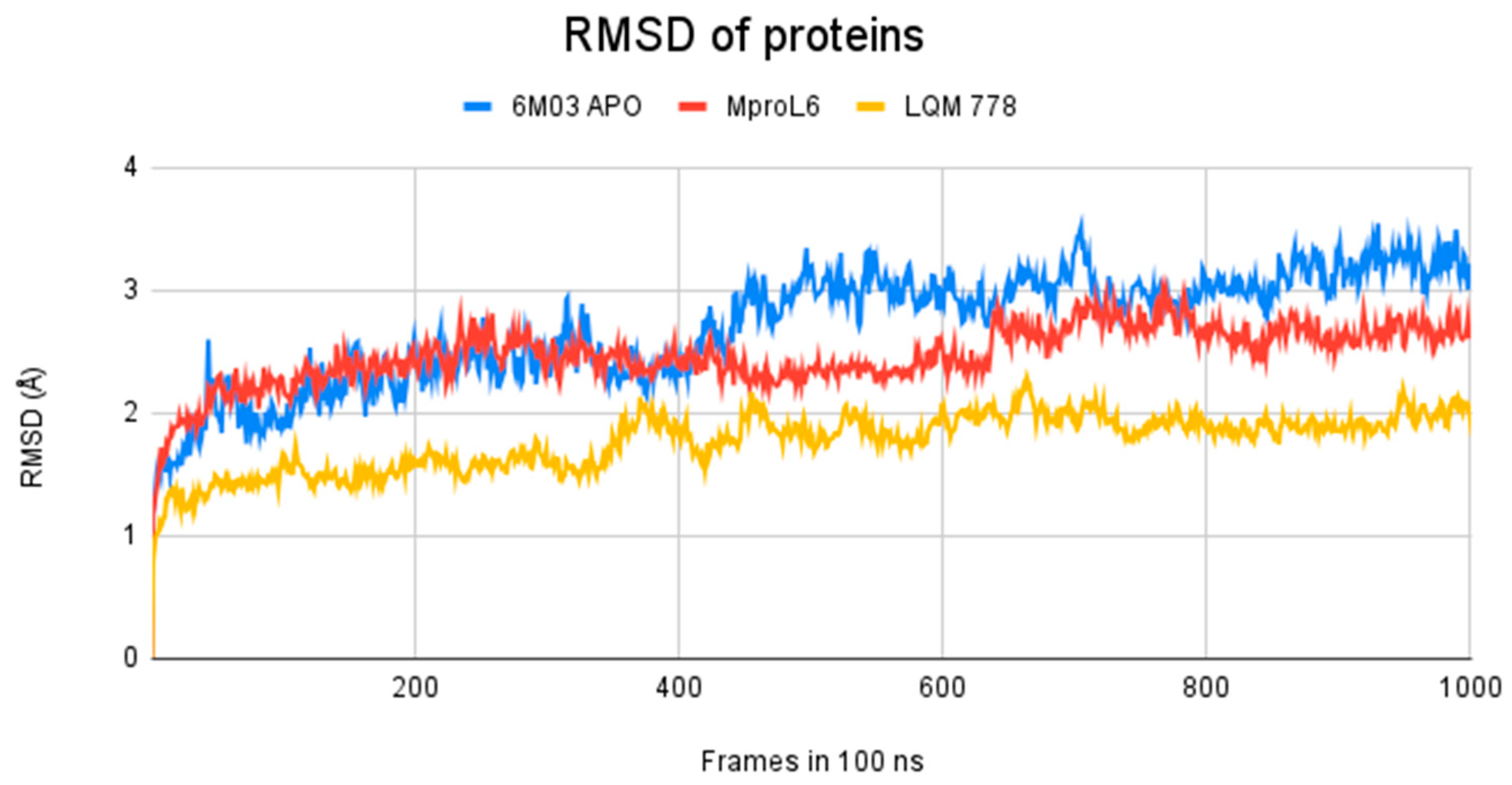
After completing the molecular dynamics simulations, the following statistical parameters were calculated for each of these systems: RMSD (root mean square deviation) (Figure 11), RMSF (root mean square fluctuation) (Figure 12), and ROG (radius of gyration) (Figure 13).
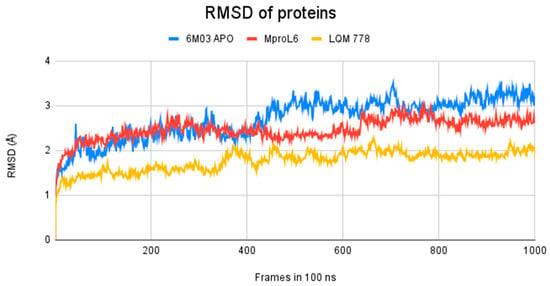
Figure 11.
RMSD of Mpro systems: apo-form (in blue) and with the ligands MproL6 (in red) and LQM 778 (in yellow).
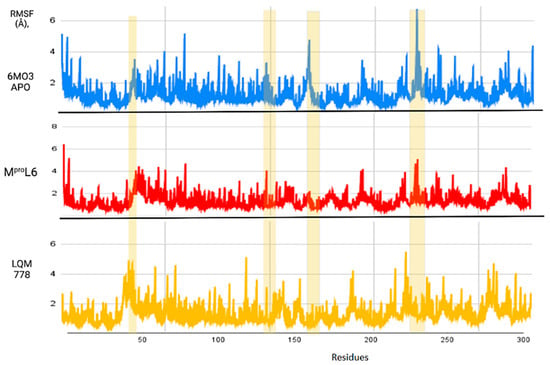
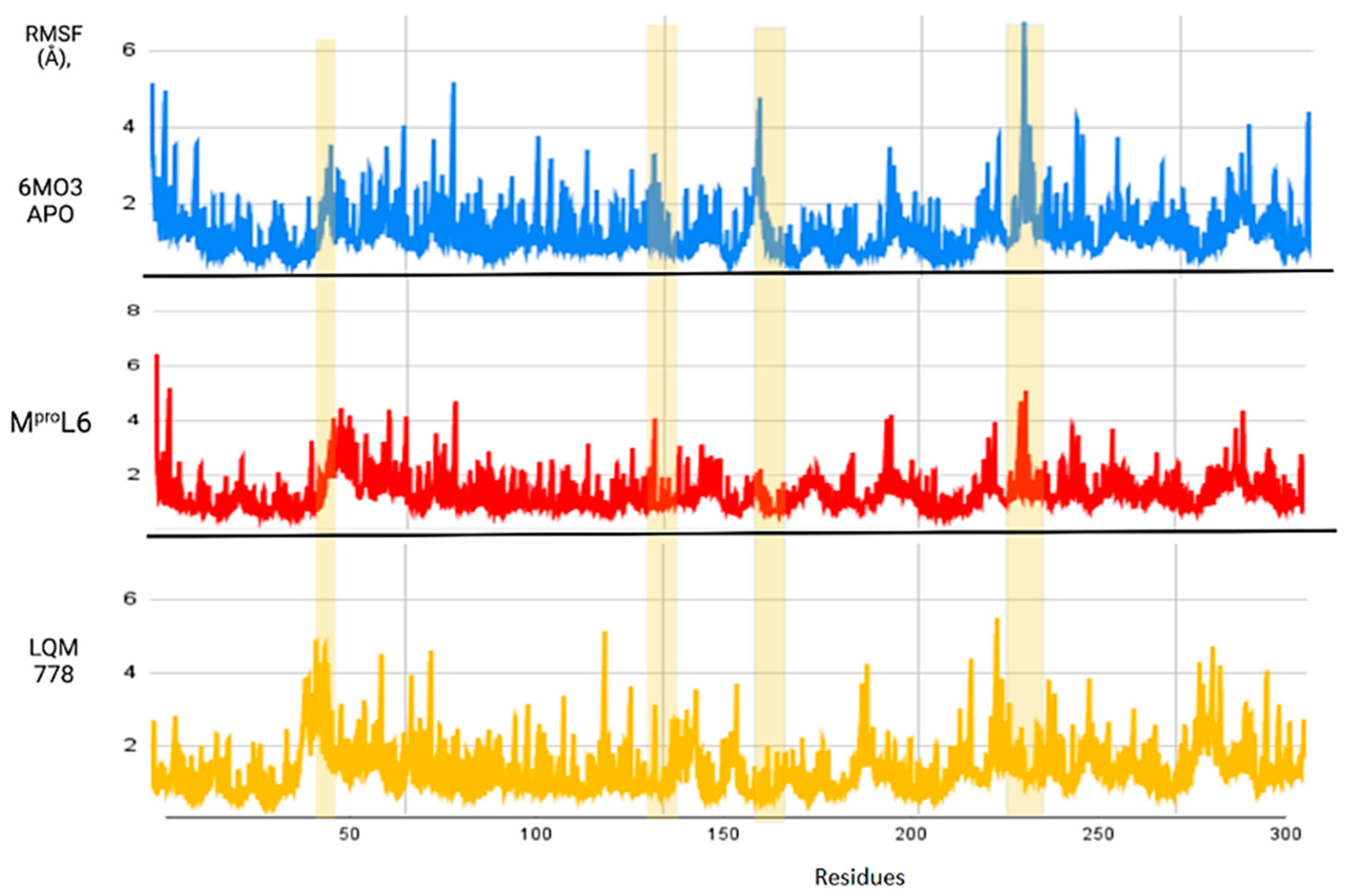
Figure 12.
RMSF of the Mpro protein in its apo-form in blue, compared to the ligands MproL6 in red and LQM 778 in purple.
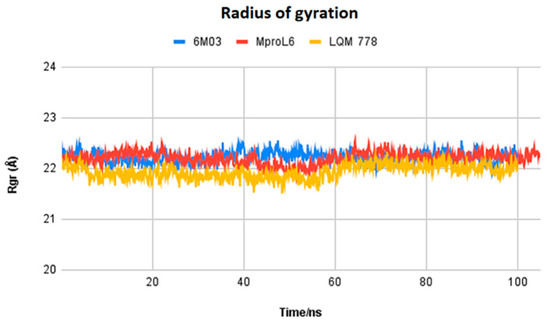
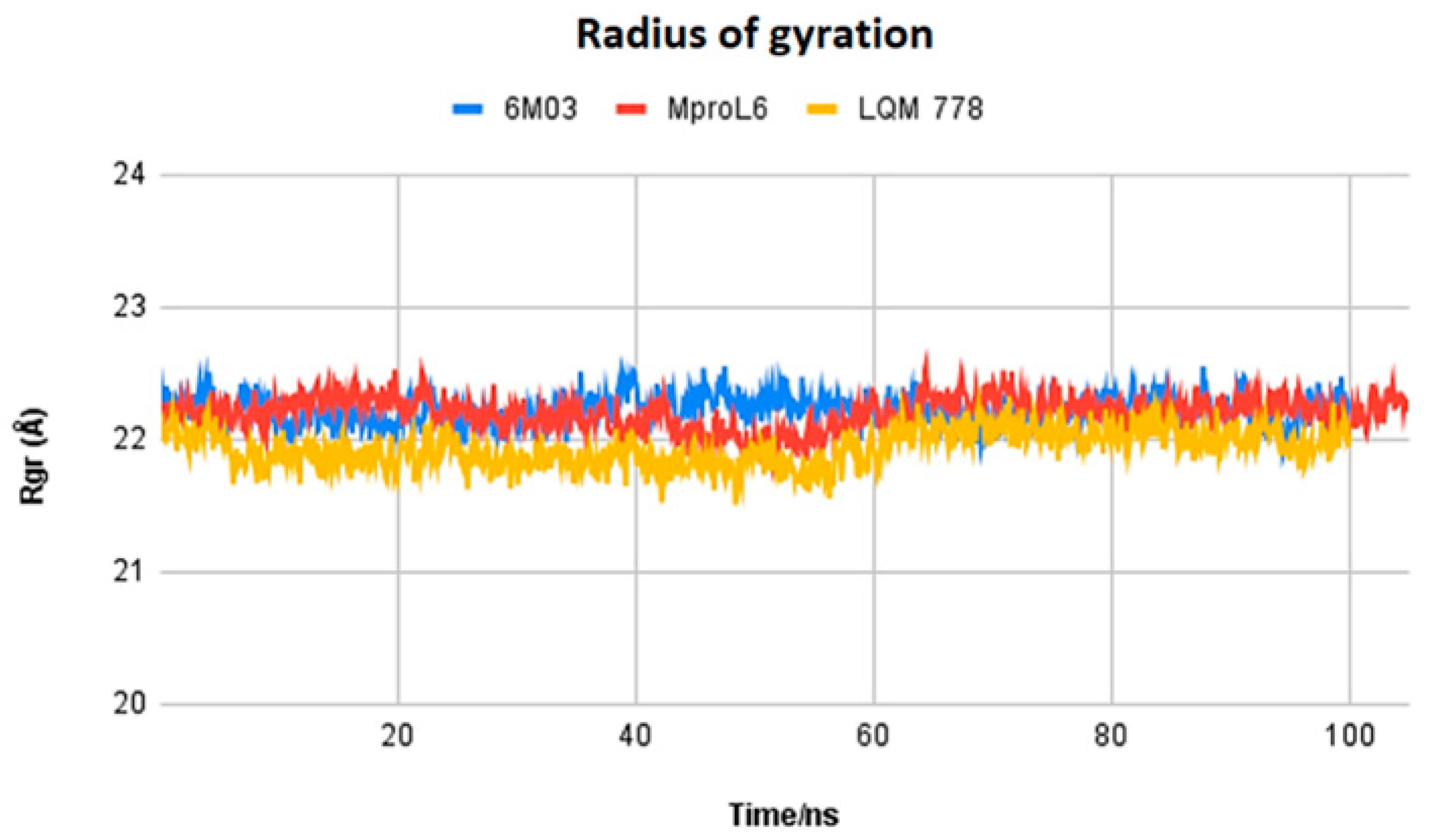
Figure 13.
Radius of gyration of the ligands MproL6 and LQM 778 and the apo-protein Mpro.
The results obtained after calculations reveal that the two systems with ligands have a lower fluctuation in the RMSD values. It is observed that the system in its apo-form reaches a maximum distance of 3.5 Å, in contrast to the system with the MproL6 ligand, which reaches a maximum distance of 3.1 Å. Finally, the system with the LQM 778 ligand has a maximum distance of 2.3 Å, suggesting that the complexes are more stable. Both ligands could maintain protein stability once they bind to the pocket.
Figure 12 shows the RMSF results, which allow us to identify the flexibility of the protein throughout the simulation, as shown in horizontal columns. The first two yellow columns indicate the region where the His41 and Cys145 residues are located. In addition, other areas are identified, such as that near residue 150, where the apo-protein showed an increase in its RMSF value, suggesting that once the ligands interact with the protein, the fluctuation of this region decreases. Finally, there is a noticeable fluctuation in the region near position 250; although it is not a significant fluctuation, it represents a distance greater than 6 Å in the apo-protein, which decreases in the plots with ligands. Therefore, it can be suggested that the protein remains in the states with more favorable stability and does not undergo deformations in its structure.
Figure 13 displays the radius of gyration of the systems: MproL6 ligand in red, LQM 778 ligand in purple, and the SARS-CoV-2 Mpro protein in blue in its apo-form. Nonhomogeneous behavior among the structures can be observed at the beginning of the simulation and up to 50 ns. However, starting at 60 ns, the proteins maintain homogeneous behavior, indicating the stability of the system.
From the results obtained thus far, based on this analysis, it can be suggested that our identified candidate drugs could potentially be effective against circulating SARS-CoV-2 variants. Currently, the antiviral activity of these compounds is being evaluated in vitro in the laboratory of Dr. L. Abrahamyan (University of Montreal, Quebec, Canada).
4. Conclusions
In this study, our group investigated one of the key molecular targets of SARS-CoV-2, the protease Mpro, of which, the PDB model 7TOB crystal structure served as a central target in our in silico molecular repositioning studies with diverse biological activities. Using a virtual screening (VS) approach, we applied the concept of repositioning using two databases, one external and one generated by our group. This approach allowed us to identify molecules with high potential as promising Mpro inhibitors.
The computational studies consisted of estimating the activity of a selection of molecules—a joint database composed of those from an in-house LQM series and the QSAR Toolbox database—against Mpro SARS-CoV-2. Two docking software were used to calculate an interaction score and, by consensus of the results, the molecules were ranked by best energy value. As a result of this process, we identified a total of thirty-two promising candidates, twenty-two from the external database and ten from our own collection of the LQM 700 series. Notably, the most relevant interactions with the protein residues His41 and Cys145 remained constant in all candidates, validating the robustness of our findings. Thus, by a comprehensive analysis of the in silico results, we determined that two molecules have significant potential as antiviral agents against SARS-CoV-2: MproL6 and LQM 778. These findings were supported by an assessment of computational integrity, including RMSD, RMSF, and radius of gyration, increasing confidence in their potential candidacy for future research and development. Furthermore, given that SARS-CoV-2 is in constant genetic evolution, looking at the latest variants revealed that the only significant amino acid substitutions found in the latest variants are in the K90R and P132H positions. This may suggest that our inhibitors identified in this study could target the Mpro of the latest SARS-CoV-2 variants, making them of great interest, especially in the context of the emergence of novel variants. With further in vitro and in vivo research, our results in this article can give rise to an arsenal of molecules against COVID-19.
Author Contributions
Conceptualization, L.A., E.A. and A.M.V.-S.; software, M.A.H.-S., V.H.V.-V. and P.A.-V.; validation, A.Y.A.-L. and L.A.C.-G.; formal analysis, M.A.H.-S., A.Y.A.-L., L.A.C.-G. and L.A.; investigation, M.A.H.-S., V.H.V.-V., L.A. and N.M.M.; resources, L.A. and E.A.; data curation, M.A.H.-S. and L.A.C.-G.; writing—original draft preparation, E.A. and L.A.; writing—review and editing, L.A., E.A., N.M.M., and J.L.M.-F.; visualization, P.A.-V., P.A.M.-S. and R.E.F.-V.; supervision, L.A. and E.A.; project administration, L.A. and E.A.; funding acquisition, L.A. and E.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Quebec-Mexico collaboration project (Groupe de travail Québec-Mexique, 2021–2023) “In silico design and evaluation of new compounds against coronaviruses including SARS-CoV-2”, supported by the Mexican Secretary of Foreign Affairs and the Quebec Ministry of International Relations and Francophonie via Dr. Enrique Angeles and Dr. Levon Abrahamyan, respectively.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Derived data supporting the findings of this study are available from the author M.A.H.S on request.
Acknowledgments
This work was partially supported by the UNAM Projects, CONACHYT Project CF-2023-I-195; PAPIIT206023; PIAPI CI2406, and LANCAD-UNAM-DGTIC-034. We thank Moises Hernández and Draucin Jiménez from FESC-UNAM for their technical assistance. This work was partially supported by the Quebec Ministry of International Relations and Francophonie via a Canadian leader of this project, Levon Abrahamyan.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zhu, H.; Wei, L.; Niu, P. The novel coronavirus outbreak in Wuhan, China. Glob. Health Res. Policy 2020, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, X.; Li, T.; Zhang, S.; Wang, L.; Wu, X.; Liu, J. The genetic sequence, origin, and diagnosis of SARS-CoV-2. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1629–1635. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. 2020, 91, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Miyah, Y.; Benjelloun, M.; Lairini, S.; Lahrichi, A. COVID-19 Impact on Public Health, Environment, Human Psychology, Global Socioeconomy, and Education. Sci. World J. 2022, 2022, 5578284. [Google Scholar] [CrossRef]
- W.H.O. Weekly Epidemiological Update on COVID-19. 15 July 2024. Available online: https://www.who.int/publications/m/item/covid-19-epidemiological-update-edition-169 (accessed on 17 July 2024).
- Weiss, S.R.; Leibowitz, J.L. Coronavirus pathogenesis. Adv. Virus Res. 2011, 81, 85–164. [Google Scholar] [CrossRef]
- Dhama, K.; Khan, S.; Tiwari, R.; Sircar, S.; Bhat, S.; Malik, Y.S.; Singh, K.P.; Chaicumpa, W.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. Coronavirus Disease 2019-COVID-19. Clin. Microbiol. Rev. 2020, 33, e00028-20. [Google Scholar] [CrossRef]
- Ding, X.L.; Jia, Q.L.; To, S.F. Human Coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). In Encyclopedia of Virology, 4th ed.; Dennis, H.B., Mark, Z., Eds.; Academic Press: Oxford, UK, 2021; pp. 428–440. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Swain, B.; Verma, R.S.; Gunthe, S.S. SARS-CoV, MERS-CoV, and 2019-nCoV viruses: An overview of origin, evolution, and genetic variations. Virusdisease 2020, 31, 411–423. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel Coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Ravi, V.; Saxena, S.; Panda, P.S. Basic virology of SARS-CoV 2. Indian J. Med. Microbiol. 2022, 40, 182–186. [Google Scholar] [CrossRef]
- Jungreis, I.; Sealfon, R.; Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nat. Commun. 2021, 12, 2642. [Google Scholar] [CrossRef]
- Redondo, N.; Zaldivar-Lopez, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front. Immunol. 2021, 12, 708264. [Google Scholar] [CrossRef] [PubMed]
- Mariano, G.; Farthing, R.J.; Lale-Farjat, S.L.M.; Bergeron, J.R.C. Structural Characterization of SARS-CoV-2: Where We Are, and Where We Need to Be. Front. Mol. Biosci. 2020, 7, 605236. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, M.; Pandey, N.; Shukla, A.; Singh, S.K. SARS coronavirus 2: From genome to infectome. Respir. Res. 2020, 21, 318. [Google Scholar] [CrossRef]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef]
- Triggle, C.R.; Bansal, D.; Ding, H.; Islam, M.M.; Farag, E.; Hadi, H.A.; Sultan, A.A. A Comprehensive Review of Viral Characteristics, Transmission, Pathophysiology, Immune Response, and Management of SARS-CoV-2 and COVID-19 as a Basis for Controlling the Pandemic. Front. Immunol. 2021, 12, 631139. [Google Scholar] [CrossRef]
- Armstrong, L.A.; Lange, S.M.; Dee Cesare, V.; Matthews, S.P.; Nirujogi, R.S.; Cole, I.; Hope, A.; Cunningham, F.; Toth, R.; Mukherjee, R.; et al. Biochemical characterization of protease activity of Nsp3 from SARS-CoV-2 and its inhibition by nanobodies. PLoS ONE 2021, 16, e0253364. [Google Scholar] [CrossRef]
- Ullrich, S.; Ekanayake, K.B.; Otting, G.; Nitsche, C. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg. Med. Chem. Lett. 2022, 62, 128629. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Hu, Q.; Xiong, Y.; Zhu, G.H.; Zhang, Y.N.; Zhang, Y.W.; Huang, P.; Ge, G.B. The SARS-CoV-2 main protease (Mpro): Structure, function, and emerging therapies for COVID-19. MedComm 2022, 3, e151. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simon-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martin-Quiros, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of COVID-19 in Nonhospitalized Patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef]
- Sheward, D.J.; Kim, C.; Fischbach, J.; Muschiol, S.; Ehling, R.A.; Bjorkstrom, N.K.; Karlsson Hedestam, G.B.; Reddy, S.T.; Albert, J.; Peacock, T.P.; et al. Evasion of neutralising antibodies by omicron sublineage BA.2.75. Lancet Infect. Dis. 2022, 22, 1421–1422. [Google Scholar] [CrossRef]
- Gandhi, S.; Klein, J.; Robertson, A.; Pena-Hernandez, M.A.; Lin, M.J.; Roychoudhury, P.; Lu, P.; Fournier, J.; Ferguson, D.; Mohamed Bakhash, S.A.; et al. De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS-CoV-2 infection in an immunocompromised patient: A case report. medRxiv 2021. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody evasion by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4 and BA.5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef]
- Morgnanesi, D.; Heinrichs, E.J.; Mele, A.R.; Wilkinson, S.; Zhou, S.; Kulp, J.L., 3rd. A computational chemistry perspective on the current status and future direction of hepatitis B antiviral drug discovery. Antiviral. Res. 2015, 123, 204–215. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Muratov, E.N.; Amaro, R.; Andrade, C.H.; Brown, N.; Ekins, S.; Fourches, D.; Isayev, O.; Kozakov, D.; Medina-Franco, J.L.; Merz, K.M.; et al. A critical overview of computational approaches employed for COVID-19 drug discovery. Chem. Soc. Rev. 2021, 50, 9121–9151. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sellner, M.; Neranjan, S.; Smiesko, M.; Lill, M.A. Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. Int. J. Mol. Sci. 2020, 21, 3626. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020, 38, 379–381. [Google Scholar] [CrossRef]
- Dimitrov, S.D.; Diderich, R.; Sobanski, T.; Pavlov, T.S.; Chankov, G.V.; Chapkanov, A.S.; Karakolev, Y.H.; Temelkov, S.G.; Vasilev, R.A.; Gerova, K.D.; et al. QSAR Toolbox—Workflow and major functionalities. SAR QSAR Environ. Res. 2016, 27, 203–219. [Google Scholar] [CrossRef]
- QSAR Toolbox. Available online: https://qsartoolbox.org (accessed on 2 October 2021).
- Anastasiu, D.C.; Karypis, G. Efficient identification of Tanimoto nearest neighbors. Int. J. Data Sci. Anal. 2017, 4, 153–172. [Google Scholar] [CrossRef]
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 M(pro): A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules 2021, 11, 607. [Google Scholar] [CrossRef]
- Chen, Y.W.; Yiu, C.B.; Wong, K.Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 2020, 9, 129. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Chemical Computing Group, ULC. Molecular Operating Environment (MOE) 2022.02; Chemical Computing Group, ULC: Montreal, QC, Canada, 2022. [Google Scholar]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Owens, J. Determining druggability. Nat. Rev. Drug Discov. 2007, 6, 187. [Google Scholar] [CrossRef]
- Michel, M.; Visnes, T.; Homan, E.J.; Seashore-Ludlow, B.; Hedenstrom, M.; Wiita, E.; Vallin, K.; Paulin, C.B.J.; Zhang, J.; Wallner, O.; et al. Computational and Experimental Druggability Assessment of Human DNA Glycosylases. ACS Omega 2019, 4, 11642–11656. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.N.; Jani, S.P.; Prasanth Kumar, S.; Modi, K.M.; Kumar, Y. Computational investigation of natural compounds as potential main protease (Mpro) inhibitors for SARS-CoV-2 virus. Comput. Biol. Med. 2022, 151, 106318. [Google Scholar] [CrossRef]
- CoVariants SARS-CoV-Mutations and Variants of Interest. Available online: https://covariants.org/ (accessed on 31 October 2023).
- Dierynck, I.; Van Marck, H.; Van Ginderen, M.; Jonckers, T.H.; Nalam, M.N.; Schiffer, C.A.; Raoof, A.; Kraus, G.; Picchio, G. TMC310911, a novel human immunodeficiency virus type 1 protease inhibitor, shows in vitro an improved resistance profile and higher genetic barrier to resistance compared with current protease inhibitors. Antimicrob. Agents Chemother. 2011, 55, 5723–5731. [Google Scholar] [CrossRef]
- Kaldor, S.W.; Kalish, V.J.; Davies, J.F., 2nd; Shetty, B.V.; Fritz, J.E.; Appelt, K.; Burgess, J.A.; Campanale, K.M.; Chirgadze, N.Y.; Clawson, D.K.; et al. Viracept (nelfinavir mesylate, AG1343): A potent, orally bioavailable inhibitor of HIV-1 protease. J. Med. Chem. 1997, 40, 3979–3985. [Google Scholar] [CrossRef]
- Islam, R.; Parves, M.R.; Paul, A.S.; Uddin, N.; Rahman, M.S.; Mamun, A.A.; Hossain, M.N.; Ali, M.A.; Halim, M.A. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 3213–3224. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schafer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O.; et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020, 11, 222. [Google Scholar] [CrossRef]
- Zanasi, A.; Mazzolini, M.; Kantar, A. A reappraisal of the mucoactive activity and clinical efficacy of bromhexine. Multidiscip. Respir. Med. 2017, 12, 7. [Google Scholar] [CrossRef]
- Hull, M.W.; Montaner, J.S. Ritonavir-boosted protease inhibitors in HIV therapy. Ann. Med. 2011, 43, 375–388. [Google Scholar] [CrossRef]
- Kiser, J.J.; Flexner, C. Direct-acting antiviral agents for hepatitis C virus infection. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 427–449. [Google Scholar] [CrossRef] [PubMed]
- Kupferschmidt, H.H.; Fattinger, K.E.; Ha, H.R.; Follath, F.; Krahenbuhl, S. Grapefruit juice enhances the bioavailability of the HIV protease inhibitor saquinavir in man. Br. J. Clin. Pharmacol. 1998, 45, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Tomita, K. Proteolytic activation of the epithelial sodium channel and therapeutic application of a serine protease inhibitor for the treatment of salt-sensitive hypertension. Clin. Exp. Nephrol. 2012, 16, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Raboisson, P.; de Kock, H.; Rosenquist, A.; Nilsson, M.; Salvador-Oden, L.; Lin, T.I.; Roue, N.; Ivanov, V.; Wahling, H.; Wickstrom, K.; et al. Structure-activity relationship study on a novel series of cyclopentane-containing macrocyclic inhibitors of the hepatitis C virus NS3/4A protease leading to the discovery of TMC435350. Bioorg. Med. Chem. Lett. 2008, 18, 4853–4858. [Google Scholar] [CrossRef] [PubMed]
- Bafna, K.; White, K.; Harish, B.; Rosales, R.; Ramelot, T.A.; Acton, T.B.; Moreno, E.; Kehrer, T.; Miorin, L.; Royer, C.A.; et al. Hepatitis C virus drugs that inhibit SARS-CoV-2 papain-like protease synergize with remdesivir to suppress viral replication in cell culture. Cell Rep. 2021, 35, 109133. [Google Scholar] [CrossRef]
- Alamri, M.A.; Altharawi, A.; Alabbas, A.B.; Alossaimi, M.A.; Alqahtani, S.M. Structure-based virtual screening and molecular dynamics of phytochemicals derived from Saudi medicinal plants to identify potential COVID-19 therapeutics. Arab J. Chem. 2020, 13, 7224–7234. [Google Scholar] [CrossRef]
- Lu, H. Drug treatment options for the 2019-new coronavirus (2019-nCoV). Biosci. Trends 2020, 14, 69–71. [Google Scholar] [CrossRef]
- Purohit, R.; Sethumadhavan, R. Structural basis for the resilience of Darunavir (TMC114) resistance major flap mutations of HIV-1 protease. Interdiscip. Sci. 2009, 1, 320–328. [Google Scholar] [CrossRef]
- Doyon, L.; Tremblay, S.; Bourgon, L.; Wardrop, E.; Cordingley, M.G. Selection and characterization of HIV-1 showing reduced susceptibility to the non-peptidic protease inhibitor tipranavir. Antiviral. Res. 2005, 68, 27–35. [Google Scholar] [CrossRef]
- Ye, G.; Wang, X.; Tong, X.; Shi, Y.; Fu, Z.F.; Peng, G. Structural Basis for Inhibiting Porcine Epidemic Diarrhea Virus Replication with the 3C-Like Protease Inhibitor GC376. Viruses 2020, 12, 240. [Google Scholar] [CrossRef]
- Kiselev, O.I.; Deeva, E.G.; Mel’nikova, T.I.; Kozeletskaia, K.N.; Kiselev, A.S.; Rusinov, V.L.; Charushin, V.N.; Chupakhin, O.N. A new antiviral drug Triazavirin: Results of phase II clinical trial. Vopr. Virusol. 2012, 57, 9–12. [Google Scholar] [PubMed]
- Khatib, M.; Pieraccini, G.; Innocenti, M.; Melani, F.; Mulinacci, N. An insight on the alkaloid content of Capparis spinosa L. root by HPLC-DAD-MS, MS/MS and 1H qNMR. J. Pharm. Biomed. Anal. 2016, 123, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Siddiqui, S.; Ahmad, R.; Mehrotra, S.; Ahmad, B.; Srivastava, A.N. Exploring nature’s bounty: Identification of Withania somnifera as a promising source of therapeutic agents against COVID-19 by virtual screening and in silico evaluation. J. Biomol. Struct. Dyn. 2022, 40, 1858–1908. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Qu, H.; Wang, S.; Yan, F.; Wang, C.; Zhang, M. Computational assessment of herbal medicine-derived compounds as potential inhibitors of SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2023, 41, 9602–9613. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. alpha-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Sharma, G.; Kumar, N.; Sharma, C.S.; Mishra, S.S. In silico guided screening of active components of C. lanceolata as 3-chymotrypsin-like protease inhibitors of novel coronavirus. 3 Biotech 2023, 13, 324. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280 e278. [Google Scholar] [CrossRef]
- Yamamoto, M.; Matsuyama, S.; Li, X.; Takeda, M.; Kawaguchi, Y.; Inoue, J.I.; Matsuda, Z. Identification of Nafamostat as a Potent Inhibitor of Middle East Respiratory Syndrome Coronavirus S Protein-Mediated Membrane Fusion Using the Split-Protein-Based Cell-Cell Fusion Assay. Antimicrob. Agents Chemother. 2016, 60, 6532–6539. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Antonopoulou, I.; Sapountzaki, E.; Rova, U.; Christakopoulos, P. Inhibition of the main protease of SARS-CoV-2 (Mpro) by repurposing/designing drug-like substances and utilizing nature’s toolbox of bioactive compounds. Comput. Struct. Biotechnol. J. 2022, 20, 1306–1344. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Henin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).