
Estimation of Two Component Activities of Binary Liquid Alloys by the Pair Potential Energy Containing a Polynomial of the Partial Radial Distribution Function

Abstract
:1. Introduction
2. Thermodynamic Model and Symmetry
2.1. Miedema Model
2.2. Regular Solution Model (RSM)
2.3. Wilson Model
2.4. Nonrandom Two-Liquid (NRTL) Model
2.5. Molecular Interaction Volume Model (MIVM)
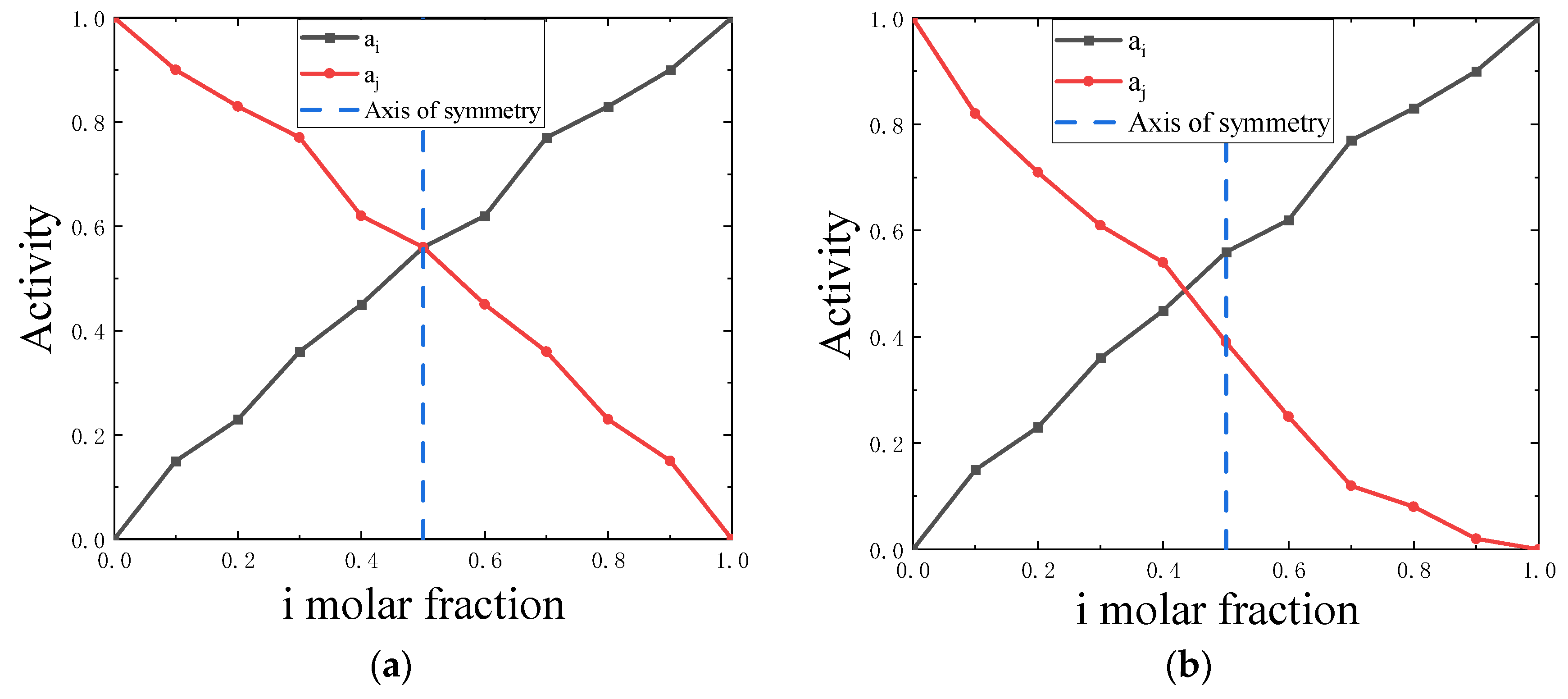
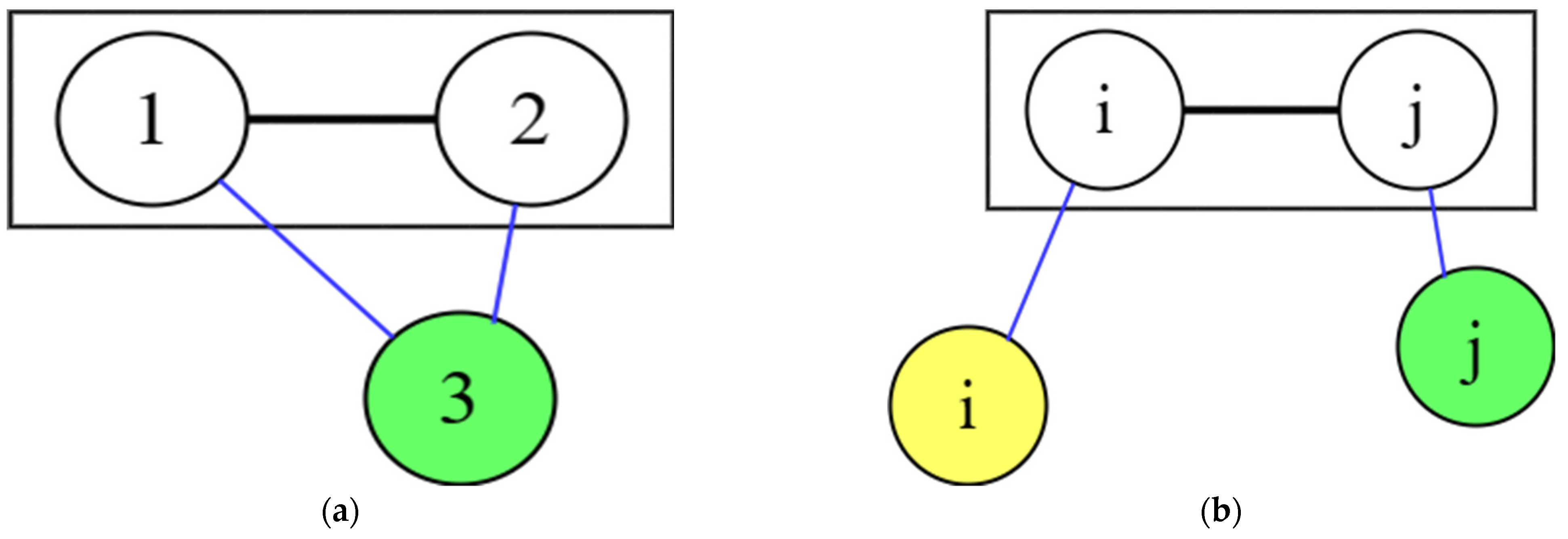
2.6. Symmetry
3. Pair Potential Energy Polynomials of the Binary Liquid
4. Result Analysis
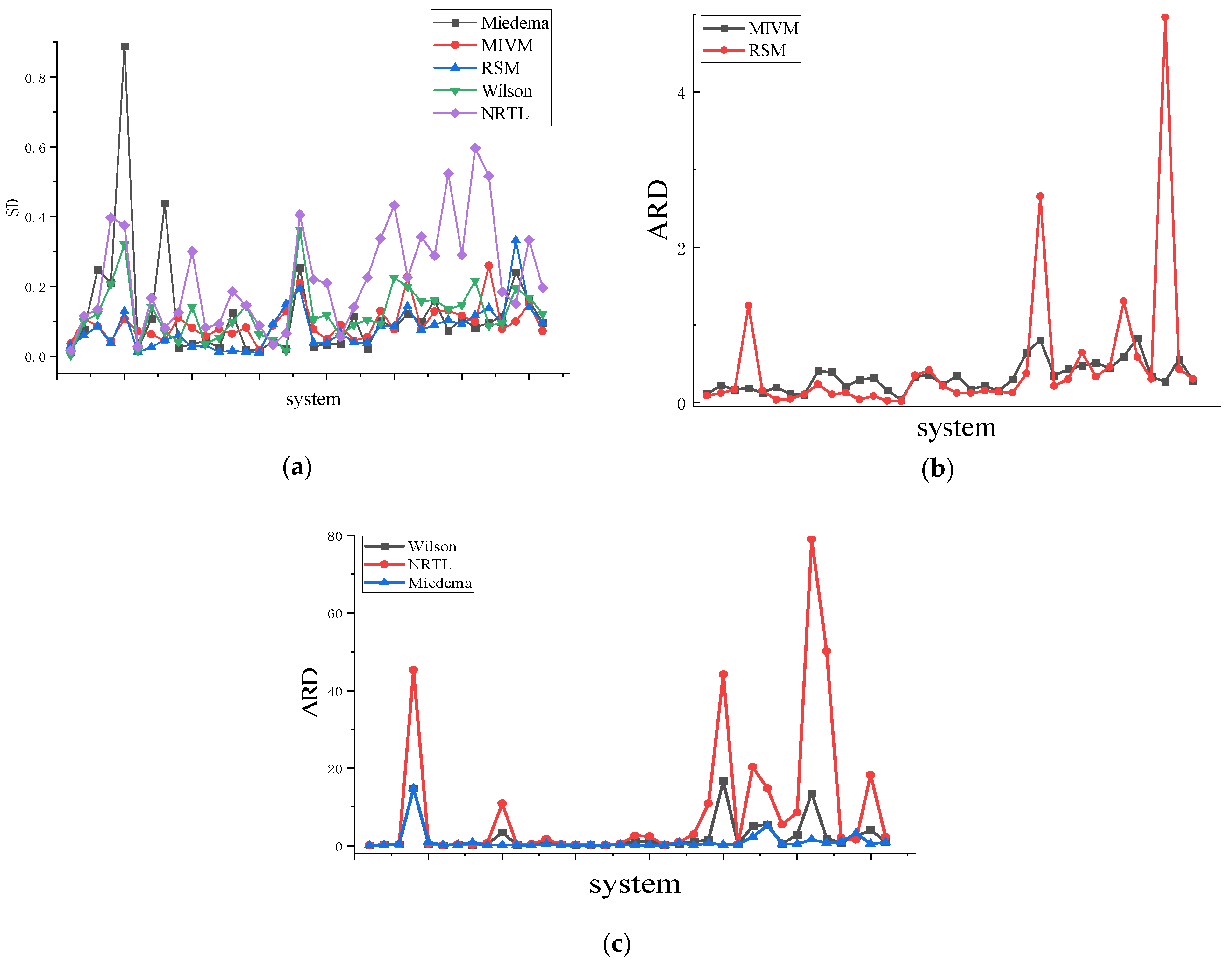
4.1. Asymmetric Method for Calculating the RDF
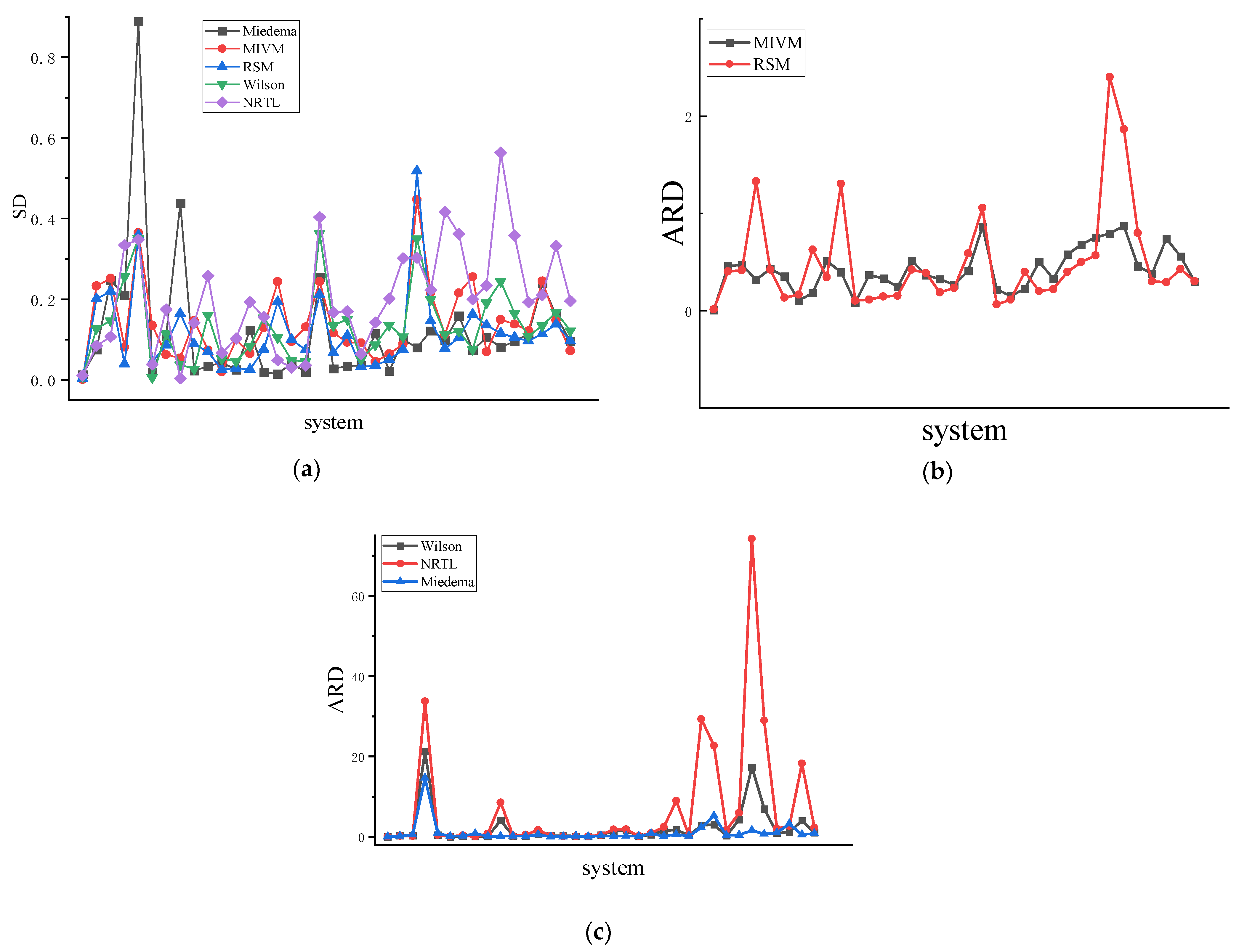
4.2. Symmetric Method for Calculating the RDF
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Xie, J.C.; Kar, T.; Xie, R.-H. An Accurate Pair Potential Function for Diatomic Systems. Chem. Phys. Lett. 2014, 591, 69–77. [Google Scholar] [CrossRef]
- Lennard-Jones, J.E. Cohesion. Proc. Phys. Soc. 1931, 43, 461. [Google Scholar] [CrossRef]
- Morse, P.M. Diatomic Molecules According to the Wave Mechanics. II. Vibrational Levels. Phys. Rev. 1929, 34, 57–64. [Google Scholar] [CrossRef]
- Born, M.; Mayer, J.E. Zur Gittertheorie der Ionenkristalle. Z. Phys. 1932, 75, 1–18. [Google Scholar] [CrossRef]
- Ter Horst, M.A.; Schatz, G.C.; Harding, L.B. Potential Energy Surface and Quasiclassical Trajectory Studies of the CN+H2 Reaction. J. Chem. Phys. 1996, 105, 558–571. [Google Scholar] [CrossRef]
- Hirst, D.M. Ab Initio Potential Energy Surfaces for Excited States of the NO2+ Molecular Ion and for the Reaction of N+ with O2. J. Chem. Phys. 2001, 115, 9320–9330. [Google Scholar] [CrossRef]
- Grandinetti, F.; Vinciguerra, V. Adducts of NF2+ with Diatomic and Simple Polyatomic Ligands: A Computational Investigation on the Structure, Stability, and Thermochemistry. Int. J. Mass Spectrom. 2002, 216, 285–299. [Google Scholar] [CrossRef]
- Fan, T.X.; Yang, G.J.; Chen, J.Q.; Zhang, D. Model Prediction of Thermodynamics Activity in Multicomponent Liquid Alloy. Key Eng. Mater. 2006, 313, 19–24. [Google Scholar] [CrossRef]
- Hildebrand, J.H. Solubility. XII. Regular Solutions. J. Am. Chem. Soc. 1929, 51, 66–80. [Google Scholar] [CrossRef]
- Hildebrand, J.H.; Prausnitz, J.M.; Scott, R.L. (Eds.) Regular and Related Solutions: The Solubility of Gases Liquids, and Solids; Van Nostrand Reinhold Company: New York, NY, USA, 1970. [Google Scholar]
- Wilson, G.M. Vapor-Liquid Equilibrium. XI. A New Expression for the Excess Free Energy of Mixing. J. Am. Chem. Soc. 1964, 86, 127–130. [Google Scholar] [CrossRef]
- Nagata, I. Correlation of ternary liquid-liquid equilibria using a modification of the wilson equation. Fluid Phase Equilibria 1992, 72, 1–14. [Google Scholar] [CrossRef]
- Renon, H.; Prausnitz, J.M. Local Compositions in Thermodynamic Excess Functions for Liquid Mixtures. AIChE J. 1968, 14, 135–144. [Google Scholar] [CrossRef]
- Tao, D.P. A New Model of Thermodynamics of Liquid Mixtures and Its Application to Liquid Alloys. Thermochim. Acta 2000, 363, 105–113. [Google Scholar] [CrossRef]
- Hill, T.L. Statistical Mechanics: Principles and Selected Applications; Courier Corporation: Chelmsford, MA, USA, 1957. [Google Scholar]
- Hu, Y. Molecular Thermodynamics of Fluids; Higher Education Press: Beijing, China, 1982. [Google Scholar]
- Wang, C.; Chen, X.; Tao, D. Estimation of Component Activities and Molar Excess Gibbs Energy of 19 Binary Liquid Alloys from Partial Pair Distribution Functions in Literature. Metals 2023, 13, 996. [Google Scholar] [CrossRef]
- Wang, C.; Chen, X.; Tao, D. Development of a Non-Integral Form of Coordination Number Equation Based on Pair Distribution Function and Gaussian Function. Metals 2023, 13, 384. [Google Scholar] [CrossRef]
- Department of Mathematics, Tongji University. Advanced Mathematics, 6th ed.; Higher Education Press: Beijing, China, 2007. [Google Scholar]
- Chanda, S.; Ahmed, A.Z.Z.; Bhuiyan, G.M.; Barman, S.K.; Sarker, S. A Test of Distribution Function Method in the Case of Liquid Transition Metals Alloys. J. Non-Cryst. Solids 2011, 357, 3774–3780. [Google Scholar] [CrossRef]
- Trybula, M.; Jakse, N.; Gasior, W.; Pasturel, A. Structural and Physicochemical Properties of Liquid Al–Zn Alloys: A Combined Study Based on Molecular Dynamics Simulations and the Quasi-Lattice Theory. J. Chem. Phys. 2014, 141, 224504. [Google Scholar] [CrossRef]
- Hongri, C.; Xiufang, B.; Hui, L.; Li, W. Molecular Dynamics Simulation on Structures of Cu–Ni Alloy. China J. Chem. Phys. 2002, 15, 288–294. [Google Scholar]
- Belova, I.V.; Ahmed, T.; Sarder, U.; Yi Wang, W.; Kozubski, R.; Liu, Z.-K.; Holland-Moritz, D.; Meyer, A.; Murch, G.E. Computer Simulation of Thermodynamic Factors in Ni–Al and Cu–Ag Liquid Alloys. Comput. Mater. Sci. 2019, 166, 124–135. [Google Scholar] [CrossRef]
- Wang, H.; Wei, B. Understanding Atomic-Scale Phase Separation of Liquid Fe–Cu Alloy. Chin. Sci. Bull. 2011, 56, 3416–3419. [Google Scholar] [CrossRef]
- Goto, R.; Shimojo, F.; Munejiri, S.; Hoshino, K. Structural and Electronic Properties of Liquid Ge–Sn Alloys: Ab Initio Molecular-Dynamics Simulations. J. Phys. Soc. Jpn. 2004, 73, 2746–2752. [Google Scholar] [CrossRef]
- Song, B.; Xu, N.; Jiang, W.; Yang, B.; Chen, X.; Xu, B.; Kong, L.; Liu, D.; Dai, Y. Study on Azeotropic Point of Pb–Sb Alloys by Ab-Initio Molecular Dynamic Simulation and Vacuum Distillation. Vacuum 2016, 125, 209–214. [Google Scholar] [CrossRef]
- Qin, J.; Pan, S.; Qi, Y.; Gu, T. The Structure and Thermodynamic Properties of Liquid Al–Si Alloys by Ab Initio Molecular Dynamics Simulation. J. Non-Cryst. Solids 2016, 433, 31–37. [Google Scholar] [CrossRef]
- Kbirou, M.; Mazroui, M.; Hasnaoui, A. Atomic Packing and Fractal Behavior of Al-Co Metallic Glasses. J. Alloys Compd. 2018, 735, 464–472. [Google Scholar] [CrossRef]
- Canales, M.; González, D.J.; González, L.E.; Padró, J.A. Static Structure and Dynamics of the Liquid Li–Na and Li–Mg Alloys. Phys. Rev. E 1998, 58, 4747–4757. [Google Scholar] [CrossRef]
- Pu, Z.; Zhang, H.; Li, Y.; Yang, B. Study on Sn-Sb Alloy by Ab-Initio Molecular Dynamic Simulation. IOP Conf. Ser. Mater. Sci. Eng. 2018, 394, 032098. [Google Scholar] [CrossRef]
- Jakse, N.; Pasturel, A. Local Order and Dynamic Properties of Liquid and Undercooled CuxZr1−x Alloys by Ab Initio Molecular Dynamics. Phys. Rev. B 2008, 78, 214204. [Google Scholar] [CrossRef]
- Korkmaz, Ş.; Korkmaz, S.D. Structure and Inter-Diffusion Coefficients of Liquid NaxK1−x Alloys. J. Phase Equilib. Diffus. 2010, 31, 15–21. [Google Scholar] [CrossRef]
- Ishii, Y.; Takanaga, T. Atomic and Electronic Structures and Dynamics in Liquid Alloys near Eutectic Point. J. Phys. Soc. Jpn. 2000, 69, 3334–3341. [Google Scholar] [CrossRef]
- Wang, C.C.; Wong, C.H. Short-to-Medium Range Order of Al–Mg Metallic Glasses Studied by Molecular Dynamics Simulations. J. Alloys Compd. 2011, 509, 10222–10229. [Google Scholar] [CrossRef]
- Korkmaz, S.D.; Korkmaz, Ş. A Study for Structure and Inter-Diffusion Coefficient of Liquid K1−xCsx Metal Alloys. Phys. Chem. Liq. 2011, 49, 801–810. [Google Scholar] [CrossRef]
- Costa Cabral, B. First Principles Molecular Dynamics of a Liquid Li–Na Alloy. J. Mol. Struct. Theochem 1999, 463, 145–149. [Google Scholar] [CrossRef]
- Bai, Y.W.; Zhao, X.L.; Bian, X.F.; Song, K.K.; Zhao, Y. Structure Evolution of Au50Cu50 Alloy from Melt to the Disordered Solid Solution. Mater. Sci. Forum 2020, 993, 273–280. [Google Scholar] [CrossRef]
- Shi, L.; Jia, L.; Ning, P.; Sun, X.; Wang, C.; Ma, Y.; Wang, F.; Qu, T.; Li, K. Vacuum Distillation and Ab Initio Molecular Dynamic Simulation of Al–Li Alloys. Vacuum 2023, 210, 111877. [Google Scholar] [CrossRef]
- Yang, S.J.; Hu, L.; Wang, L.; Wei, B. Molecular Dynamics Simulation of Liquid Structure for Undercooled Zr-Nb Alloys Assisted with Electrostatic Levitation Experiments. Chem. Phys. Lett. 2018, 701, 109–114. [Google Scholar] [CrossRef]
- Özdemir Kart, S.; Tomak, M.; Uludoğan, M.; Çağın, T. Liquid Properties of Pd–Ni Alloys. J. Non-Cryst. Solids 2004, 337, 101–108. [Google Scholar] [CrossRef]
- Liu, D.; Qin, J.Y.; Gu, T.K. The Structure of Liquid Mg–Cu Binary Alloys. J. Non-Cryst. Solids 2010, 356, 1587–1592. [Google Scholar] [CrossRef]
- Rao, R.V.G.; Venkatesh, R. Investigations of the Dynamic Properties of the Liquid Phase of Glassy Ca–Al Alloys. Phys. Stat. Sol. B 1992, 170, 39–46. [Google Scholar] [CrossRef]
- Mendelev, M.I.; Kramer, M.J.; Hao, S.G.; Ho, K.M.; Wang, C.Z. Development of Interatomic Potentials Appropriate for Simulation of Liquid and Glass Properties of NiZr2 Alloy. Philos. Mag. 2012, 92, 4454–4469. [Google Scholar] [CrossRef]
- Roik, O.S.; Yakovenko, O.M.; Kazimirov, V.P.; Sokol’skii, V.E.; Golovataya, N.V.; Kashirina, Y.O. Structure of Liquid Al Sn Alloys. J. Mol. Liq. 2021, 330, 115570. [Google Scholar] [CrossRef]
- Roik, O.S.; Samsonnikov, O.V.; Kazimirov, V.P.; Sokolskii, V.E.; Galushko, S.M. Medium-Range Order in Al-Based Liquid Binary Alloys. J. Mol. Liq. 2010, 151, 42–49. [Google Scholar] [CrossRef]
- Chen, F.; Cao, C.; Zhong, Q.; Liu, J.; Yang, L.; Chen, Z. Ab Initio Molecular Dynamics Study on Local Structure and Dynamic Properties of Liquid Ni62Nb38 Alloy. Mater. Today Commun. 2021, 27, 102207. [Google Scholar] [CrossRef]
- Gruner, S.; Kaban, I.; Kleinhempel, R.; Hoyer, W.; Jóvári, P.; Delaplane, R.G. Short-Range Order and Atomic Clusters in Liquid Cu–Sn Alloys. J. Non-Cryst. Solids 2005, 351, 3490–3496. [Google Scholar] [CrossRef]
- Jakse, N.; Nguyen, T.L.T.; Pasturel, A. Local Order and Dynamic Properties of Liquid Au x Si1−x Alloys by Molecular Dynamics Simulations. J. Chem. Phys. 2012, 137, 204504. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-Y.; Kim, H.; Hwang, G.S. A Comparative First-Principles Study of the Structure, Energetics, and Properties of Li–M (M = Si, Ge, Sn) Alloys. J. Phys. Chem. C 2011, 115, 20018–20026. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Qin, J.; Dong, B.; Pan, S. The Reassessment of the Structural Transition Regions along the Liquidus of Fe–Si Alloys and a Possible Liquid–Liquid Structural Transition in FeSi2 Alloy. Phys. Lett. A 2018, 382, 2655–2661. [Google Scholar] [CrossRef]
- Bhuiyan, G.M.; Ali, I.; Rahman, S.M.M. Atomic Transport Properties of AgIn Liquid Binary Alloys. Phys. B Condens. Matter 2003, 334, 147–159. [Google Scholar] [CrossRef]
- Weber, H.; Schumacher, M.; Jóvári, P.; Tsuchiya, Y.; Skrotzki, W.; Mazzarello, R.; Kaban, I. Experimental and Ab Initio Molecular Dynamics Study of the Structure and Physical Properties of Liquid GeTe. Phys. Rev. B 2017, 96, 054204. [Google Scholar] [CrossRef]
- Peng, H.L.; Voigtmann, T.; Kolland, G.; Kobatake, H.; Brillo, J. Structural and Dynamical Properties of Liquid Al–Au Alloys. Phys. Rev. B 2015, 92, 184201. [Google Scholar] [CrossRef]
- Guo, F.; Tian, Y.; Qin, J.; Xu, R.; Zhang, Y.; Zheng, H.; Lv, T.; Qin, X.; Tian, X.; Sun, Y. Structure of Liquid Cu–Sb Alloys by Ab Initio Molecular Dynamics Simulations, High Temperature X-ray Diffraction, and Resistivity. J Mater Sci 2013, 48, 4438–4445. [Google Scholar] [CrossRef]
- Franke, P.; Neuschütz, D. (Eds.) Binary Systems. Part 1_Elements and Binary Systems from Ag-Al to Au-Tl; Lehrstuhl für Theoretische Hüttenkunde, Rheinisch-Westfälische Technische Hochschule Aachen; Landolt-Börnstein—Group IV Physical Chemistry; Springer: Berlin/Heidelberg, Germany, 2002; Volume 19B1, ISBN 978-3-540-65327-1. [Google Scholar]
- Franke, P.; Neuschütz, D. (Eds.) Binary Systems. Part 2: Elements and Binary Systems from B–C to Cr–Zr; Landolt-Börnstein—Group IV Physical Chemistry; Springer: Berlin/Heidelberg, Germany, 2004; Volume 19B2, ISBN 978-3-540-20205-9. [Google Scholar]
- Franke, P.; Neuschütz, D. (Eds.) Binary Systems. Part 3: Binary Systems from Cs-K to Mg-Zr; Landolt-Börnstein—Group IV Physical Chemistry; Springer: Berlin/Heidelberg, Germany, 2005; Volume 19B3, ISBN 978-3-540-23119-6. [Google Scholar]
- Franke, P.; Neuschütz, D. (Eds.) Binary Systems. Part 4: Binary Systems from Mn-Mo to Y-Zr; Springer: Berlin/Heidelberg, Germany, 2006; Volume 19B4. [Google Scholar]
- Franke, P.; Neuschütz, D. (Eds.) Binary Systems. Part 5: Binary Systems Supplement 1: Phase Diagrams, Phase Transition Data, Integral and Partial Quantities of Alloys; Landolt-Börnstein—Group IV Physical Chemistry; Springer: Berlin/Heidelberg, Germany, 2007; Volume 19B5, ISBN 978-3-540-45279-9. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Co-Ni | Al-Zn | Cu-Ni | Al-Ni | Cu-Fe | Ge-Sn | Ag-Cu | Pb-Sb | Al-Si | Al-Co |
| 0 | 0 | 0.0028 | 0.0034 | 0.0048 | 0.007 | 0.0088 | 0.0096 | 0.0102 | 0.0114 | |
| System | Li-Mg | Sb-Sn | Cu-Zr | K-Na | Pb-Sn | Al-Mg | Cs-K | Li-Na | Au-Cu | Al-Li |
| 0.0114 | 0.0115 | 0.0119 | 0.0136 | 0.0158 | 0.0177 | 0.0228 | 0.0336 | 0.0364 | 0.0452 | |
| System | Nb-Zr | Ni-Pd | Cu-Mg | Al-Ca | Ni-Zr | Al-Sn | Al-Cu | Nb-Ni | Cu-Sn | Au-Si |
| 0.0473 | 0.0494 | 0.0597 | 0.0724 | 0.0807 | 0.0823 | 0.1116 | 0.1334 | 0.1342 | 0.139 | |
| System | Li-Sn | Fe-Si | Ag-In | Ge-Te | Al-Au | Cu-Sb | ||||
| 0.14 | 0.1454 | 0.1483 | 0.1548 | 0.160 | 0.208 |
| System | Co-Ni [20] | Al-Zn [21] | Cu-Ni [22] | Al-Ni [23] | Cu-Fe [24] | Ge-Sn [25] | Ag-Cu [23] | Pb-Sb [26] | Al-Si [27] | Al-Co [28] |
| System | Li-Mg [29] | Sb-Sn [30] | Cu-Zr [31] | K-Na [32] | Pb-Sn [33] | Al-Mg [34] | Cs-K [35] | Li-Na [36] | Au-Cu [37] | Al-Li [38] |
| System | Nb-Zr [39] | Ni-Pd [40] | Cu-Mg [41] | Al-Ca [42] | Ni-Zr [43] | Al-Sn [44] | Al-Cu [45] | Nb-Ni [46] | Cu-Sn [47] | Au-Si [48] |
| System | Li-Sn [49] | Fe-Si [50] | Ag-In [51] | Ge-Te [52] | Al-Au [53] | Cu-Sb [54] |
| System | MIVM | RSM | Wilson | NRTL | |||
|---|---|---|---|---|---|---|---|
| Co-Ni | 0.964 | 0.977 | 0.343 | 0.964 | 0.977 | −0.036 | −0.023 |
| Al-Zn | 1.246 | 0.743 | 0.423 | 1.166 | 0.795 | 0.220 | −0.297 |
| Cu-Ni | 1.086 | 0.863 | 0.374 | 0.999 | 0.938 | 0.082 | −0.147 |
| Al-Ni | 1.493 | 1.649 | −5.203 | 0.991 | 2.485 | 0.401 | 0.500 |
| Cu-Fe | 0.943 | 0.805 | 1.512 | 0.943 | 0.805 | −0.059 | −0.217 |
| Ge-Sn | 0.740 | 1.273 | 0.266 | 1.214 | 0.776 | −0.302 | 0.241 |
| Ag-Cu | 0.675 | 1.194 | 1.225 | 0.471 | 1.709 | −0.394 | 0.177 |
| Pb-Sb | 1.558 | 0.517 | 1.046 | 1.457 | 0.553 | 0.443 | −0.660 |
| Al-Si | 1.405 | 1.077 | −1.856 | 1.226 | 1.235 | 0.340 | 0.074 |
| Al-Co | 1.112 | 1.884 | −4.213 | 0.802 | 2.612 | 0.106 | 0.633 |
| Li-Mg | 1.357 | 0.914 | −1.095 | 1.469 | 0.844 | 0.305 | −0.090 |
| Sb-Sn | 0.769 | 1.545 | −0.847 | 0.735 | 1.618 | −0.262 | 0.435 |
| Cu-Zr | 0.908 | 1.669 | −2.277 | 1.782 | 0.851 | −0.096 | 0.512 |
| K-Na | 0.700 | 1.176 | 1.018 | 0.366 | 2.252 | −0.357 | 0.162 |
| Pb-Sn | 1.044 | 0.912 | 0.267 | 0.932 | 1.021 | 0.043 | −0.092 |
| Al-Mg | 0.820 | 1.123 | 0.459 | 1.162 | 0.793 | −0.198 | 0.116 |
| Cs-K | 1.204 | 0.635 | 1.310 | 0.801 | 0.955 | 0.185 | −0.454 |
| Li-Na | 0.764 | 0.999 | 1.347 | 1.416 | 0.539 | −0.270 | −0.001 |
| Au-Cu | 1.163 | 1.444 | −2.877 | 0.812 | 2.068 | 0.151 | 0.368 |
| Al-Li | 1.136 | 1.379 | −2.356 | 1.485 | 1.055 | 0.128 | 0.321 |
| Nb-Zr | 0.814 | 1.255 | −0.110 | 1.048 | 0.975 | −0.206 | 0.227 |
| Ni-Pd | 1.168 | 1.087 | −1.307 | 1.590 | 0.798 | 0.153 | 0.080 |
| Cu-Mg | 1.312 | 1.258 | −2.781 | 2.575 | 0.641 | 0.272 | 0.230 |
| Al-Ca | 2.202 | 1.259 | −5.761 | 5.826 | 0.476 | 0.789 | 0.230 |
| Ni-Zr | 1.522 | 1.730 | −5.373 | 3.247 | 0.811 | 0.420 | 0.548 |
| Al-Sn | 0.624 | 1.440 | 0.598 | 1.024 | 0.877 | −0.471 | 0.365 |
| Al-Cu | 1.568 | 1.530 | −5.209 | 2.192 | 1.152 | 0.476 | 0.450 |
| Nb-Ni | 1.772 | 1.071 | −3.558 | 1.070 | 1.774 | 0.572 | 0.069 |
| Cu-Sn | 1.987 | 0.849 | −2.904 | 0.874 | 1.932 | 0.687 | −0.163 |
| Au-Si | 1.224 | 1.684 | −3.129 | 1.034 | 1.993 | 0.202 | 0.521 |
| Li-Sn | 3.396 | 2.186 | −10.225 | 4.263 | 1.742 | 1.223 | 0.782 |
| Fe-Si | 5.678 | 1.776 | −9.822 | 4.695 | 2.148 | 1.737 | 0.574 |
| Ag-In | 1.595 | 0.925 | −2.227 | 2.476 | 0.596 | 0.467 | −0.078 |
| Ge-Te | 0.409 | 1.891 | 1.285 | 0.912 | 0.848 | −0.894 | 0.637 |
| Al-Au | 1.680 | 1.660 | −5.740 | 2.287 | 1.219 | 0.519 | 0.507 |
| Cu-Sb | 1.804 | 0.881 | −2.315 | 0.757 | 2.098 | 0.590 | −0.127 |
| System | Miedema | MIVM | RSM | Wilson | NRTL | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| SD | ARD/% | SD | ARD/% | SD | ARD/% | SD | ARD/% | SD | ARD/% | |
| Co-Ni | 0.013 | 3.852 | 0.036 | 10.8 | 0.029 | 8.8 | 0.003 | 0.8 | 0.015 | 4.4 |
| Al-Zn | 0.075 | 15.536 | 0.108 | 22 | 0.058 | 12.1 | 0.100 | 20.5 | 0.116 | 23.7 |
| Cu-Ni | 0.247 | 49.697 | 0.085 | 16.5 | 0.085 | 16.5 | 0.120 | 23.1 | 0.134 | 25.7 |
| Al-Ni | 0.210 | 1467.533 | 0.044 | 18.3 | 0.037 | 125 | 0.208 | 1472 | 0.398 | 4520 |
| Cu-Fe | 0.889 | 93.800 | 0.106 | 12.5 | 0.128 | 15.1 | 0.321 | 37.5 | 0.375 | 44 |
| Ge-Sn | 0.019 | 5.384 | 0.071 | 19.4 | 0.011 | 3.1 | 0.016 | 4.1 | 0.027 | 7.5 |
| Ag-Cu | 0.108 | 19.924 | 0.062 | 10.8 | 0.025 | 4.6 | 0.143 | 26.1 | 0.167 | 30.5 |
| Pb-Sb | 0.438 | 79.951 | 0.044 | 9.9 | 0.047 | 10.5 | 0.070 | 15.9 | 0.080 | 18.1 |
| Al-Si | 0.023 | 10.017 | 0.110 | 40.5 | 0.058 | 23.2 | 0.041 | 18.5 | 0.125 | 63.3 |
| Al-Co | 0.034 | 10.163 | 0.080 | 39.2 | 0.027 | 10.5 | 0.142 | 341 | 0.300 | 1085 |
| Li-Mg | 0.043 | 17.882 | 0.056 | 20.7 | 0.032 | 12.3 | 0.035 | 14.2 | 0.082 | 34.9 |
| Sb-Sn | 0.025 | 6.367 | 0.078 | 29.3 | 0.012 | 3.6 | 0.054 | 24.5 | 0.093 | 44.2 |
| Cu-Zr | 0.124 | 53.790 | 0.064 | 31.6 | 0.015 | 8.4 | 0.098 | 74.4 | 0.185 | 162 |
| K-Na | 0.019 | 3.619 | 0.081 | 15.3 | 0.012 | 2 | 0.144 | 27.8 | 0.145 | 28 |
| Pb-Sn | 0.015 | 1.847 | 0.016 | 3.2 | 0.009 | 1.7 | 0.063 | 14.3 | 0.087 | 19.6 |
| Al-Mg | 0.042 | 14.076 | 0.087 | 32.9 | 0.092 | 34.9 | 0.048 | 17 | 0.033 | 11.2 |
| Cs-K | 0.020 | 4.475 | 0.129 | 36 | 0.148 | 41.7 | 0.016 | 3.3 | 0.066 | 16.9 |
| Li-Na | 0.255 | 24.451 | 0.210 | 22.9 | 0.192 | 21.3 | 0.364 | 40.5 | 0.405 | 45.2 |
| Au-Cu | 0.028 | 7.697 | 0.076 | 34.5 | 0.038 | 12.1 | 0.105 | 101 | 0.219 | 255 |
| Al-Li | 0.034 | 17.993 | 0.048 | 17 | 0.037 | 12.2 | 0.118 | 115 | 0.209 | 236 |
| Nb-Zr | 0.036 | 8.347 | 0.089 | 21.1 | 0.066 | 15.1 | 0.058 | 13 | 0.056 | 12.4 |
| Ni-Pd | 0.115 | 72.225 | 0.044 | 15 | 0.039 | 14.1 | 0.090 | 53.7 | 0.140 | 91 |
| Cu-Mg | 0.022 | 8.778 | 0.054 | 30.2 | 0.038 | 12.4 | 0.104 | 103 | 0.226 | 286 |
| Al-Ca | 0.100 | 56.098 | 0.129 | 64.3 | 0.087 | 37.4 | 0.094 | 144 | 0.338 | 1081 |
| Ni-Zr | 0.080 | 20.755 | 0.076 | 80.2 | 0.085 | 266 | 0.225 | 1661 | 0.433 | 4415 |
| Al-Sn | 0.122 | 18.737 | 0.226 | 34.4 | 0.142 | 21.3 | 0.199 | 30.4 | 0.226 | 34.5 |
| Al-Cu | 0.099 | 228.621 | 0.076 | 43 | 0.075 | 30.1 | 0.158 | 516 | 0.343 | 2031 |
| Nb-Ni | 0.159 | 517.211 | 0.128 | 47.2 | 0.089 | 64 | 0.161 | 540 | 0.288 | 1478 |
| Cu-Sn | 0.073 | 38.233 | 0.131 | 51.2 | 0.103 | 33.3 | 0.135 | 48 | 0.524 | 544 |
| Au-Si | 0.106 | 43.309 | 0.116 | 44 | 0.090 | 45.6 | 0.147 | 279 | 0.290 | 847 |
| Li-Sn | 0.081 | 159.055 | 0.098 | 59.1 | 0.117 | 130 | 0.216 | 1347 | 0.596 | 7906 |
| Fe-Si | 0.095 | 67.563 | 0.259 | 82.9 | 0.137 | 58.3 | 0.088 | 186 | 0.516 | 5003 |
| Ag-In | 0.114 | 97.817 | 0.077 | 32.8 | 0.095 | 30.3 | 0.095 | 76 | 0.185 | 191 |
| Ge-Te | 0.240 | 319.161 | 0.099 | 27.3 | 0.333 | 496 | 0.195 | 239 | 0.150 | 148 |
| Al-Au | 0.159 | 53.722 | 0.149 | 55.9 | 0.139 | 42.7 | 0.168 | 405 | 0.333 | 1822 |
| Cu-Sb | 0.095 | 80.468 | 0.072 | 28 | 0.096 | 30.4 | 0.122 | 104 | 0.196 | 229 |
| Ave | 0.121 | 102.7 | 0.095 | 32.2 | 0.078 | 47.4 | 0.124 | 226 | 0.225 | 911 |
| System | MIVM | RSM | Wilson | NRTL | |||
|---|---|---|---|---|---|---|---|
| Co-Ni | 1.089 | 0.900 | 0.118 | 1.089 | 0.900 | 0.085 | −0.106 |
| Al-Zn | 1.317 | 0.938 | −1.162 | 1.232 | 1.003 | 0.275 | −0.064 |
| Cu-Ni | 1.391 | 0.877 | −1.138 | 1.280 | 0.953 | 0.330 | −0.132 |
| Al-Ni | 1.155 | 2.099 | −5.114 | 0.766 | 3.163 | 0.144 | 0.742 |
| Cu-Fe | 0.890 | 1.145 | −0.103 | 0.890 | 1.145 | −0.117 | 0.135 |
| Ge-Sn | 0.562 | 1.573 | 0.545 | 0.923 | 0.958 | −0.576 | 0.453 |
| Ag-Cu | 1.309 | 0.582 | 1.538 | 0.914 | 0.833 | 0.269 | −0.542 |
| Pb-Sb | 1.231 | 0.810 | 0.017 | 1.151 | 0.866 | 0.207 | −0.211 |
| Al-Si | 1.534 | 1.140 | −2.499 | 1.338 | 1.307 | 0.428 | 0.131 |
| Al-Co | 0.830 | 1.774 | −2.218 | 0.551 | 2.673 | −0.186 | 0.573 |
| Li-Mg | 1.087 | 0.988 | −0.363 | 1.177 | 0.912 | 0.083 | −0.012 |
| Sb-Sn | 0.825 | 1.576 | −1.288 | 0.788 | 1.650 | −0.192 | 0.455 |
| Cu-Zr | 1.206 | 1.206 | 1.206 | 1.206 | 1.206 | 1.206 | 1.206 |
| K-Na | 0.562 | 1.364 | 1.390 | 0.293 | 2.613 | −0.577 | 0.311 |
| Pb-Sn | 1.601 | 0.830 | −1.549 | 1.430 | 0.929 | 0.471 | −0.186 |
| Al-Mg | 0.808 | 1.126 | 0.528 | 1.144 | 0.796 | −0.213 | 0.119 |
| Cs-K | 1.642 | 0.660 | −0.391 | 1.092 | 0.992 | 0.496 | −0.416 |
| Li-Na | 0.728 | 1.069 | 1.245 | 1.350 | 0.577 | −0.317 | 0.067 |
| Au-Cu | 2.423 | 0.515 | −1.229 | 1.692 | 0.737 | 0.885 | −0.664 |
| Al-Li | 0.813 | 1.374 | −0.580 | 1.062 | 1.051 | −0.208 | 0.318 |
| Nb-Zr | 0.671 | 1.414 | 0.279 | 0.864 | 1.098 | −0.399 | 0.346 |
| Ni-Pd | 1.174 | 1.105 | 11.250 | −1.445 | 1.599 | 0.811 | 0.159 |
| Cu-Mg | 0.882 | 1.538 | −1.689 | 1.731 | 0.783 | −0.126 | 0.430 |
| Al-Ca | 2.423 | 0.515 | −1.229 | 1.692 | 0.737 | 0.885 | −0.664 |
| Ni-Zr | 2.150 | 4.152 | −12.151 | 4.587 | 1.947 | 0.766 | 1.424 |
| Al-Sn | 0.649 | 1.393 | 0.566 | 1.065 | 0.849 | −0.433 | 0.332 |
| Al-Cu | 1.963 | 1.534 | −6.647 | 1.472 | 2.180 | 0.714 | 0.453 |
| Nb-Ni | 3.602 | 1.064 | −7.456 | 2.174 | 1.763 | 1.281 | 0.062 |
| Cu-Sn | 4.758 | 0.617 | −5.972 | 2.091 | 1.403 | 1.560 | −0.484 |
| Au-Si | 1.438 | 0.875 | −0.992 | 1.214 | 1.036 | 0.363 | −0.134 |
| Li-Sn | 2.077 | 2.672 | −8.741 | 2.608 | 2.129 | 0.731 | 0.983 |
| Fe-Si | 1.534 | 1.843 | −4.414 | 1.268 | 2.228 | 0.428 | 0.611 |
| Ag-In | 1.066 | 1.467 | −2.560 | 1.654 | 0.946 | 0.064 | 0.384 |
| Ge-Te | 0.636 | 3.025 | −3.272 | 1.419 | 1.356 | −0.452 | 1.107 |
| Al-Au | 1.903 | 0.844 | −2.370 | 0.799 | 2.011 | 0.643 | −0.169 |
| Cu-Sb | 1.743 | 1.017 | −3.207 | 1.743 | 1.017 | 0.556 | 0.017 |
| System | Miedema | MIVM | RSM | Wilson | NRTL | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| SD | ARD/% | SD | ARD/% | SD | ARD/% | SD | ARD/% | SD | ARD/% | |
| Co-Ni | 0.013 | 3.852 | 0.002 | 0.4 | 0.004 | 1.1 | 0.007 | 2.1 | 0.011 | 3.3 |
| Al-Zn | 0.075 | 15.536 | 0.233 | 45.8 | 0.201 | 40 | 0.128 | 26 | 0.085 | 17.5 |
| Cu-Ni | 0.247 | 49.697 | 0.252 | 46.9 | 0.220 | 41.3 | 0.147 | 28.3 | 0.107 | 20.8 |
| Al-Ni | 0.210 | 1467.533 | 0.081 | 31.7 | 0.039 | 133 | 0.257 | 2130 | 0.335 | 3370 |
| Cu-Fe | 0.889 | 93.800 | 0.365 | 42.8 | 0.359 | 42 | 0.351 | 41.2 | 0.347 | 40.7 |
| Ge-Sn | 0.019 | 5.384 | 0.135 | 35.4 | 0.045 | 13.2 | 0.007 | 1.6 | 0.039 | 10.8 |
| Ag-Cu | 0.108 | 19.924 | 0.063 | 10.2 | 0.087 | 16.3 | 0.115 | 21.2 | 0.175 | 31.8 |
| Pb-Sb | 0.438 | 79.951 | 0.054 | 17.9 | 0.164 | 62.6 | 0.038 | 13.3 | 0.004 | 0.9 |
| Al-Si | 0.023 | 10.017 | 0.147 | 50.8 | 0.089 | 34.2 | 0.027 | 12.1 | 0.142 | 72.6 |
| Al-Co | 0.034 | 10.163 | 0.074 | 39.6 | 0.070 | 130 | 0.161 | 411 | 0.258 | 852 |
| Li-Mg | 0.043 | 17.882 | 0.020 | 7.9 | 0.026 | 10.2 | 0.052 | 21.7 | 0.067 | 28.5 |
| Sb-Sn | 0.025 | 6.367 | 0.100 | 36.5 | 0.029 | 11.3 | 0.046 | 20.7 | 0.103 | 49.6 |
| Cu-Zr | 0.124 | 53.790 | 0.065 | 33 | 0.026 | 14.4 | 0.084 | 62.1 | 0.193 | 171 |
| K-Na | 0.019 | 3.619 | 0.130 | 24.3 | 0.076 | 15.2 | 0.154 | 29.6 | 0.156 | 29.9 |
| Pb-Sn | 0.015 | 1.847 | 0.244 | 51.4 | 0.194 | 42 | 0.105 | 23.7 | 0.049 | 11 |
| Al-Mg | 0.042 | 14.076 | 0.095 | 36.2 | 0.101 | 38.4 | 0.049 | 17.5 | 0.031 | 10.7 |
| Cs-K | 0.020 | 4.475 | 0.132 | 32.3 | 0.074 | 18.8 | 0.046 | 11.5 | 0.036 | 8.9 |
| Li-Na | 0.255 | 24.451 | 0.244 | 26.7 | 0.211 | 23.4 | 0.363 | 40.4 | 0.404 | 45.1 |
| Au-Cu | 0.028 | 7.697 | 0.117 | 40.7 | 0.067 | 58.8 | 0.135 | 137 | 0.168 | 181 |
| Al-Li | 0.034 | 17.993 | 0.093 | 86.2 | 0.110 | 106 | 0.151 | 156 | 0.171 | 182 |
| Nb-Zr | 0.036 | 8.347 | 0.092 | 21.6 | 0.032 | 6.3 | 0.052 | 11.5 | 0.065 | 14.7 |
| Ni-Pd | 0.115 | 72.225 | 0.046 | 15.2 | 0.036 | 11.3 | 0.088 | 52.4 | 0.143 | 93.2 |
| Cu-Mg | 0.022 | 8.778 | 0.065 | 22.3 | 0.051 | 40 | 0.136 | 144 | 0.202 | 245 |
| Al-Ca | 0.100 | 56.098 | 0.089 | 50.3 | 0.075 | 20 | 0.107 | 175 | 0.301 | 901 |
| Ni-Zr | 0.080 | 20.755 | 0.248 | 75.6 | 0.098 | 35.8 | 0.140 | 86.2 | 0.573 | 6633.0 |
| Al-Sn | 0.122 | 18.737 | 0.216 | 32.8 | 0.147 | 21.9 | 0.200 | 30.6 | 0.224 | 34.3 |
| Al-Cu | 0.099 | 228.621 | 0.113 | 57.6 | 0.078 | 39.9 | 0.114 | 289 | 0.417 | 2930 |
| Nb-Ni | 0.159 | 517.211 | 0.216 | 67.7 | 0.104 | 49.9 | 0.121 | 302 | 0.363 | 2270 |
| Cu-Sn | 0.073 | 38.233 | 0.255 | 75.4 | 0.163 | 56.7 | 0.076 | 28 | 0.200 | 167 |
| Au-Si | 0.106 | 43.309 | 0.070 | 79 | 0.136 | 240 | 0.192 | 425 | 0.234 | 591 |
| Li-Sn | 0.081 | 159.055 | 0.150 | 86.9 | 0.116 | 187 | 0.244 | 1730 | 0.564 | 7420 |
| Fe-Si | 0.095 | 67.563 | 0.138 | 45.8 | 0.105 | 79.9 | 0.165 | 693 | 0.358 | 2900 |
| Ag-In | 0.114 | 97.817 | 0.122 | 37.9 | 0.097 | 30.1 | 0.109 | 90.8 | 0.193 | 204 |
| Ge-Te | 0.240 | 319.161 | 0.245 | 73.9 | 0.114 | 29.1 | 0.135 | 118 | 0.211 | 268 |
| Al-Au | 0.159 | 53.722 | 0.149 | 55.9 | 0.139 | 42.7 | 0.168 | 405 | 0.333 | 1820 |
| Cu-Sb | 0.095 | 80.468 | 0.072 | 29.9 | 0.097 | 30 | 0.122 | 105 | 0.196 | 228 |
| Ave | 0.121 | 102.7 | 0.137 | 42.35 | 0.105 | 49.24 | 0.128 | 219.2 | 0.207 | 884.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hang, J.; Tao, D. Estimation of Two Component Activities of Binary Liquid Alloys by the Pair Potential Energy Containing a Polynomial of the Partial Radial Distribution Function. Metals 2023, 13, 1773. https://doi.org/10.3390/met13101773
Hang J, Tao D. Estimation of Two Component Activities of Binary Liquid Alloys by the Pair Potential Energy Containing a Polynomial of the Partial Radial Distribution Function. Metals. 2023; 13(10):1773. https://doi.org/10.3390/met13101773
Chicago/Turabian StyleHang, Jiulong, and Dongping Tao. 2023. "Estimation of Two Component Activities of Binary Liquid Alloys by the Pair Potential Energy Containing a Polynomial of the Partial Radial Distribution Function" Metals 13, no. 10: 1773. https://doi.org/10.3390/met13101773
APA StyleHang, J., & Tao, D. (2023). Estimation of Two Component Activities of Binary Liquid Alloys by the Pair Potential Energy Containing a Polynomial of the Partial Radial Distribution Function. Metals, 13(10), 1773. https://doi.org/10.3390/met13101773