Genetic Diversity of Dengue Vector Aedes albopictus Collected from South Korea, Japan, and Laos

 , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
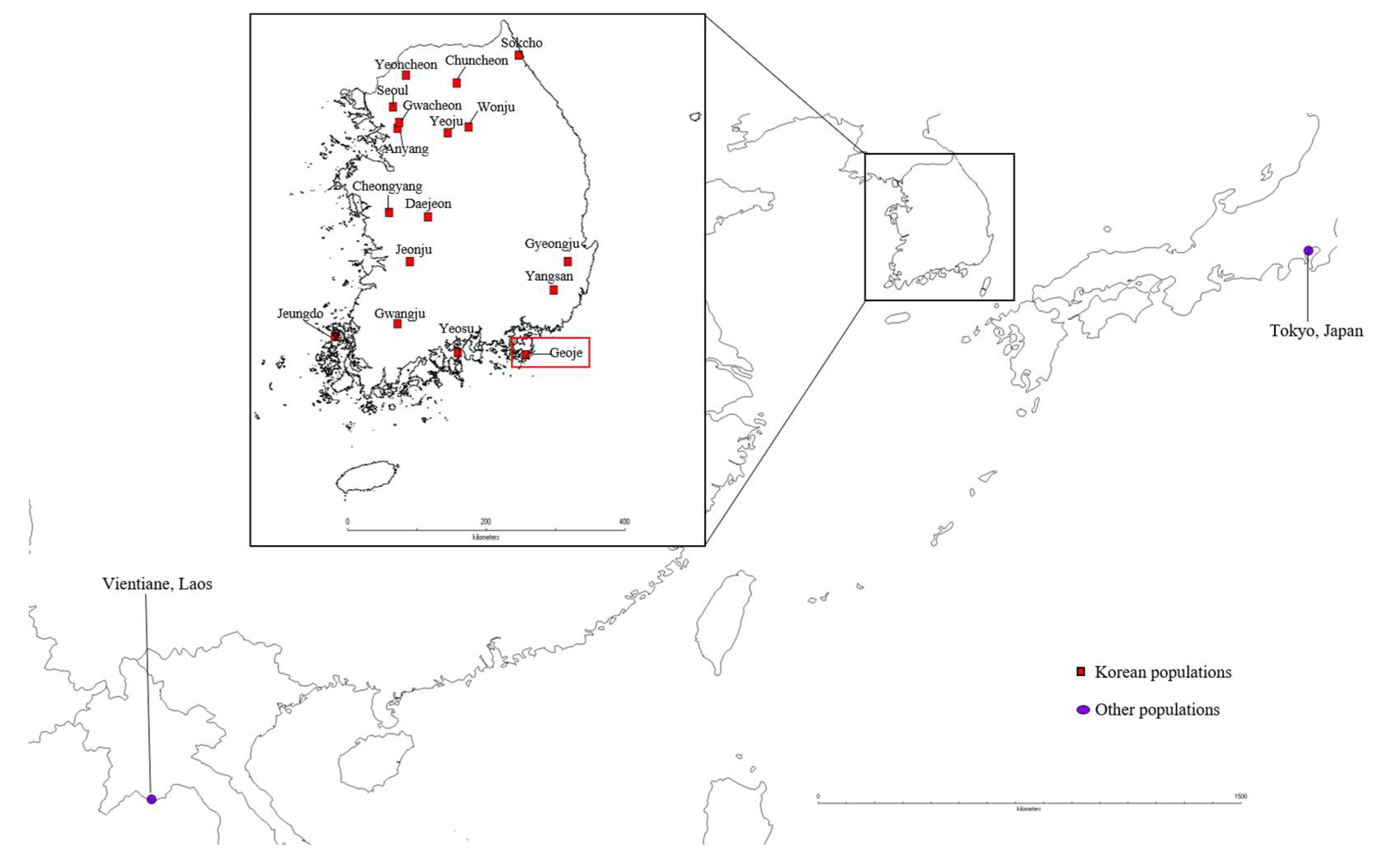
2.1. Sampling and DNA Extraction
2.2. Molecular Methods (Polymerase Chain Reaction [PCR])
2.3. Mitochondrial DNA Data Analysis for Genetic Diversity and Gene Flow
2.4. Microsatellite Data Analyses
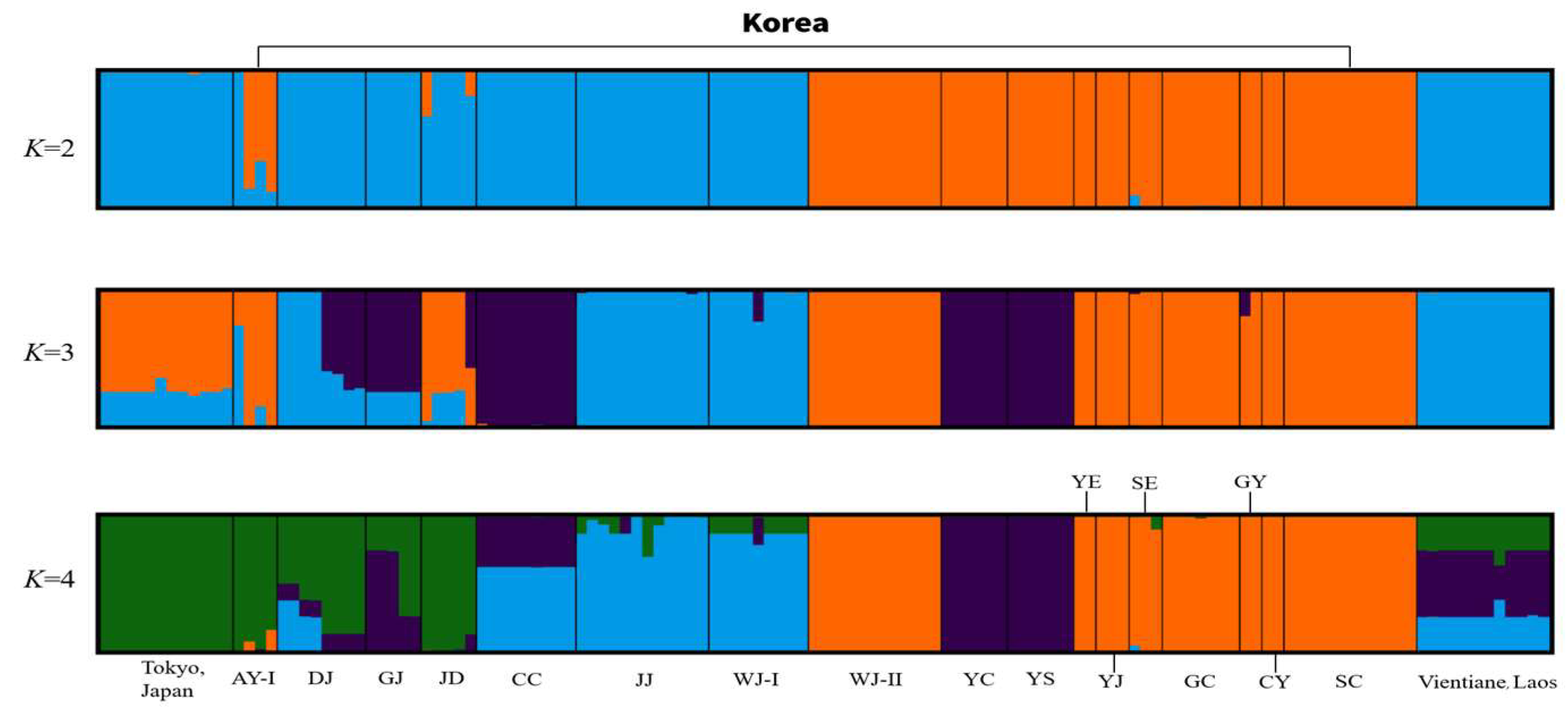
3. Results
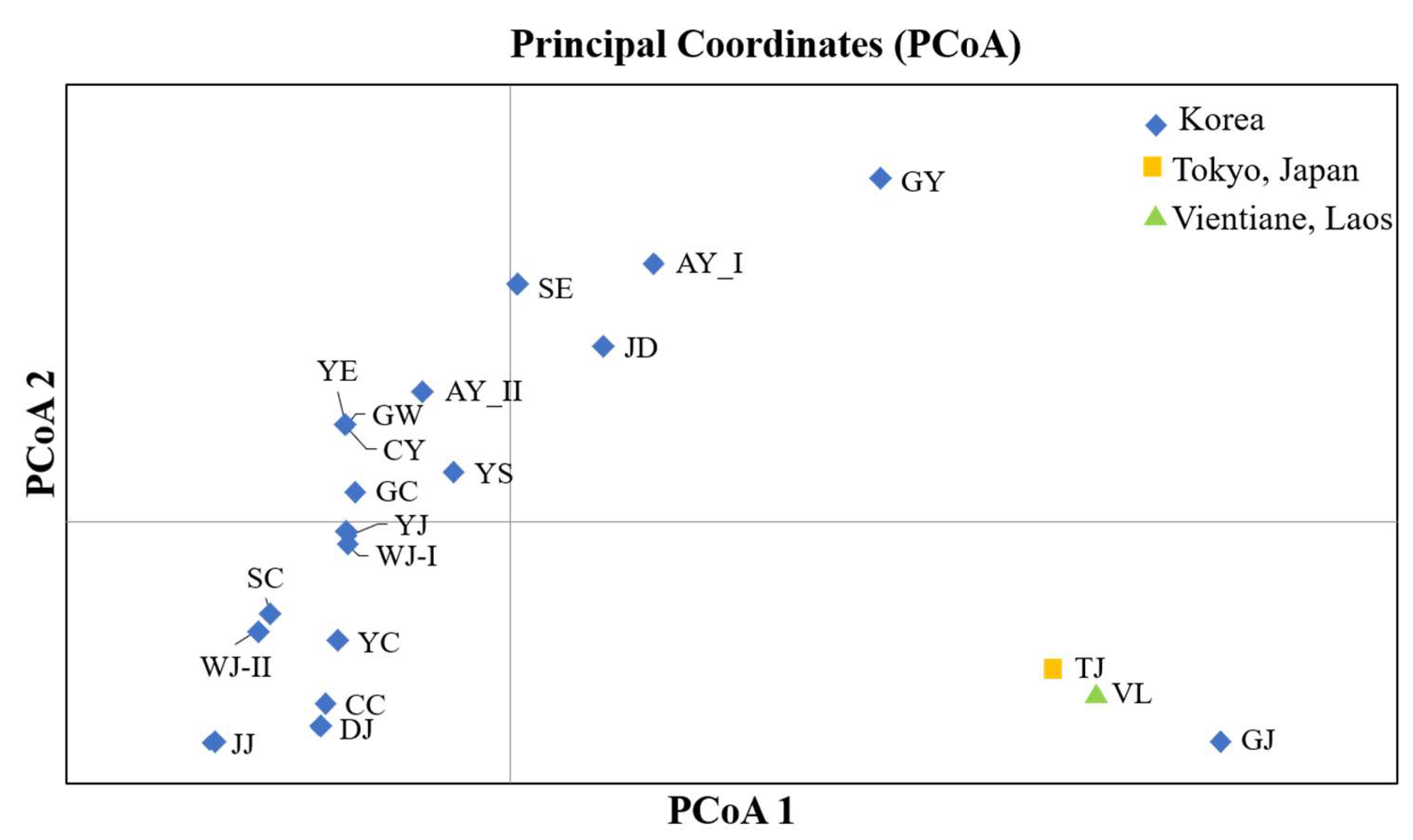
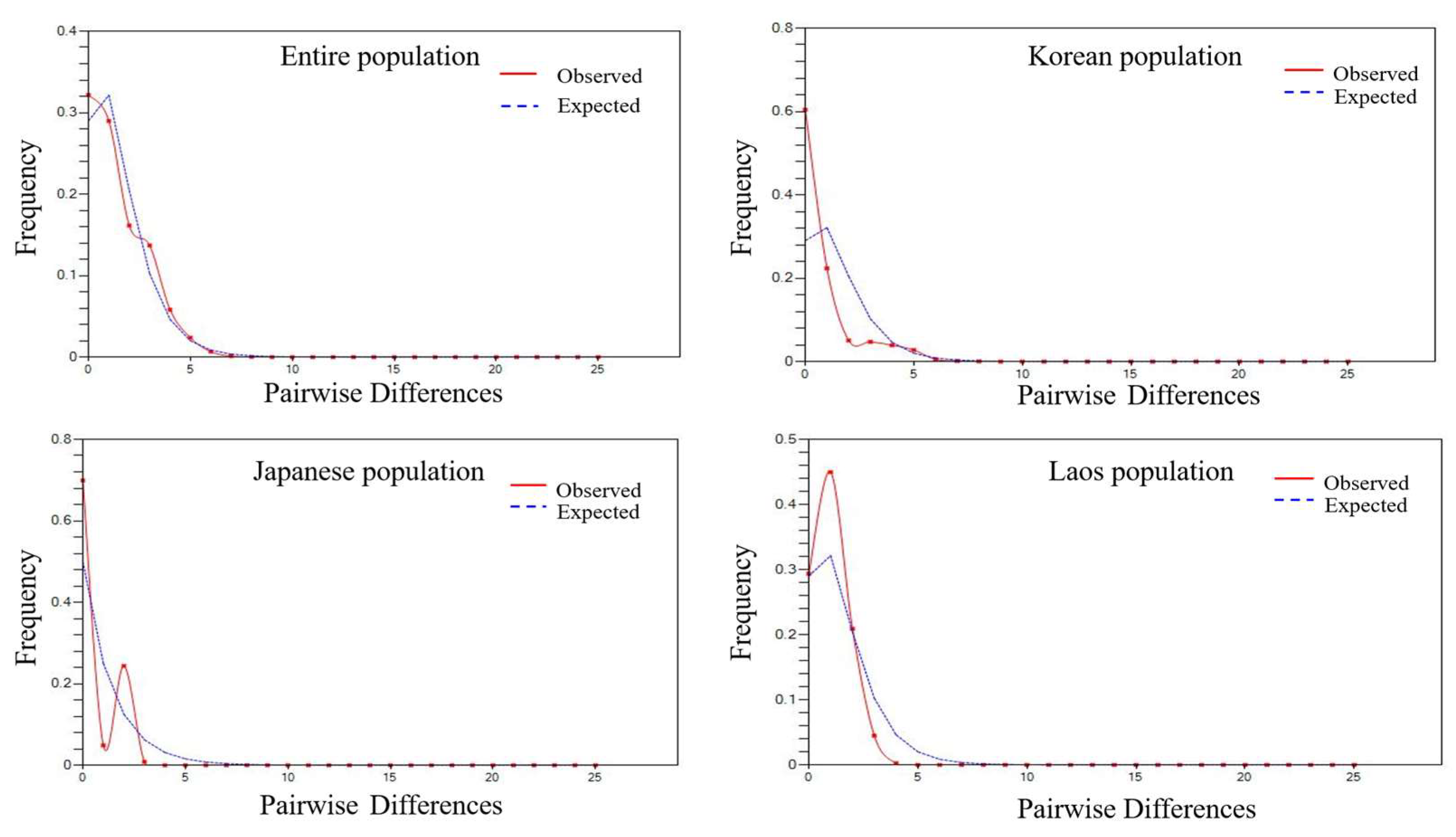
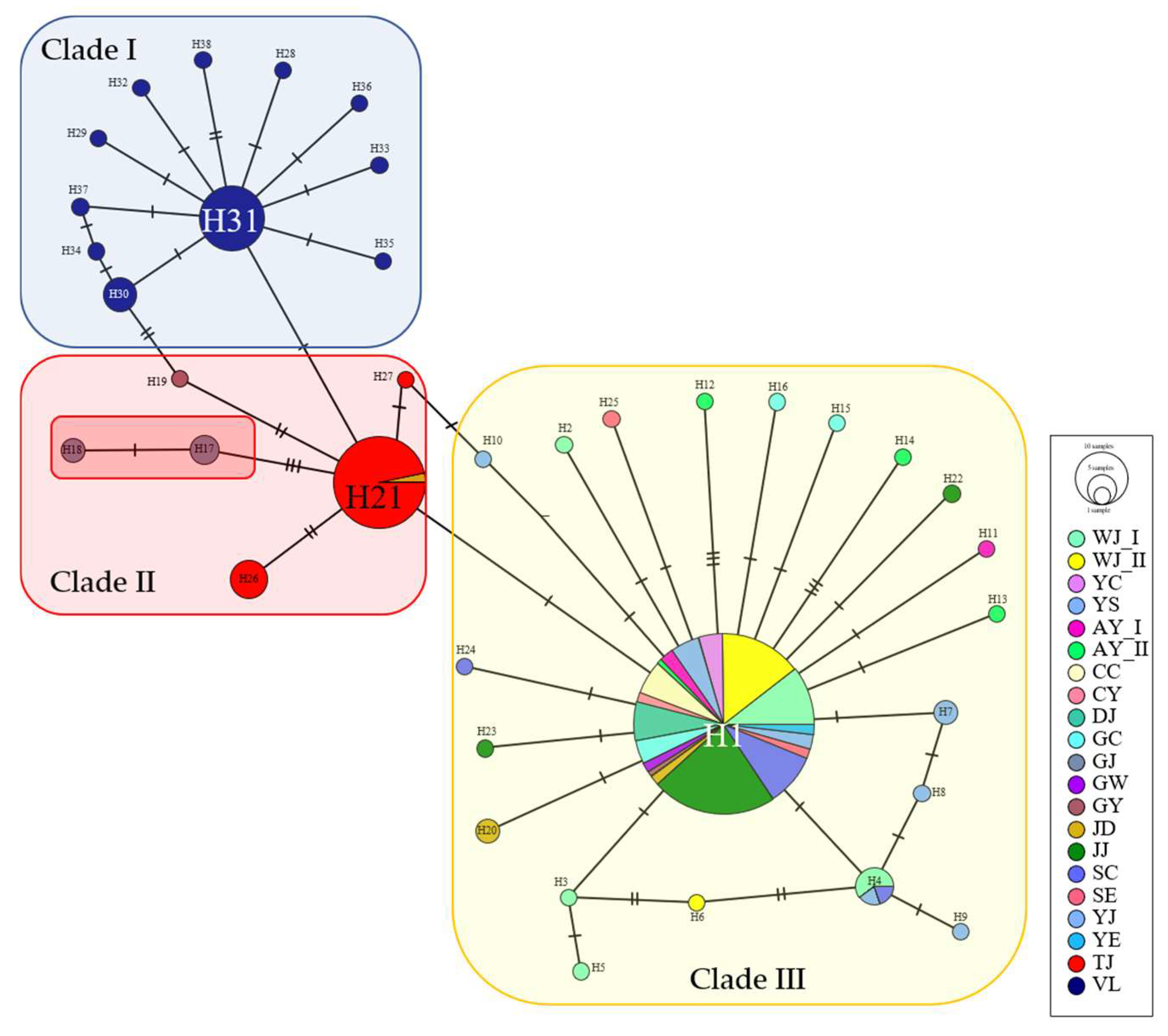
3.1. Genetic Diversity and Population Structure of Mitochondrial DNA
3.2. Genetic Diversity and Population Structure of Ae. albopictus Based on Microsatellite Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Randolph, S.E.; Rogers, D.J. The arrival, establishment and spread of exotic diseases: Patterns and predictions. Nat. Rev. Microbiol. 2010, 8, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Hulme, P.E. Trade, transport and trouble: Managing invasive species pathways in an era of globalization. J. Appl. Ecol. 2009, 46, 10–18. [Google Scholar] [CrossRef]
- Kenis, M.; Auger-Rozenberg, M.-A.; Roques, A.; Timms, L.; Péré, C.; Cock, M.J.; Settele, J.; Augustin, S.; Lopez-Vaamonde, C. Ecological effects of invasive alien insects. Biol. Invasions 2009, 11, 21–45. [Google Scholar] [CrossRef]
- Juliano, S.A.; Philip Lounibos, L. Ecology of invasive mosquitoes: Effects on resident species and on human health. Ecol. Lett 2005, 8, 558–574. [Google Scholar] [CrossRef] [PubMed]
- Demeulemeester, J.; Deblauwe, I.; De Witte, J.C.; Jansen, F.; Hendy, A.; Madder, M. First interception of Aedes (Stegomyia) albopictus in Lucky bamboo shipments in Belgium. J. Eur. Mosq. Control. Assoc. 2014, 32, 14–16. [Google Scholar]
- Ibáñez-Justicia, A.; Koenraadt, C.J.; Stroo, A.; Van Lammeren, R.; Takken, W. Risk-based and adaptive invasive mosquito surveillance at lucky bamboo and used tire importers in the netherlands. J. Am. Mosq. Control. Assoc. 2020, 36, 89–98. [Google Scholar] [CrossRef]
- Tatem, A.J.; Hay, S.I.; Rogers, D.J. Global traffic and disease vector dispersal. Proc. Natl. Acad. Sci. USA 2006, 103, 6242–6247. [Google Scholar] [CrossRef]
- Reiter, P. Climate change and mosquito-borne disease. Env. Health Perspect. 2001, 109, 141–161. [Google Scholar]
- Boyer, S.; Marcombe, S.; Yean, S.; Fontenille, D. High diversity of mosquito vectors in Cambodian primary schools and consequences for arbovirus transmission. PloS ONE 2020, 15, e0233669. [Google Scholar] [CrossRef]
- Levin, M.L. Medical entomology for students. Emerg. Infect. Dis. 2014, 20, 1428. [Google Scholar] [CrossRef]
- Morens, D.M.; Folkers, G.K.; Fauci, A.S. The challenge of emerging and re-emerging infectious diseases. Nature 2004, 430, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Medlock, J.; Hansford, K.; Versteirt, V.; Cull, B.; Kampen, H.; Fontenille, D.; Hendrickx, G.; Zeller, H.; Van Bortel, W.; Schaffner, F. An entomological review of invasive mosquitoes in Europe. Bull. Entomol. Res. 2015, 105, 637–663. [Google Scholar] [CrossRef] [PubMed]
- Petrić, D.; Bellini, R.; Scholte, E.-J.; Rakotoarivony, L.M.; Schaffner, F. Monitoring population and environmental parameters of invasive mosquito species in Europe. Parasites Vectors 2014, 7, 187. [Google Scholar] [CrossRef]
- Motoki, M.T.; Fonseca, D.M.; Miot, E.F.; Demari-Silva, B.; Thammavong, P.; Chonephetsarath, S.; Phommavanh, N.; Hertz, J.C.; Kittayapong, P.; Brey, P.T. Population genetics of Aedes albopictus (Diptera: Culicidae) in its native range in Lao People’s Democratic Republic. Parasites Vectors 2019, 12, 477. [Google Scholar] [CrossRef] [PubMed]
- Kutsuna, S.; Kato, Y.; Moi, M.L.; Kotaki, A.; Ota, M.; Shinohara, K.; Kobayashi, T.; Yamamoto, K.; Fujiya, Y.; Mawatari, M. Autochthonous dengue fever, Tokyo, Japan, 2014. Emerg. Infect. Dis. 2015, 21, 517. [Google Scholar] [CrossRef]
- Miki, S.; Lee, W.-C.; Lee, M.-J. A comparative study of the trends of imported Dengue cases in Korea and Japan 2011-2015. J. Clin. Med. Res. 2017, 9, 650. [Google Scholar] [CrossRef]
- Gloria-Soria, A.; Ayala, D.; Bheecarry, A.; Calderon-Arguedas, O.; Chadee, D.D.; Chiappero, M.; Coetzee, M.; Elahee, K.B.; Fernandez-Salas, I.; Kamal, H.A. Global genetic diversity of Aedes aegypti. Mol. Ecol. 2016, 25, 5377–5395. [Google Scholar] [CrossRef]
- Alphey, L.; McKemey, A.; Nimmo, D.; Neira Oviedo, M.; Lacroix, R.; Matzen, K.; Beech, C. Genetic control of Aedes mosquitoes. Pathog. Glob. Health 2013, 107, 170–179. [Google Scholar] [CrossRef]
- Kang, S.; Jung, J.; Kim, W. Population genetic structure of the malaria vector Anopheles sinensis (Diptera: Culicidae) sensu stricto and evidence for possible introgression in the Republic of Korea. J. Med. Entomol 2015, 52, 1270–1281. [Google Scholar] [CrossRef]
- Lee, E.; Yang, S.-C.; Kim, T.-K.; Noh, B.-E.; Lee, H.S.; Kim, H.; Roh, J.Y.; Lee, W.-G. Geographical Genetic Variation and Sources of Korean Aedes albopictus (Diptera: Culicidae) Populations. J. Med. Entomol. 2020, 57, 1057–1068. [Google Scholar] [CrossRef]
- Porretta, D.; Mastrantonio, V.; Bellini, R.; Somboon, P.; Urbanelli, S. Glacial history of a modern invader: Phylogeography and species distribution modelling of the Asian tiger mosquito Aedes albopictus. PLoS ONE 2012, 7, e44515. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Jung, Y.; Min, G.-S.; Kim, W. Analysis of the population genetic structure of the malaria vector Anopheles sinensis in South Korea based on mitochondrial sequences. Am. J. Trop Med. Hyg. 2007, 77, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Nam, K.W.; Jeong, J.Y.; Yoo, S.J.; Koh, Y.-S.; Lee, S.; Heo, S.T.; Seong, S.-Y.; Lee, K.H. The effects of climate change and globalization on mosquito vectors: Evidence from Jeju Island, South Korea on the potential for Asian tiger mosquito (Aedes albopictus) influxes and survival from Vietnam rather than Japan. PloS ONE 2013, 8, e68512. [Google Scholar] [CrossRef]
- Yang, S.; Lee, E.; Lee, W.; Shin-Hyeong, C. Geographical distribution of Aedes albopictus around urban areas in Korea. Public Health Wkly. Rep. 2018, 11, 463–468. [Google Scholar]
- KCDC. Infectious Diseases Surveillance Yearbook. 2019. Available online: https://www.kdca.go.kr/npt/biz/npp/portal/nppPblctDtaView.do?pblctDtaSeAt=1&pblctDtaSn=2139 (accessed on 29 June 2020).
- Yeom, J.-S. Current status and outlook of mosquito-borne diseases in Korea. J. Korean Med. Assoc. 2017, 60, 468–474. [Google Scholar] [CrossRef]
- Park, J.-H.; Lee, D.-W. Dengue fever in South Korea, 2006–2010. Emerg. Infect. Dis. 2012, 18, 1525. [Google Scholar] [CrossRef] [PubMed]
- Maynard, A.J.; Ambrose, L.; Cooper, R.D.; Chow, W.K.; Davis, J.B.; Muzari, M.O.; van den Hurk, A.F.; Hall-Mendelin, S.; Hasty, J.M.; Burkot, T.R. Tiger on the prowl: Invasion history and spatio-temporal genetic structure of the Asian tiger mosquito Aedes albopictus (Skuse 1894) in the Indo-Pacific. PLoS Negl. Trop. Dis. 2017, 11, e0005546. [Google Scholar] [CrossRef]
- Pless, E.; Gloria-Soria, A.; Evans, B.R.; Kramer, V.; Bolling, B.G.; Tabachnick, W.J.; Powell, J.R. Multiple introductions of the dengue vector, Aedes aegypti, into California. PLoS Negl. Trop. Dis. 2017, 11, e0005718. [Google Scholar] [CrossRef]
- Elnour, M.-A.B.; Gloria-Soria, A.; Azrag, R.S.; Alkhaibari, A.M.; Powell, J.R.; Salim, B. Population genetic analysis of Aedes aegypti mosquitoes from Sudan revealed recent independent colonization events by the two subspecies. Front. Genet. 2022, 104, 825652. [Google Scholar] [CrossRef]
- Kamau, L.; Mukabana, W.; Hawley, W.; Lehmann, T.; Irungu, L.; Orago, A.; Collins, F. Analysis of genetic variability in Anopheles arabiensis and Anopheles gambiae using microsatellite loci. Insect Mol. Biol. 1999, 8, 287–297. [Google Scholar] [CrossRef]
- Carrazco-Montalvo, A.; Ponce, P.; Villota, S.D.; Quentin, E.; Muñoz-Tobar, S.; Coloma, J.; Cevallos, V. Establishment, Genetic Diversity, and Habitat Suitability of Aedes albopictus Populations from Ecuador. Insects 2022, 13, 305. [Google Scholar] [CrossRef] [PubMed]
- Lv, R.-C.; Zhu, C.-Q.; Wang, C.-H.; Ai, L.-L.; Lv, H.; Zhang, B.; Li, C.-M.; An, J.; Wang, P.-G.; Hu, D. Genetic diversity and population structure of Aedes aegypti after massive vector control for dengue fever prevention in Yunnan border areas. Sci. Rep. 2020, 10, 12731. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Jung, J. Comparative population genetics of the invasive mosquito Aedes albopictus and the native mosquito Aedes flavopictus in the Korean peninsula. Parasites Vectors 2021, 14, 377. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, M.U.; Sinka, M.E.; Duda, K.A.; Mylne, A.Q.; Shearer, F.M.; Barker, C.M.; Moore, C.G.; Carvalho, R.G.; Coelho, G.E.; Van Bortel, W. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. eLife 2015, 4, e08347. [Google Scholar] [CrossRef]
- López-Pujol, J. The Importance of Biological Interactions in the Study of Biodiversity; BoD–Books on Demand; IntechOpen Limited: London, UK, 2011. [Google Scholar]
- Swan, T.; Russell, T.L.; Staunton, K.M.; Field, M.A.; Ritchie, S.A.; Burkot, T.R. A literature review of dispersal pathways of Aedes albopictus across different spatial scales: Implications for vector surveillance. Parasites Vectors 2022, 15, 303. [Google Scholar] [CrossRef]
- Rašić, G.; Filipović, I.; Weeks, A.R.; Hoffmann, A.A. Genome-wide SNPs lead to strong signals of geographic structure and relatedness patterns in the major arbovirus vector, Aedes aegypti. BMC Genom. 2014, 15, 275. [Google Scholar] [CrossRef]
- Sherpa, S.; Tutagata, J.; Gaude, T.; Laporte, F.; Kasai, S.; Ishak, I.H.; Guo, X.; Shin, J.; Boyer, S.; Marcombe, S. Genomic Shifts, Phenotypic Clines, and Fitness Costs Associated With Cold Tolerance in the Asian Tiger Mosquito. Mol. Biol. Evol. 2022, 39, msac104. [Google Scholar] [CrossRef]
- Birungi, J.; Munstermann, L.E. Genetic structure of Aedes albopictus (Diptera: Culicidae) populations based on mitochondrial ND5 sequences: Evidence for an independent invasion into Brazil and United States. Ann. Entomol. Soc. Am. 2002, 95, 125–132. [Google Scholar] [CrossRef]
- Beebe, N.W.; Ambrose, L.; Hill, L.A.; Davis, J.B.; Hapgood, G.; Cooper, R.D.; Russell, R.C.; Ritchie, S.A.; Reimer, L.J.; Lobo, N.F. Tracing the tiger: Population genetics provides valuable insights into the Aedes (Stegomyia) albopictus invasion of the Australasian Region. PLoS Negl. Trop. Dis. 2013, 7, e2361. [Google Scholar] [CrossRef]
- Manni, M.; Gomulski, L.M.; Aketarawong, N.; Tait, G.; Scolari, F.; Somboon, P.; Guglielmino, C.R.; Malacrida, A.R.; Gasperi, G. Molecular markers for analyses of intraspecific genetic diversity in the Asian Tiger mosquito, Aedes albopictus. Parasites Vectors 2015, 8, 188. [Google Scholar] [CrossRef]
- Porretta, D.; Gargani, M.; Bellini, R.; Calvitti, M.; Urbanelli, S. Isolation of microsatellite markers in the tiger mosquito Aedes albopictus (Skuse). Mol. Ecol. Notes 2006, 6, 880–881. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.-X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Zhong, D.; Lo, E.; Hu, R.; Metzger, M.E.; Cummings, R.; Bonizzoni, M.; Fujioka, K.K.; Sorvillo, T.E.; Kluh, S.; Healy, S.P. Genetic analysis of invasive Aedes albopictus populations in Los Angeles County, California and its potential public health impact. PLoS ONE 2013, 8, e68586. [Google Scholar] [CrossRef]
- ŽITKO, T.; KOVAČIĆ, A.; Desdevises, Y.; Puizina, J. Genetic variation in East-Adriatic populations of the Asian tiger mosquito, Aedes albopictus (Diptera: Culicidae), inferred from NADH5 and COI sequence variability. Eur. J. Entomol. 2011, 108, 501–508. [Google Scholar] [CrossRef]
- Raharimalala, F.N.; Ravaomanarivo, L.H.; Ravelonandro, P.; Rafarasoa, L.S.; Zouache, K.; Tran-Van, V.; Mousson, L.; Failloux, A.-B.; Hellard, E.; Moro, C.V. Biogeography of the two major arbovirus mosquito vectors, Aedes aegypti and Aedes albopictus (Diptera, Culicidae), in Madagascar. Parasites Vectors 2012, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Lischer, H.E.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2012, 28, 298–299. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Goudet, J. FSTAT (version 1.2): A computer program to calculate F-statistics. J. Hered 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornuet, J.-M. BOTTLENECK: A program for detecting recent effective population size reductions from allele data frequencies. J. Hered 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Bjorkman, C.; Niemela, P. Climate Change and Insect Pests; CABI: Wallingford, UK, 2015; Volume 8. [Google Scholar]
- Bale, J.S.; Masters, G.J.; Hodkinson, I.D.; Awmack, C.; Bezemer, T.M.; Brown, V.K.; Butterfield, J.; Buse, A.; Coulson, J.C.; Farrar, J. Herbivory in global climate change research: Direct effects of rising temperature on insect herbivores. Glob. Chang. Biol. 2002, 8, 1–16. [Google Scholar] [CrossRef]
- Kingsolver, J.G.; Arthur Woods, H.; Buckley, L.B.; Potter, K.A.; MacLean, H.J.; Higgins, J.K. Complex life cycles and the responses of insects to climate change. Integr. Comp. Biol. 2011, 51, 719–732. [Google Scholar] [CrossRef]
- Sherpa, S.; Blum, M.G.; Capblancq, T.; Cumer, T.; Rioux, D.; Després, L. Unravelling the invasion history of the Asian tiger mosquito in Europe. Mol. Ecol. 2019, 28, 2360–2377. [Google Scholar] [CrossRef]
- Castonguay-Vanier, J.; Klitting, R.; Sengvilaipaseuth, O.; Piorkowski, G.; Baronti, C.; Sibounheuang, B.; Vongsouvath, M.; Chanthongthip, A.; Thongpaseuth, S.; Mayxay, M. Molecular epidemiology of dengue viruses in three provinces of Lao PDR, 2006–2010. PLoS Negl. Trop. Dis. 2018, 12, e0006203. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.H.; Park, J.A.; Kim, J.S.; Hur, Y.J.; Song, M.S.; Hwang, T.G.; Choi, Y. A case of an imported dengue hemorrhagic fever with spontaneous bleeding: Case report and review of the literature. Korean J. Pediatr. Infect. Dis. 2011, 18, 207–211. [Google Scholar] [CrossRef]
- Moreira, L.A.; Iturbe-Ormaetxe, I.; Jeffery, J.A.; Lu, G.; Pyke, A.T.; Hedges, L.M.; Rocha, B.C.; Hall-Mendelin, S.; Day, A.; Riegler, M. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, Chikungunya, and Plasmodium. Cell 2009, 139, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Rasgon, J.L.; Cornel, A.J.; Scott, T.W. Evolutionary history of a mosquito endosymbiont revealed through mitochondrial hitchhiking. Proc. R. Soc. B 2006, 273, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Moo-Llanes, D.A.; López-Ordóñez, T.; Torres-Monzón, J.A.; Mosso-González, C.; Casas-Martínez, M.; Samy, A.M. Assessing the potential distributions of the invasive mosquito vector Aedes albopictus and its natural Wolbachia infections in Mexico. Insects 2021, 12, 143. [Google Scholar] [CrossRef]
- Park, C.H.; Lim, H.; Kim, H.; Lee, W.G.; Roh, J.Y.; Park, M.Y.; Shin, E.-H. High prevalence of Wolbachia infection in Korean populations of Aedes albopictus (Diptera: Culicidae). J. Asia-Pacif. Entomol. 2016, 19, 191–194. [Google Scholar] [CrossRef]
- Armbruster, P.; Damsky Jr, W.E.; Giordano, R.; Birungi, J.; Munstermann, L.E.; Conn, J.E. Infection of New-and Old-World Aedes albopictus (Diptera: Culicidae) by the intracellular parasite Wolbachia: Implications for host mitochondrial DNA evolution. J. Med. Entomol. 2003, 40, 356–360. [Google Scholar] [CrossRef]
- Schuler, H.; Köppler, K.; Daxböck-Horvath, S.; Rasool, B.; Krumböck, S.; Schwarz, D.; Hoffmeister, T.S.; Schlick-Steiner, B.C.; Steiner, F.M.; Telschow, A. The hitchhiker’s guide to Europe: The infection dynamics of an ongoing Wolbachia invasion and mitochondrial selective sweep in Rhagoletis cerasi. Mol. Ecol. 2016, 25, 1595–1609. [Google Scholar] [CrossRef]
- Atyame, C.M.; Delsuc, F.; Pasteur, N.; Weill, M.; Duron, O. Diversification of Wolbachia endosymbiont in the Culex pipiens mosquito. Mol. Biol. Evol. 2011, 28, 2761–2772. [Google Scholar] [CrossRef]
- Medlock, J.M.; Hansford, K.M.; Schaffner, F.; Versteirt, V.; Hendrickx, G.; Zeller, H.; Bortel, W.V. A review of the invasive mosquitoes in Europe: Ecology, public health risks, and control options. Vector Borne Zoonotic Dis. 2012, 12, 435–447. [Google Scholar] [CrossRef]
- Kamgang, B.; Wilson-Bahun, T.A.; Irving, H.; Kusimo, M.O.; Lenga, A.; Wondji, C.S. Geographical distribution of Aedes aegypti and Aedes albopictus (Diptera: Culicidae) and genetic diversity of invading population of Ae. albopictus in the Republic of the Congo. Wellcome Open Res. 2018, 3, 79. [Google Scholar] [CrossRef] [PubMed]
- Namiki, T.; Komine-Aizawa, S.; Takada, K.; Hayakawa, S. Asian tiger mosquitos (Aedes albopictus) in urban Tokyo, Japan show low cytochrome c oxidase subunit 1 diversity. Entomol. Sci. 2021, 24, 48–54. [Google Scholar] [CrossRef]
- Thu, H.M.; Aye, K.M.; Thein, S. The effect of temperature and humidity on dengue virus propagation in Aedes aegypti mosquitos. Southeast. Asian J. Trop. Med. Public Health 1998, 29, 280–284. [Google Scholar]
- Lee, H.; Kim, J.E.; Lee, S.; Lee, C.H. Potential effects of climate change on dengue transmission dynamics in Korea. PLoS ONE 2018, 13, e0199205. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Size of population and breeding structure in relation to evolution. Science 1938, 87, 430–431. [Google Scholar]
- Battaglia, V.; Gabrieli, P.; Brandini, S.; Capodiferro, M.R.; Javier, P.A.; Chen, X.-G.; Achilli, A.; Semino, O.; Gomulski, L.M.; Malacrida, A.R. The worldwide spread of the tiger mosquito as revealed by mitogenome haplogroup diversity. Front. Genet. 2016, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Duong, C.-V.; Kang, J.-H.; Nguyen, V.-V.; Bae, Y.-J. Genetic diversity and population structure of the Asian tiger mosquito (Aedes albopictus) in Vietnam: Evidence for genetic differentiation by climate region. Genes 2021, 12, 1579. [Google Scholar] [CrossRef]
- Kotsakiozi, P.; Richardson, J.B.; Pichler, V.; Favia, G.; Martins, A.J.; Urbanelli, S.; Armbruster, P.A.; Caccone, A. Population genomics of the Asian tiger mosquito, Aedes albopictus: Insights into the recent worldwide invasion. Ecol. Evol. 2017, 7, 10143–10157. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, H.-D.; Guo, X.-X.; Xing, D.; Dong, Y.-D.; Lan, C.-J.; Wang, G.; Li, C.-J.; Li, C.-X.; Zhao, T.-Y. Dispersal patterns and population genetic structure of Aedes albopictus (Diptera: Culicidae) in three different climatic regions of China. Parasites Vectors 2021, 14, 12. [Google Scholar] [CrossRef]
- Liu, P.; Lu, L.; Jiang, J.; Guo, Y.; Yang, M.; Liu, Q. The expanding pattern of Aedes aegypti in southern Yunnan, China: Insights from microsatellite and mitochondrial DNA markers. Parasites Vectors 2019, 12, 561. [Google Scholar] [CrossRef]
- Salgueiro, P.; Serrano, C.; Gomes, B.; Alves, J.; Sousa, C.A.; Abecasis, A.; Pinto, J. Phylogeography and invasion history of Aedes aegypti, the Dengue and Zika mosquito vector in Cape Verde islands (West Africa). Evol. Appl. 2019, 12, 1797–1811. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.W.; Meece, J.K.; McGowan, J.A.; Lovin, D.D.; Hemme, R.R.; Chadee, D.D.; McAbee, K.; Brown, S.E.; Knudson, D.L.; Severson, D.W. Microsatellite isolation and linkage group identification in the yellow fever mosquito Aedes aegypti. J. Hered 2007, 98, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Guglielmino, C.R.; Scolari, F.; Vega-Rúa, A.; Failloux, A.-B.; Somboon, P.; Lisa, A.; Savini, G.; Bonizzoni, M.; Gomulski, L.M. Genetic evidence for a worldwide chaotic dispersion pattern of the arbovirus vector, Aedes albopictus. PLoS Negl. Trop. Dis. 2017, 11, e0005332. [Google Scholar] [CrossRef] [PubMed]
- Khamis, F.; Karam, N.; Ekesi, S.; De Meyer, M.; Bonomi, A.; Gomulski, L.; Scolari, F.; Gabrieli, P.; Siciliano, P.; Masiga, D. Uncovering the tracks of a recent and rapid invasion: The case of the fruit fly pest Bactrocera invadens (Diptera: Tephritidae) in Africa. Mol. Ecol. 2009, 18, 4798–4810. [Google Scholar] [CrossRef] [PubMed]
- Kolar, C.S.; Lodge, D.M. Progress in invasion biology: Predicting invaders. Trends Ecol. Evol. 2001, 16, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, J.L.; Cassey, P.; Blackburn, T. The role of propagule pressure in explaining species invasions. Trends Ecol. Evol. 2005, 20, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Schottenhammer, A. The “China Seas” in world history: A general outline of the role of Chinese and East Asian maritime space from its origins to c. 1800. J. Mar. Isl. Cult. 2012, 1, 63–86. [Google Scholar] [CrossRef]
- Kwak, B.O.; Kwon, Y.S.; Hong, Y.J.; Nahm, C.H.; Jang, W.; Uh, Y.; Cho, Y.G.; Kim, J.; Kim, M.; Kim, D.H. Seroprevalence of neutralizing antibodies against Japanese encephalitis virus among adolescents and adults in Korea: A prospective multicenter study. Vaccines 2020, 8, 328. [Google Scholar] [CrossRef]
- Ryu, J.; Choi, K.S. Species diversity of the Culex pipiens complex in the Republic of Korea. Entomol. Res. 2022, 52, 376–381. [Google Scholar] [CrossRef]
- Akintola, A.A.; Park, B.; Choi, E.H.; Hwang, U.W. Complete mitochondrial genome of a malaria vector mosquito Anopheles sinensis from South Korea. Mitochondrial DNA Part. B 2022, 7, 881–883. [Google Scholar] [CrossRef]
- Hong, J.-W.; Hong, J.; Kwon, E.E.; Yoon, D. Temporal dynamics of urban heat island correlated with the socio-economic development over the past half-century in Seoul, Korea. Env. Pollut. 2019, 254, 112934. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, P.; Jeon, J.-Y.; Kim, H.W.; Oh, H.-S.; Adhikari, P.; Seo, C. Northward range expansion of southern butterflies according to climate change in South Korea. J. Clim. 2020, 11, 643–656. [Google Scholar] [CrossRef]
- Yang, S.C.; Lee, H.i.; Kim, H.; Lee, W.G. Transmission ability of Zika virus with artificially infected Aedes albopictus in Korea. Entomol Res. 2021, 51, 413–420. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locality | Sample ID | Geographic Coordinates | Date | Sample Size | No. Haplotypes | S | k | Hd | π | Tajima’s D | Fu’s Fs |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Wonju (2017) | WJ-I | 37°22′52.6″ N 127°53′36.1″ E | 2017.07 | 18 | 5 | 4 | 0.725 | 0.549 | 0.00067 | −1.12822 | −2.0958 |
| Wonju (2020) | WJ-II | 37°22′52.6″ N 127°53′36.1″ E | 2017.07 | 18 | 2 | 3 | 0.333 | 0.111 | 0.00031 | −1.71304 (p = 0.022) | 0.65061 |
| Yeoncheon | YC | 37°58′35.6″ N 127°04′05.3″ E | 2017.07 | 5 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Yangsan | YS | 35°31′13.9″ N 129°00′40.1″ E | 2017.07 | 12 | 6 | 4 | 1.152 | 0.758 | 0.00107 | −0.45947 | −2.89747 |
| Anyang (2018) | AY-I | 37°22′01.7″ N 126°57′39.0″ E | 2018.06 | 4 | 2 | 1 | 0.5 | 0.5 | 0.00046 | −0.61237 | 0.17185 |
| Anyang (2020) | AY-II | 37°22′01.7″ N 126°57′39.0"E | 2020.08 | 4 | 4 | 6 | 3.167 | 1 | 0.00293 | −0.31446 | −1.15708 |
| Chuncheon | CC | 37°53′15.0″ N 127°44′02.9″ E | 2017.08 | 7 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cheongyang | CY | 36°24′28.5″ N 126°51′04.8″ E | 2017.09 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Daejeon | DJ | 36°21′02.3″ N 127°21′35.5″ E | 2017.09 | 8 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Gwacheon | GC | 37°26′00.0″ N 126°59′00.1″ E | 2020.07 | 7 | 3 | 2 | 0.571 | 0.524 | 0.00053 | −1.23716 | −0.9218 |
| Geoje | GJ | 34°46′43.8″ N 128°38′42.1″ E | 2017.07 | 5 | 2 | 1 | 0.6 | 0.6 | 0.00056 | 1.22474 | 0.62615 |
| Gwangju | GW | 35°07′53.0″ N 126°57′30.0″ E | 2017.09 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Gyeongju | GY | 35°50′41.4″ N 129°11′43.4″ E | 2017.07 | 2 | 2 | 3 | 3 | 1 | 0.00278 | 0 | 1.09861 |
| Jeungdo | JD | 34°59′15.8″ N 126°08′07.3"E | 2017.07 | 5 | 3 | 2 | 1 | 0.8 | 0.00093 | 0.24314 | −0.47542 |
| Jeonju | JJ | 35°50′37.2″ N 127°07′09.0″ E | 2017.08 | 28 | 3 | 2 | 0.143 | 0.14 | 0.00013 | −1.5106 (p = 0.045) | −2.26798 |
| Sokcho | SC | 38°12′10.5″ N 128°33′07.5″ E | 2020.09 | 13 | 3 | 2 | 0.308 | 0.295 | 0.00028 | −1.46801 | −1.4015 |
| Seoul | SE | 37°36′32.9″ N 126°54′06.5″ E | 2020.07 | 3 | 2 | 1 | 0.667 | 0.667 | 0.00062 | 0 | 0.20067 |
| Yeoju | YJ | 37°18′43.5″ N 127°36′52.8″ E | 2020.07 | 3 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Yeosu | YE | 34°47′57.0″ N 127°44′52.4″ E | 2020.06 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Tokyo, Japan | TJ | 35°33′01.2″ N 139°44′16.5″ E | 2018.09 | 35 | 3 | 3 | 0.561 | 0.301 | 0.00052 | −0.51759 | 0.39764 |
| Vientiane, Laos | VL | 17°57′42.6″ N 102°36′30.2″ E | 2018.07 | 28 | 11 | 10 | 1.013 | 0.706 | 0.00094 | −1.94835 (p = 0.010) | −8.85951 |
| Total/average | 211 | 38 | 41 | 1.426 | 0.678 | 0.00132 | −0.44959 | −0.80624 |
| Source of Variation | df | Sum of Squares | Variance Components | Percentage of Variation (%) | F-Index * |
|---|---|---|---|---|---|
| Among groups | 2 | 67.077 | 0.58191 | 56.02 | 0.56024 |
| Among populations within groups | 18 | 27.71 | 0.16776 | 16.15 | 0.36729 |
| Within populations. | 190 | 54.91 | 0.28900 | 27.82 | 0.72176 |
| Total | 210 | 149.697 | 1.03867 |
| Source of Variation | df | Sum of Squares | Variance Components | Percentage of Variation (%) | F-Index |
|---|---|---|---|---|---|
| Among groups | 2 | 58.357 | −0.15670 | −2.78 | −0.02780 |
| Among populations within groups | 16 | 336.027 | 1.33962 | 23.77 | 0.23124 |
| Within populations | 243 | 1082.238 | 4.45365 | 79.01 | 0.20987 |
| Total | 261 | 1476.622 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, J.; Rahman, M.-M.; Kim, J.; Marcombe, S.; Jung, J. Genetic Diversity of Dengue Vector Aedes albopictus Collected from South Korea, Japan, and Laos. Insects 2023, 14, 297. https://doi.org/10.3390/insects14030297
Shin J, Rahman M-M, Kim J, Marcombe S, Jung J. Genetic Diversity of Dengue Vector Aedes albopictus Collected from South Korea, Japan, and Laos. Insects. 2023; 14(3):297. https://doi.org/10.3390/insects14030297
Chicago/Turabian StyleShin, Jiyeong, Md-Mafizur Rahman, Juil Kim, Sébastien Marcombe, and Jongwoo Jung. 2023. "Genetic Diversity of Dengue Vector Aedes albopictus Collected from South Korea, Japan, and Laos" Insects 14, no. 3: 297. https://doi.org/10.3390/insects14030297
APA StyleShin, J., Rahman, M.-M., Kim, J., Marcombe, S., & Jung, J. (2023). Genetic Diversity of Dengue Vector Aedes albopictus Collected from South Korea, Japan, and Laos. Insects, 14(3), 297. https://doi.org/10.3390/insects14030297