
Rare Causes of Arterial Hypertension and Thoracic Aortic Aneurysms—A Case-Based Review

 , and
, and
Abstract
1. Introduction
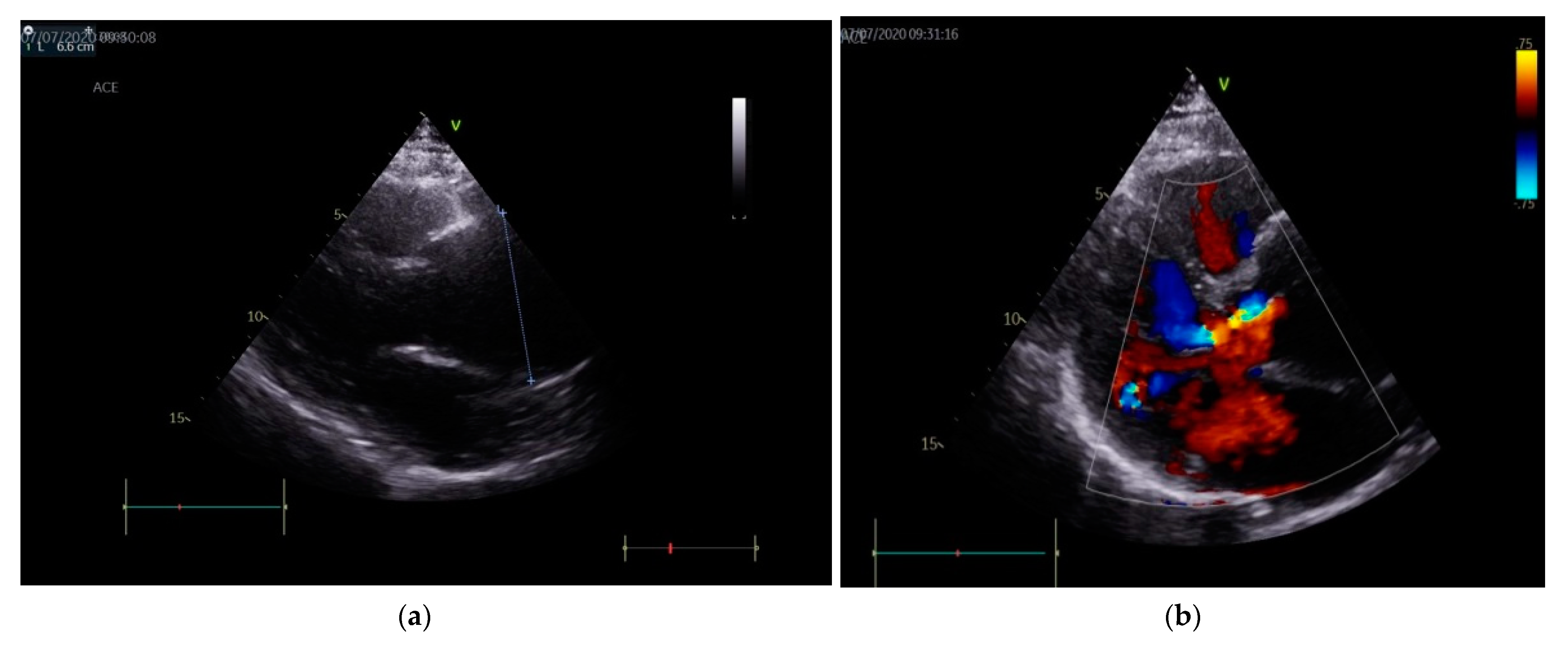
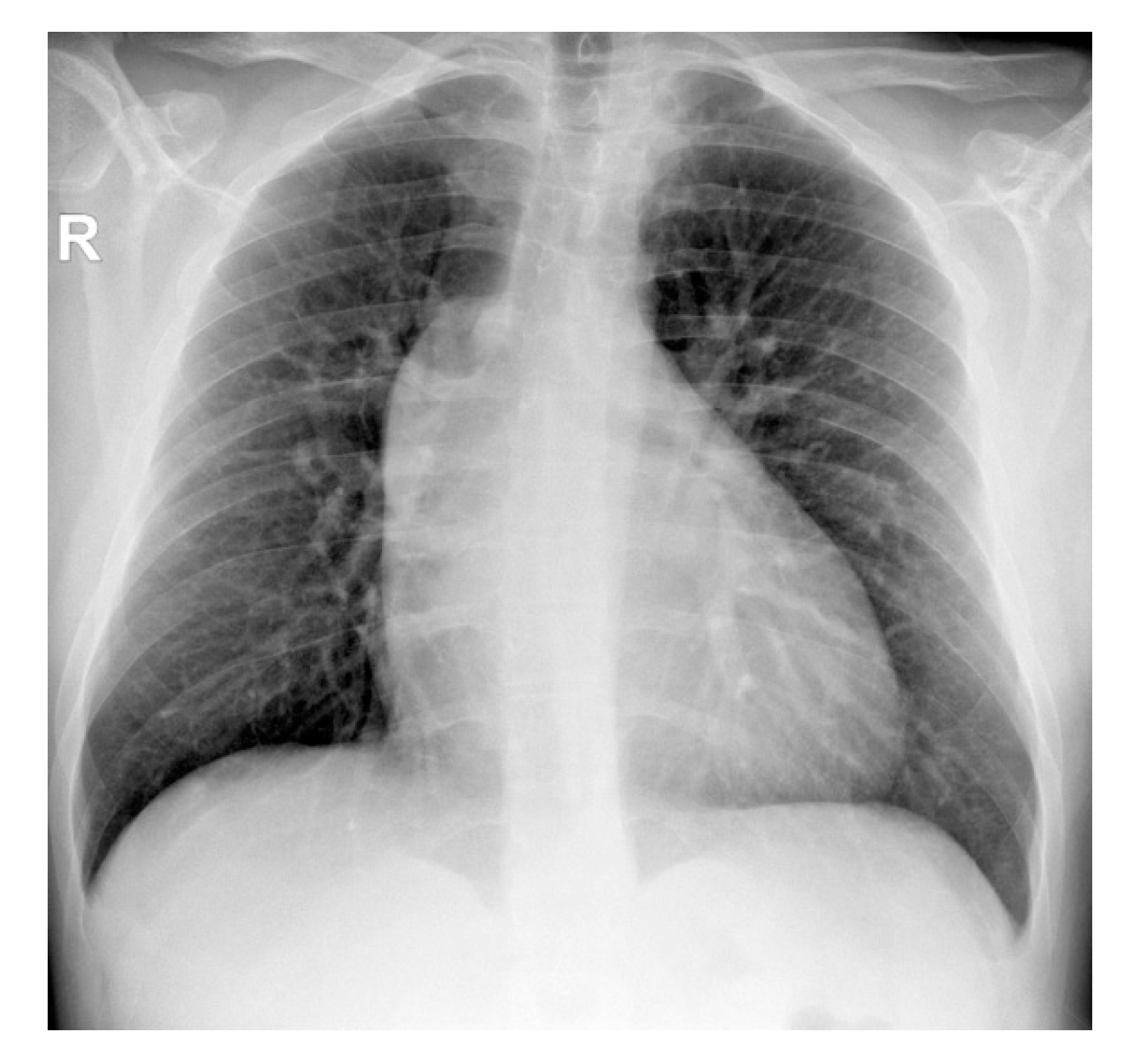
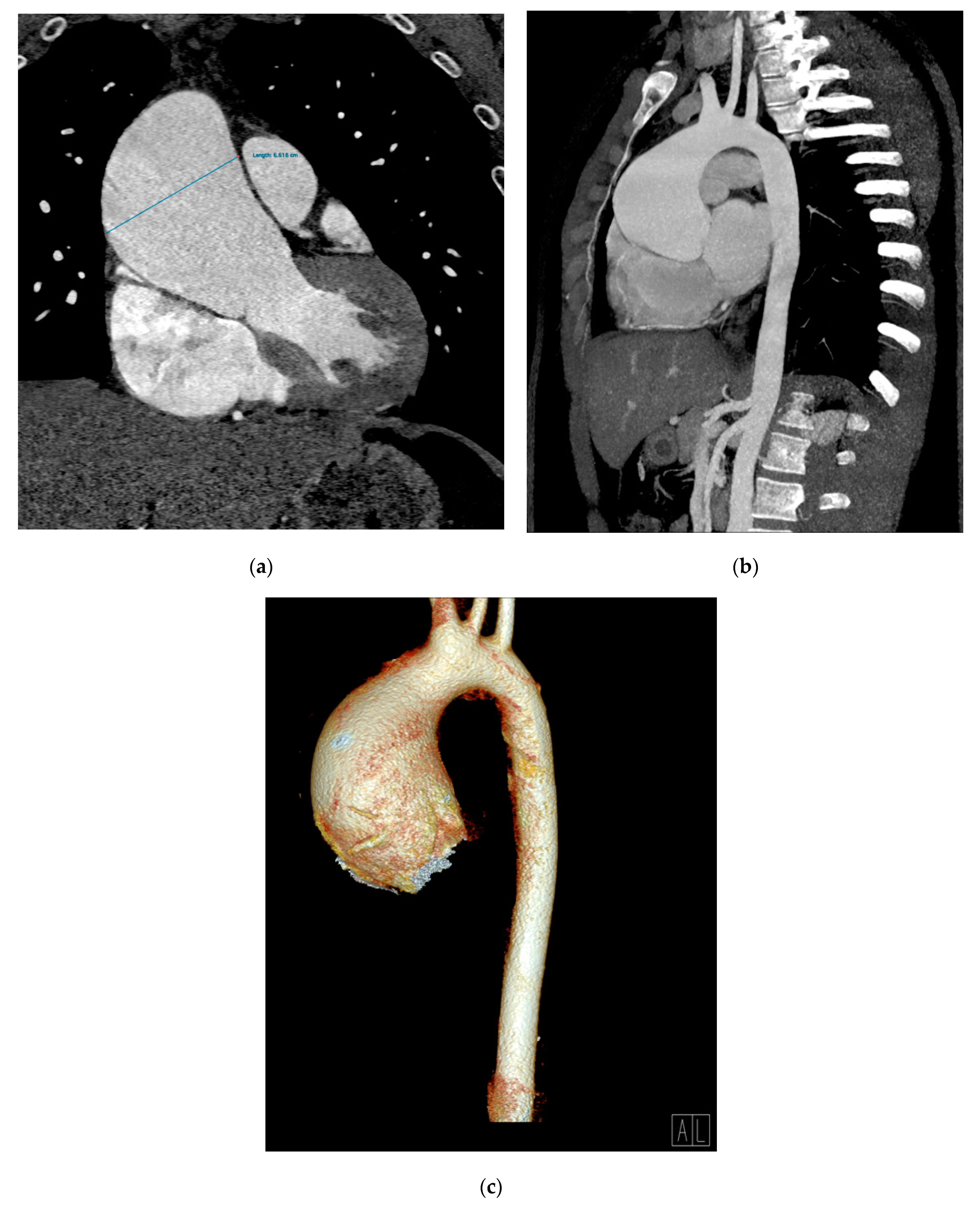
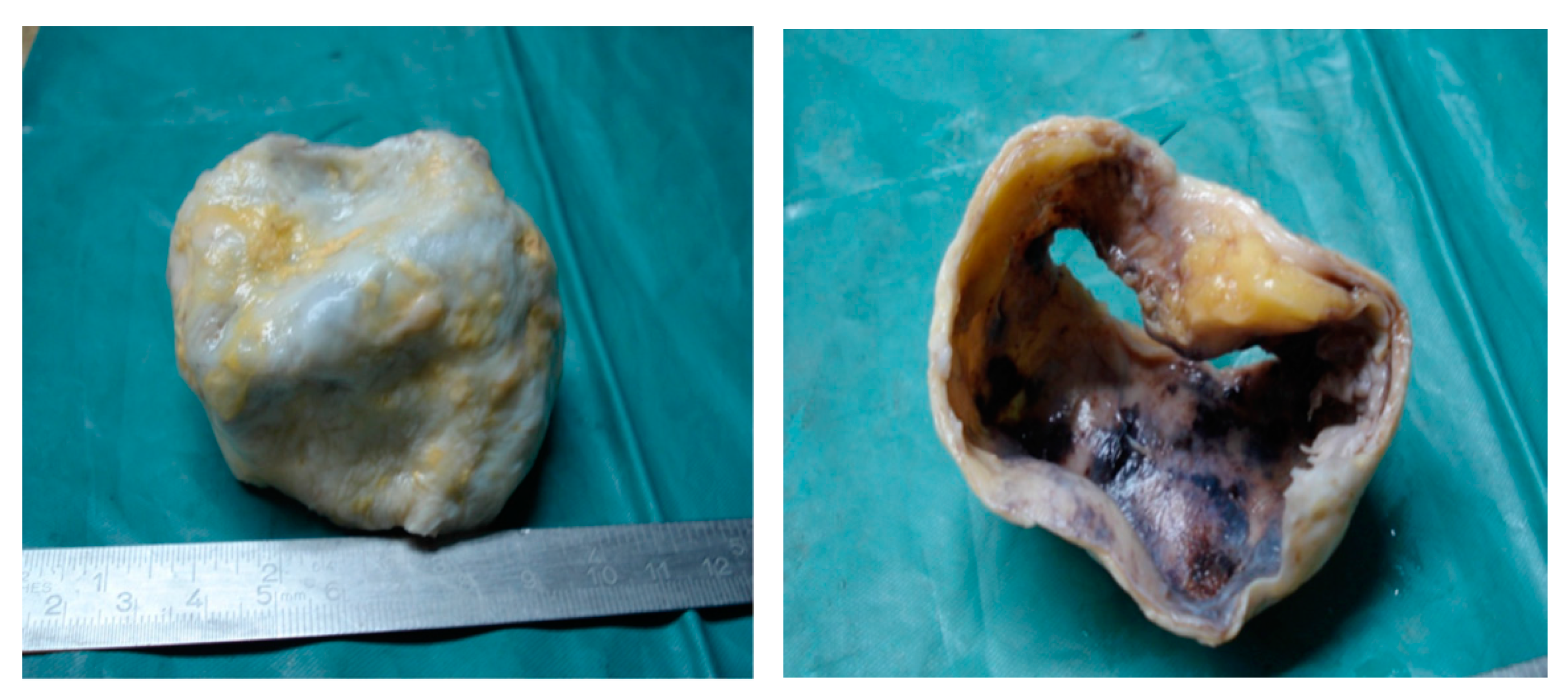
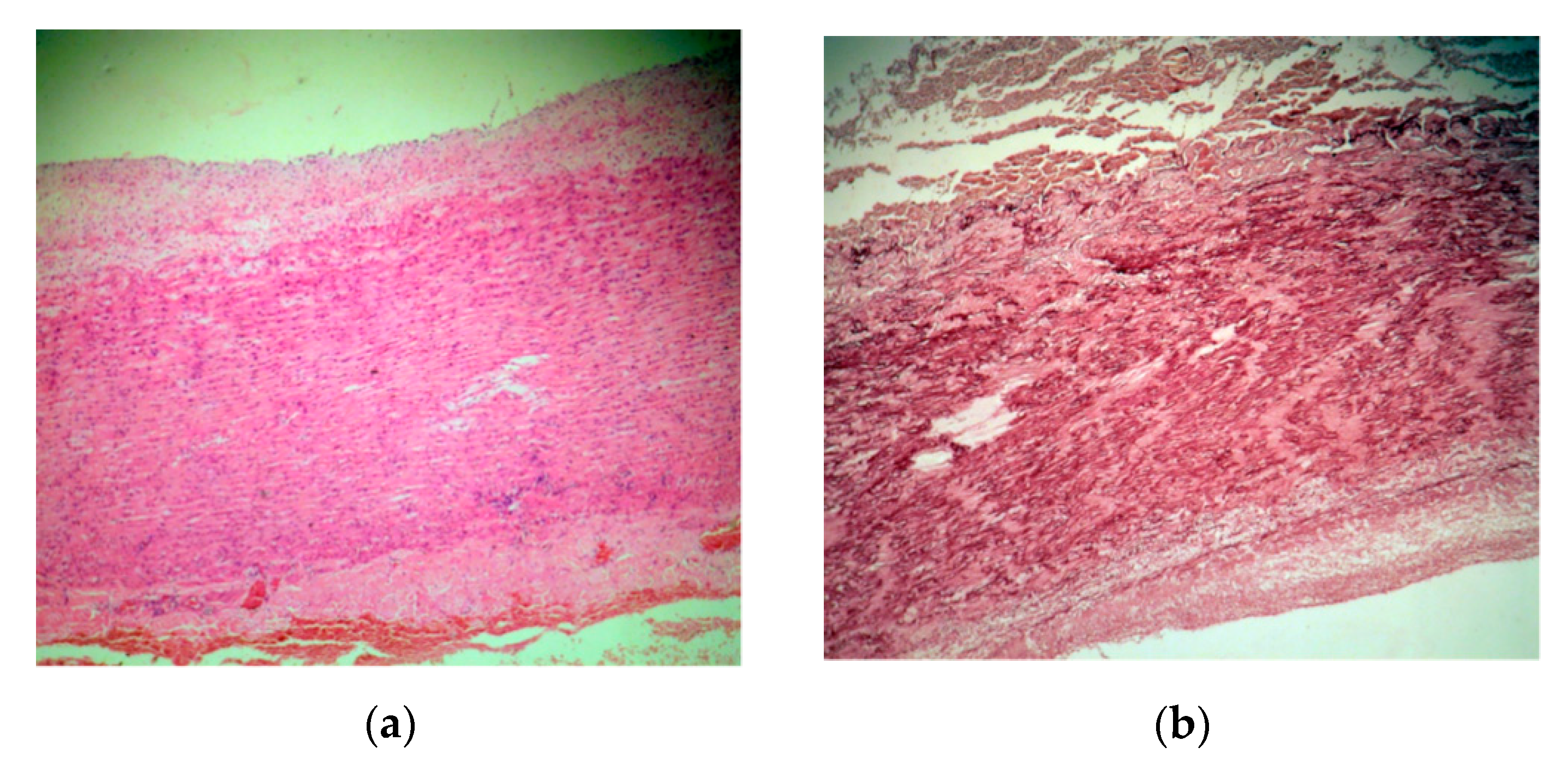
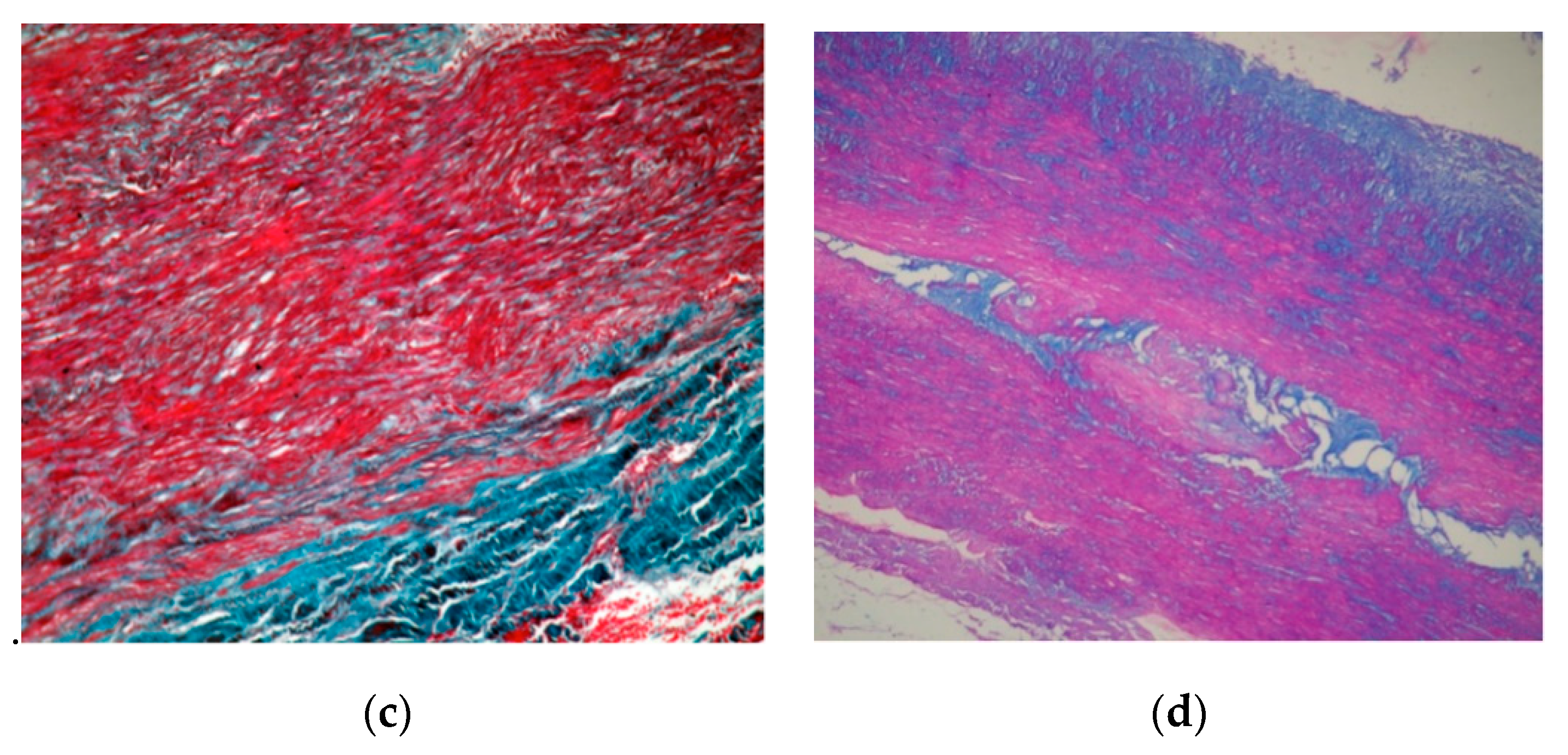
2. Case Presentation
3. Discussions
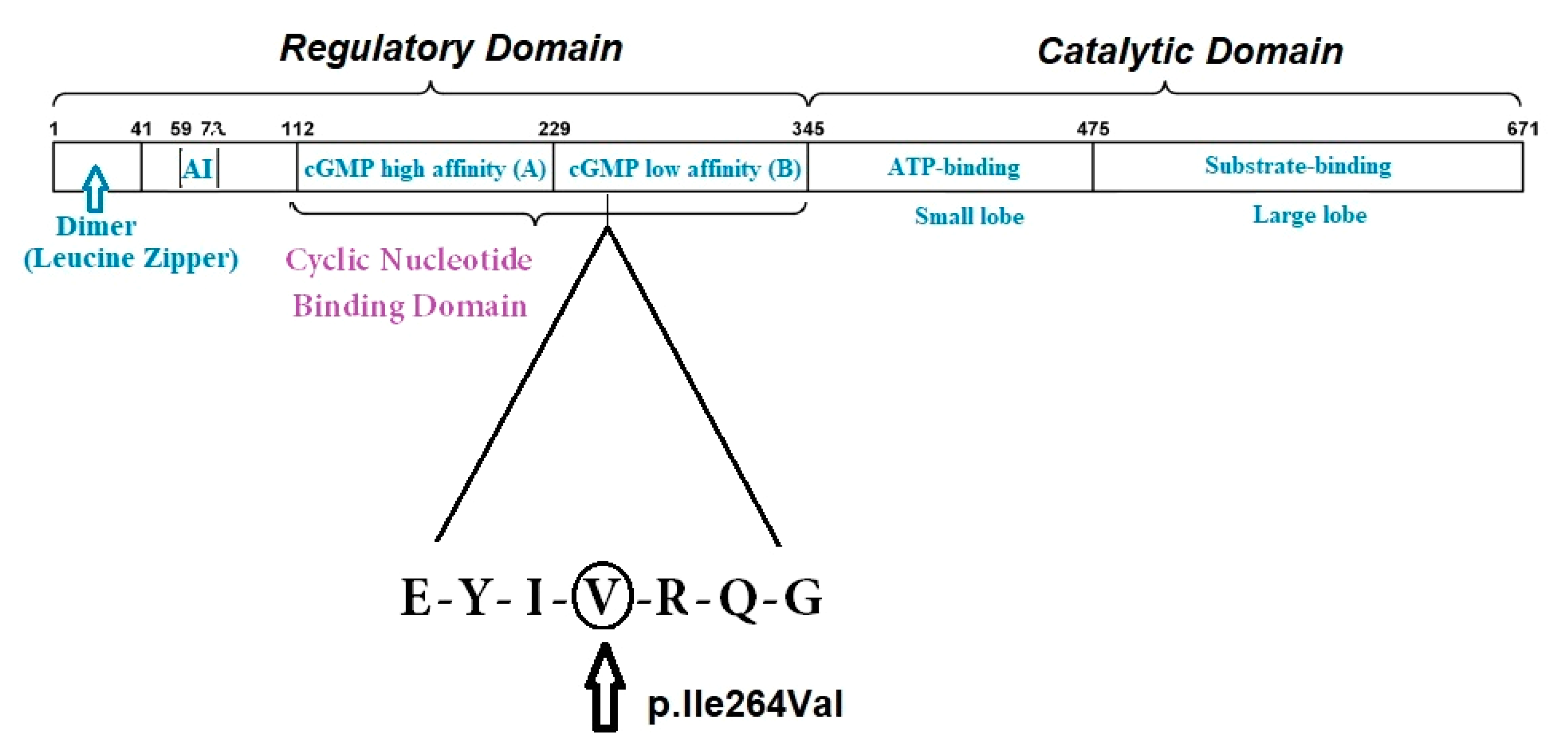
3.1. Genetic Testing in TAA: The Role of PRKG1
3.2. Hypertension in TAA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism | Effector | Molecular Interactions | References |
|---|---|---|---|
| Vasoconstriction | Renin–angiotensin system | Ang-II signaling activated by mutations decreasing VSMC contraction; | [4] |
| PKG1 | Key mediator of the NO/cGMP signaling pathways in VSMC contraction; Suppression of RGS, phosphorylation of sarcomere proteins; Control of TGF-β downstream events, regulation of transient receptor potential canonical channels and mechanosensing; Dysregulation of hypoxia-inducible factor -1 (HIF-1α)/ VEGF signaling; | [48] [50] [31] | |
| Endothelial cell migration and apoptosis | PKG1 | Regulation of endothelial cell migration; | [47] |
| Fibrillin-1 | Induction of EC apoptosis; inhibition of EC proliferation; Involvement in vascular rarefaction in chronic renal injury; | [46] | |
| Vascular smooth muscle cell migration, apoptosis and phenotype switch | PKG1 | Reduction of VSMC migration by focal adhesion disassembly; Regulation of VSMC apoptosis, proliferation, differentiation and phenotype switch from contractile to synthetic; | [8,31,49,50] |
| Osteopontin | Induction of medial thickening and neo-intimal formation; | [40] | |
| Extracellular matrix remodeling | Defective VSMC contraction | FBN1, ACTA2, MYH11, MYLK and PRKG1 activate TGF-β signaling | [4] |
| Fibrillin-1 | Important in elastin deposition, anchoring and load bearing, associated with aortic and arterial stiffness | [45] [46] | |
| Metalloproteinases | MMP2 activation by hyperactive PKG1; Increased MMP2 involved in TAA and hypertension; | [31,57,58,59] | |
| Epigenetic regulators | miRNA 145 activates TGF-β and increases OPN and collagen expression; | [1] | |
| Oxidative stress and inflammation | PRKG1 | Formation of reactive oxygen species and oxidative stress by activating JNK and NOX4; | [31,52] |
| Osteopontin | Upregulation by oxidative stress; | [40] | |
| Epigenetic regulators | miR-155-p used by TNF-α to induce VSMC phenotype and function alteration; | [50,55] | |
| Pro-inflammatory cytokines | IL-6 contributes to angiotensin II—induced microvascular dysfunction; IL-6 associated with aortic dimensions in TAA | [54] [60] |
3.3. Risk of Acute Thoracic Aortic Syndrome in TAA According to Etiology
3.4. Therapy of PRKG1-Associated TAA
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F., Jr.; Casey, D.E.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the Diagnosis and Management of Patients With Thoracic Aortic Disease: Executive Summary. Catheter. Cardiovasc. Interv. 2010, 76, 43–86. [Google Scholar] [CrossRef] [PubMed]
- Ostberg, N.P.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules 2020, 10, 182. [Google Scholar] [CrossRef]
- Sherifova, S.; Holzapfel, G.A. Biomechanics of aortic wall failure with a focus on dissection and aneurysm: A review. Acta Biomater. 2019, 99, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases. Circ. Res. 2019, 124, 588–606. [Google Scholar] [CrossRef]
- Jana, S.; Hu, M.; Shen, M.; Kassiri, Z. Extracellular matrix, regional heterogeneity of the aorta, and aortic aneurysm. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Maleszewski, J.J. Inflammatory ascending aortic disease: Perspectives from pathology. J. Thorac. Cardiovasc. Surg. 2015, 149, 176–183. [Google Scholar] [CrossRef]
- Gago-Díaz, M.; Blanco-Verea, A.; Teixidó, G.; Huguet, F.; Gut, M.; Laurie, S.; Gut, I.; Carracedo, Á.; Evangelista, A.; Brion, M. PRKG1and genetic diagnosis of early-onset thoracic aortic disease. Eur. J. Clin. Investig. 2016, 46, 787–794. [Google Scholar] [CrossRef]
- Rainer, P.P.; Kass, D.A. Old dog, new tricks: Novel cardiac targets and stress regulation by protein kinase G. Cardiovasc. Res. 2016, 111, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Tomoaia, R.; Molnar, A.; Beyer, R. Dădârlat-Pop, A.Ș.; Frîngu, F.; Gurzau, D.; Simu, G.; Minciună, I.A.; Caloian, B.; Zdrenghea, D.; et al. The role of multimodal imaging in the diagnosis of an asymptomatic patient with congenital anomaly. Med. Ultrason. 2020, 2020, 1–4. [Google Scholar] [CrossRef]
- Verhagen, J.M.; Kempers, M.; Cozijnsen, L.; Bouma, B.J.; Duijnhouwer, A.L.; Post, J.G.; Hilhorst-Hofstee, Y.; Bekkers, S.C.; Kerstjens-Frederikse, W.S.; Van Brakel, T.J.; et al. Expert consensus recommendations on the cardiogenetic care for patients with thoracic aortic disease and their first-degree relatives. Int. J. Cardiol. 2018, 258, 243–248. [Google Scholar] [CrossRef]
- El-Hamamsy, I.; Ouzounian, M.; Demers, P.; McClure, S.; Hassan, A.; Dagenais, F.; Chu, M.W.; Pozeg, Z.; Bozinovski, J.; Peterson, M.D.; et al. State-of-the-Art Surgical Management of Acute Type A Aortic Dissection. Can. J. Cardiol. 2016, 32, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J. Possible Mechanical Roles of Glycosaminoglycans in Thoracic Aortic Dissection and Associations with Dysregulated Transforming Growth Factor-β. J. Vasc. Res. 2013, 50, 1–10. [Google Scholar] [CrossRef]
- Halushka, M.K.; Angelini, A.; Bartoloni, G.; Basso, C.; Batoroeva, L.; Bruneval, P.; Buja, L.M.; Butany, J.; D’Amati, G.; Fallon, J.T.; et al. Consensus statement on surgical pathology of the aorta from the Society for Cardiovascular Pathology and the Association For European Cardiovascular Pathology: II. Noninflammatory degenerative diseases—Nomenclature and diagnostic criteria. Cardiovasc. Pathol. 2016, 25, 247–257. [Google Scholar] [CrossRef]
- Karimi, A.; Milewicz, D.M. Structure of the Elastin-Contractile Units in the Thoracic Aorta and How Genes That Cause Thoracic Aortic Aneurysms and Dissections Disrupt This Structure. Can. J. Cardiol. 2016, 32, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D.; Schwartz, M.A.; Tellides, G.; Milewicz, D.M. Role of Mechanotransduction in Vascular Biology. Circ. Res. 2015, 116, 1448–1461. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Trybus, K.M.; Guo, D.-C.; Sweeney, H.L.; Regalado, E.; Kamm, K.; Stull, J.T. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arter. Thromb. Vasc. Biol. 2017, 37, 26–34. [Google Scholar] [CrossRef]
- Shen, Y.H.; Lemaire, S.A.; Webb, N.R.; Cassis, L.A.; Daugherty, A.; Lu, H.S. Aortic Aneurysms and Dissections Series. Arter. Thromb. Vasc. Biol. 2020, 40, e37–e46. [Google Scholar] [CrossRef]
- Clément, M.; Chappell, J.; Raffort, J.; Lareyre, F.; Vandestienne, M.; Taylor, A.L.; Finigan, A.; Harrison, J.; Bennett, M.R.; Bruneval, P.; et al. Vascular Smooth Muscle Cell Plasticity and Autophagy in Dissecting Aortic Aneurysms. Arter. Thromb. Vasc. Biol. 2019, 39, 1149–1159. [Google Scholar] [CrossRef]
- Roccabianca, S.; Ateshian, G.A.; Humphrey, J.D. Biomechanical roles of medial pooling of glycosaminoglycans in thoracic aortic dissection. Biomech. Model. Mechanobiol. 2014, 13, 13–25. [Google Scholar] [CrossRef]
- Roccabianca, S.; Bellini, C.; Humphrey, J.D. Computational modelling suggests good, bad and ugly roles of glycosaminoglycans in arterial wall mechanics and mechanobiology. J. R. Soc. Interface 2014, 11, 20140397. [Google Scholar] [CrossRef] [PubMed]
- Bedeleanu, D.; Coman, C.; Encica, S.; Hagiu, R.; Molnar, A.; Capalneanu, R. Stanford Type A Aortic Dissection in a Hypertensive Patient with Atherosclerosis of Aorta and Aortitis. Echocardiography 2000, 17, 181–185. [Google Scholar] [CrossRef]
- Guo, D.-C.; Regalado, E.; Casteel, D.E.; Santos-Cortez, R.L.; Gong, L.; Kim, J.J.; Dyack, S.; Horne, S.G.; Chang, G.; Jondeau, G.; et al. Recurrent Gain-of-Function Mutation in PRKG1 Causes Thoracic Aortic Aneurysms and Acute Aortic Dissections. Am. J. Hum. Genet. 2013, 93, 398–404. [Google Scholar] [CrossRef]
- Pisano, C.; Balistreri, C.R.; Ricasoli, A.; Ruvolo, G. Cardiovascular Disease in Ageing: An Overview on Thoracic Aortic Aneurysm as an Emerging Inflammatory Disease. Mediat. Inflamm. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dinesh, N.E.H.; Reinhardt, D.P. Inflammation in thoracic aortic aneurysms. Herz 2019, 44, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Aurelian, S.V.; Adrian, M.; Andercou, O.; Bruno, S.; Alexandru, O.; Catalin, T.; Dan, B. Neutrophil-to-Lymphocyte Ratio: A Comparative Study of Rupture to Nonruptured Infrarenal Abdominal Aortic Aneurysm. Ann. Vasc. Surg. 2019, 58, 270–275. [Google Scholar] [CrossRef]
- Lareyre, F.; Raffort, J.; Le, D.; Chan, H.L.; Le Houerou, T.; Cochennec, F.; Touma, J.; Desgranges, P. High Neutrophil to Lymphocyte Ratio Is Associated With Symptomatic and Ruptured Thoracic Aortic Aneurysm. Angiology 2018, 69, 686–691. [Google Scholar] [CrossRef]
- Wan, Y.; Aragon-Martin, J.; Collins, L.; Guo, D.; Saggar-Malik, A.; Jahangiri, M.; Millewicz, D.; Child, A.H. Screening of the PRKG1 gene in a British cohort of Thoracic Aortic Aneurysm and Dissection (TAAD) patients. Conf. Am. Soc. Hum. Genet. 2014, 14–15. [Google Scholar] [CrossRef]
- Wolford, B.N.; Hornsby, W.E.; Guo, D.; Zhou, W.; Lin, M.; Farhat, L.; McNamara, J.; Driscoll, A.; Wu, X.; Schmidt, E.M.; et al. Clinical Implications of Identifying Pathogenic Variants in Individuals With Thoracic Aortic Dissection. HHS Public Access 2020, 12. [Google Scholar] [CrossRef]
- Guo, J.; Cai, L.; Jia, L.; Li, X.; Xi, X.; Zheng, S.; Liu, X.; Piao, C.; Liu, T.; Sun, Z.; et al. Wide mutation spectrum and frequent variant Ala27Thr of FBN1 identified in a large cohort of Chinese patients with sporadic TAAD. Sci. Rep. 2015, 5, 13115. [Google Scholar] [CrossRef]
- Isselbacher, E.M.; Cardenas, C.L.L.; Lindsay, M.E. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation 2016, 133, 2516–2528. [Google Scholar] [CrossRef]
- Schwaerzer, G.K.; Kalyanaraman, H.; Casteel, D.E.; Dalton, N.D.; Gu, Y.; Lee, S.; Zhuang, S.; Wahwah, N.; Schilling, J.M.; Patel, H.H.; et al. Aortic pathology from protein kinase G activation is prevented by an antioxidant vitamin B12 analog. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Shalhub, S.; Regalado, E.S.; Guo, D.-C.; Milewicz, D.M. The natural history of type B aortic dissection in patients with PRKG1 mutation c.530G>A (p.Arg177Gln). J. Vasc. Surg. 2019, 70, 718–723. [Google Scholar] [CrossRef]
- Zhang, W.; Han, Q.; Liu, Z.; Zhou, W.; Cao, Q.; Zhou, W. Exome sequencing reveals a de novo PRKG1 mutation in a sporadic patient with aortic dissection. BMC Med. Genet. 2018, 19, 218. [Google Scholar] [CrossRef] [PubMed]
- Kwartler, C.S.; Gong, L.; Chen, J.; Wang, S.; Kulmacz, R.; Duan, X.-Y.; Janda, A.; Huang, J.; Kamm, K.E.; Stull, J.T.; et al. Variants of Unknown Significance in Genes Associated with Heritable Thoracic Aortic Disease Can Be Low Penetrant “Risk Variants”. Am. J. Hum. Genet. 2018, 103, 138–143. [Google Scholar] [CrossRef]
- Portelli, S.S.; Robertson, E.N.; Malecki, C.; Liddy, K.A.; Hambly, B.D.; Jeremy, R.W. Epigenetic influences on genetically triggered thoracic aortic aneurysm. Biophys. Rev. 2018, 10, 1241–1256. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Manel, N. Aicardi–Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015, 15, 429–440. [Google Scholar] [CrossRef]
- Hou, C.L.; Li, B.; Cheng, Y.J.; Li, M.; De Yang, Z. Upregulation of cGMP-dependent protein kinase (PRKG1) in the development of adolescents idhiopatic scoliosis. Orthop. Surg. 2020, 4, 1261–1269. [Google Scholar] [CrossRef]
- Molnar, A.; Săcui, D.; Scridon, T. Risk factors influencing the surgical outcome in 138 consecutive patients with infrarenal aortic aneurysm: Experience at the Cluj-Napoca Cardiovascular Surgery Center. Chirurgia 2014, 109, 223–228. [Google Scholar] [PubMed]
- Nierenberg, J.L.; Li, C.; He, J.; Gu, D.; Chen, J.; Lu, X.; Li, J.; Wu, X.; Gu, C.C.; Hixson, J.E.; et al. Blood Pressure Genetic Risk Score Predicts Blood Pressure Responses to Dietary Sodium and Potassium. Hypertension 2017, 70, 1106–1112. [Google Scholar] [CrossRef]
- Caesar, C.; Lyle, A.N.; Joseph, G.; Weiss, D.; Alameddine, F.M.F.; Lassègue, B.; Griendling, K.K.; Taylor, W.R. Cyclic Strain and Hypertension Increase Osteopontin Expression in the Aorta. Cell. Mol. Bioeng. 2017, 10, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Tagetti, A.; Bonafini, S.; Ohlsson, T.; Engström, G.; Almgren, P.; Minuz, P.; Smith, G.; Melander, O.; Fava, C. A genetic risk score for hypertension is associated with risk of thoracic aortic aneurysm. J. Hum. Hypertens. 2019, 33, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Brautbar, A.; LeMaire, S.A.; Franco, L.M.; Coselli, J.S.; Milewicz, D.M.; Belmont, J.W. FBN1 Mutations in Patients with Descending Thoracic Aortic Dissections. Am. J. Med. Genet. 2010, 152, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Iakoubova, O.A.; Tong, C.H.; Rowland, C.M.; Luke, M.M.; García, V.E.; Catanese, J.J.; Moomiaie, R.M.; Sotonyi, P.; Ascady, G.; Nikas, D.; et al. Genetic Variants in FBN-1 and Risk for Thoracic Aortic Aneurysm and Dissection. PLoS ONE 2014, 9, e91437. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, J.; Berg, N.D.; Torp-Pedersen, C.; Hansen, T.W.; Linneberg, A.; Fenger, M. Fibrillin-1 genotype and risk of prevalent hypertension: A study in two independent populations. Blood Press. 2012, 21, 273–280. [Google Scholar] [CrossRef]
- Medley, T.L.; Cole, T.J.; Gatzka, C.D.; Wang, W.Y.; Dart, A.M.; Kingwell, B.A. Fibrillin-1 genotype is associated with aortic stiffness and disease severity in patients with coronary artery disease. Circulation 2002, 105, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liao, J.; Yuan, Q.; Hong, X.; Li, J.; Peng, Y.; He, M.; Zhu, H.; Zhu, M.; Hou, F.F.; et al. Fibrillin-1–enriched microenvironment drives endothelial injury and vascular rarefaction in chronic kidney disease. Sci. Adv. 2021, 7, eabc7170. [Google Scholar] [CrossRef]
- Brown, I.A.; Diederich, L.; Good, M.E.; DeLalio, L.J.; Murphy, S.A.; Cortese-Krott, M.M.; Hall, J.L.; Le, T.H.; Isakson, B.E. Vascular Smooth Muscle Remodeling in Conductive and Resistance Arteries in Hypertension: VSMC in hypertension. Arter. Thromb. Vasc. Biol. 2018, 38, 1969–1985. [Google Scholar] [CrossRef]
- Liu, K.; Liu, Z.; Qi, H.; Liu, B.; Wu, J.; Liu, Y.; Zhang, J.; Cao, H.; Yan, Y.; He, Y.; et al. Genetic Variation in SLC8A1 Gene Involved in Blood Pressure Responses to Acute Salt Loading. Am. J. Hypertens. 2017, 31, 415–421. [Google Scholar] [CrossRef]
- Sellak, H.; Choi, C.-S.; Dey, N.B.; Lincoln, T.M. Transcriptional and post-transcriptional regulation of cGMP-dependent protein kinase (PKG-I): Pathophysiological significance. Cardiovasc. Res. 2013, 97, 200–207. [Google Scholar] [CrossRef][Green Version]
- Choi, S.; Park, M.; Kim, J.; Park, W.; Kim, S.; Lee, D.-K.; Hwang, J.Y.; Choe, J.; Won, M.-H.; Ryoo, S.; et al. TNF-α elicits phenotypic and functional alterations of vascular smooth muscle cells by miR-155-5p–dependent down-regulation of cGMP-dependent kinase 1. J. Biol. Chem. 2018, 293, 14812–14822. [Google Scholar] [CrossRef]
- Pei, H.; Tian, C.; Sun, X.; Qian, X.; Liu, P.; Liu, W.; Chang, Q. Overexpression of MicroRNA-145 Promotes Ascending Aortic Aneurysm Media Remodeling through TGF-β1. Eur. J. Vasc. Endovasc. Surg. 2015, 49, 52–59. [Google Scholar] [CrossRef]
- Yang, P.; Schmit, B.M.; Fu, C.; DeSart, K.; Oh, S.P.; Berceli, S.A.; Jiang, Z. Smooth muscle cell-specific Tgfbr1 deficiency promotes aortic aneurysm formation by stimulating multiple signaling events. Sci. Rep. 2016, 6, 35444. [Google Scholar] [CrossRef]
- Michel, J.-B.; Jondeau, G.; Milewicz, D.M. From genetics to response to injury: Vascular smooth muscle cells in aneurysms and dissections of the ascending aorta. Cardiovasc. Res. 2018, 114, 578–589. [Google Scholar] [CrossRef]
- Senchenkova, E.Y.; Russell, J.; Yildirim, A.; Granger, D.N.; Gavins, F.N. Novel Role of T Cells and IL-6 (Interleukin-6) in Angiotensin II–Induced Microvascular Dysfunction. Hypertension 2019, 73, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Palterer, B.; Vitiello, G.; Carraresi, A.; Giudizi, M.G.; Cammelli, D.; Parronchi, P. Bench to bedside review of myositis autoantibodies. Clin. Mol. Allergy 2018, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, R.; Altinbas, L.; Jäger, M.; Zaradzki, M.; Lehmann, D.; Timmermann, B.; Clayton, N.P.; Zhu, Y.; Kallenbach, K.; Kararigas, G.; et al. An integrative systems approach identifies novel candidates in Marfan syndrome-related pathophysiology. J. Cell. Mol. Med. 2019, 23, 2526–2535. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Fujimaki, T.; Yoshida, T.; Oguri, M.; Yajima, K.; Hibino, T.; Murohara, T. Impact of matrix metalloproteinase-2 levels on long-term outcome following pharmacological or electrical cardioversion in patients with atrial fibrillation. Europace 2009, 11, 332–337. [Google Scholar] [CrossRef]
- Odenbach, J.; Wang, X.; Cooper, S.; Chow, F.L.; Oka, T.; Lopaschuk, G.; Kassiri, Z.; Fernandez-Patron, C. MMP-2 Mediates Angiotensin II–Induced Hypertension Under the Transcriptional Control of MMP-7 and TACE. Hypertension 2011, 57, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, R.; Tscheuschler, A.; Laschinski, P.; Uffelmann, X.; Discher, P.; Fuchs, J.; Kreibich, M.; Peyronnet, R.; Kari, F.A. A potential key mechanism in ascending aortic aneurysm development: Detection of a linear relationship between MMP-14/TIMP-2 ratio and active MMP-2. PLoS ONE 2019, 14, e0212859. [Google Scholar] [CrossRef]
- Fujita, D.; Preiss, L.; Aizawa, K.; Asch, F.; Eagle, K.; Suzuki, T.; GenTAC Registry Investigators. Circulating interleukin-6 (IL-6) levels are associated with aortic dimensions in genetic aortic conditions. PLoS ONE 2019, 14, e0214084. [Google Scholar] [CrossRef] [PubMed]
- Bossone, E.; LaBounty, T.M.; Eagle, K.A. Acute aortic syndromes: Diagnosis and management, an update. Eur. Heart J. 2018, 39, 739–749d. [Google Scholar] [CrossRef]
- Sawada, H.; Chen, J.Z.; Wright, B.C.; Sheppard, M.B.; Lu, H.S.; Daugherty, A. Heterogeneity of aortic smooth muscle cells: A determinant for regional characteristics of thoracic aortic aneurysms? J. Transl. Intern. Med. 2018, 6, 93–96. [Google Scholar] [CrossRef]
- Vapnik, J.S.; Kim, J.B.; Isselbacher, E.M.; Ghoshhajra, B.B.; Cheng, Y.; Sundt, T.M.; MacGillivray, T.E.; Cambria, R.P.; Lindsay, M.E. Characteristics and Outcomes of Ascending Versus Descending Thoracic Aortic Aneurysms. Am. J. Cardiol. 2016, 117, 1683–1690. [Google Scholar] [CrossRef]
- Isselbacher, E.M. Thoracic and abdominal aortic aneurysms. Circulation 2005, 111, 816–828. [Google Scholar] [CrossRef]
- Goldfinger, J.Z.; Halperin, J.L.; Marin, M.L.; Stewart, A.S.; Eagle, K.A.; Fuster, V. Thoracic Aortic Aneurysm and Dissection. J. Am. Coll. Cardiol. 2014, 64, 1725–1739. [Google Scholar] [CrossRef] [PubMed]
- Pape, L.A.; Awais, M.; Woznicki, E.M.; Suzuki, T.; Trimarchi, S.; Evangelista, A.; Myrmel, T.; Larsen, M.; Harris, K.M.; Greason, K.; et al. Presentation, Diagnosis, and Outcomes of Acute Aortic Dissection. J. Am. Coll. Cardiol. 2015, 66, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, R.; Aatre, R.D.; Kanthi, Y. Diagnostic approach and management of genetic aortopathies. Vasc. Med. 2020, 25, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Safal, S.; Tyagi, D. Aortitis and aortic aneurysm in systemic vasculitis. Indian J. Thorac. Cardiovasc. Surg. 2019, 35, 47–56. [Google Scholar] [CrossRef]
- Evans, J.M.; Bowles, C.A.; Bjornsson, J.; Mullany, C.J.; Hunder, G.G. Thoracic aortic aneurysm and rupture in giant cell arteritis. A descriptive study of 41 cases. Arthritis Rheum. 1994, 37, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-P.; Zhu, P. Clinical features of aortic dissection associated with Takayasu’s arteritis. J. Geriatr. Cardiol. 2017, 14, 485–487. [Google Scholar]
- Laco, J.; Steiner, I.; Holubec, T.; Dominik, J.; Holubcova, Z.; Vojacek, J. Isolated thoracic aortitis: Clinicopathological and immunohistochemical study of 11 cases. Cardiovasc. Pathol. 2011, 20, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-M.; Ma, W.-G.; Peterss, S.; Wang, L.-F.; Qiao, Z.-Y.; Ziganshin, B.A.; Zheng, J.; Liu, Y.-M.; Elefteriades, J.A.; Sun, L.-Z. Aortic Dissection in Pregnancy: Management Strategy and Outcomes. Ann. Thorac. Surg. 2017, 103, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Sharp, R.P.; Gales, B.J. Nebivolol versus other beta blockers in patients with hypertension and erectile dysfunction. Ther. Adv. Urol. 2017, 9, 59–63. [Google Scholar] [CrossRef]
- Giusti, B.; Nistri, S.; Sticchi, E.; De Cario, R.; Abbate, R.; Gensini, G.F.; Pepe, G. A Case Based Approach to Clinical Genetics of Thoracic Aortic Aneurysm/Dissection. BioMed Res. Int. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Aurelian, S.V.; Adrian, M.; Dan, B.; Bruno, S.; Alexandru, O.; Catalin, T.; Octavian, A.; Aurel, A. Giant Infrarenal Aortic Aneurysm Rupture Preceded by Left Lower Limb Motor Deficit. Ann. Vasc. Surg. 2017, 43, 317. [Google Scholar] [CrossRef]







| Feature | Mutation or Missense Variant | Pathogenicity | References |
|---|---|---|---|
| Thoracic aneurysm | c.530G>A (p.R177Q) c.477C>T (p.Thr159Thr), c.993T>C (p.Val331Val), c.790A>G, p.Ile264Val | P VUS | [7,22,27], CC |
| Abdominal dissection | c.1108G>A (p.Gly370Ser) | P | [33] |
| Arterial tortuosity | c.530G>A (p.R177Q), c.1108G>A (p.Gly370Ser) | P | [22,33] |
| Hypertension | c.530G>A (p.R177Q), c.1108G>A (p.Gly370Ser) c.790A>G, p.Ile264Val | P VUS | [22,33] CC |
| Scoliosis | c.530G>A (p.R177Q) c.790A>G, p.Ile264Val | P VUS | [7] CC |
| Wrist and thumb sign | c.530G>A (p.R177Q) | P | [7] |
| Arachnodactily | c.993T>C (p.Val331Val) | VUS | [27] |
| Pectus carinatus/excavatum | c.530G>A (p.R177Q) c.993T>C (p.Val331Val) | P VUS | [7] [27] |
| Miopy | c.530G>A (p.R177Q) c.477C>T (p.Thr159Thr) | P VUS | [7] [27] |
| Keratoconus | c.1108G>A (p.Gly370Ser) | P | [33] |
| Small deep set eyes | c.993T>C (p.Val331Val), c.790A>G, p.Ile264Val | VUS | [27], CC |
| Skin striae | c.530G>A (p.R177Q) c.477C>T (p.Thr159Thr), c.993T>C (p.Val331Val) | P VUS | [7] [27] |
| Keloid scars | c.477C>T (p.Thr159Thr) | VUS | [27] |
| Premature aging | c.993T>C (p.Val331Val) | VUS | [27] |
| Subluxating patellae | c.790A>G, p.Ile264Val | VUS | CC |
| Joint pain | c.993T>C (p.Val331Val) | VUS | [27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Encica, S.; Molnar, A.; Manole, S.; Filan, T.; Oprița, S.; Bursașiu, E.; Vulturar, R.; Damian, L. Rare Causes of Arterial Hypertension and Thoracic Aortic Aneurysms—A Case-Based Review. Diagnostics 2021, 11, 446. https://doi.org/10.3390/diagnostics11030446
Encica S, Molnar A, Manole S, Filan T, Oprița S, Bursașiu E, Vulturar R, Damian L. Rare Causes of Arterial Hypertension and Thoracic Aortic Aneurysms—A Case-Based Review. Diagnostics. 2021; 11(3):446. https://doi.org/10.3390/diagnostics11030446
Chicago/Turabian StyleEncica, Svetlana, Adrian Molnar, Simona Manole, Teodora Filan, Simona Oprița, Eugen Bursașiu, Romana Vulturar, and Laura Damian. 2021. "Rare Causes of Arterial Hypertension and Thoracic Aortic Aneurysms—A Case-Based Review" Diagnostics 11, no. 3: 446. https://doi.org/10.3390/diagnostics11030446
APA StyleEncica, S., Molnar, A., Manole, S., Filan, T., Oprița, S., Bursașiu, E., Vulturar, R., & Damian, L. (2021). Rare Causes of Arterial Hypertension and Thoracic Aortic Aneurysms—A Case-Based Review. Diagnostics, 11(3), 446. https://doi.org/10.3390/diagnostics11030446