
Mechanisms of Neuronal Damage in Acute Hepatic Porphyrias



{kind=link}
Abstract
1. Introduction
Experimental Models for Studying AHPs
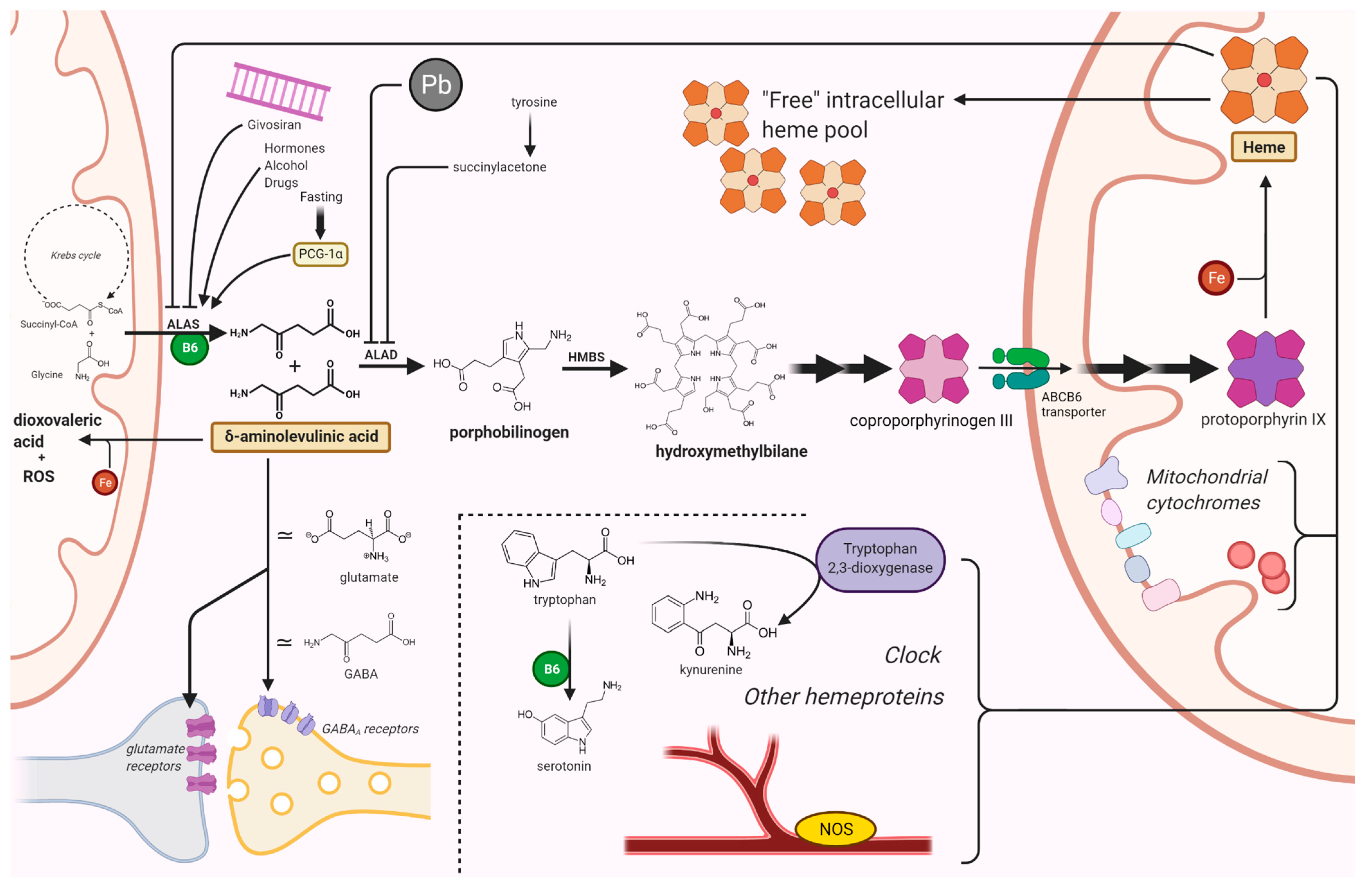
2. δ-Aminolevulinic Acid Toxicity
2.1. Mechanisms of Transport and Uptake of δ-ALA in the CNS
2.2. Endogenous Production of δ-ALA in the CNS
2.3. Oxidative Damage, Mitochondrial Alterations and Effects on Iron Homeostasis
2.4. Neurotransmitter Balance Disruption
2.5. Other Effects of δ-Aminolevulinic Acid
3. Heme Deficiency-Induced Dysfunction
3.1. Alterations in Heme-Dependent Signal Transduction
3.2. Cytochrome Dysfunction
3.3. Effects on Tryptophan and Glucose Metabolism
3.3.1. Effect on Melatonin and Circadian Cycles
3.4. Effects on Nitric Oxide Synthase
4. Other Proposed Mechanisms of Toxicity
4.1. Cataplerosis Induced by Succinyl-CoA Deficiency
4.2. Pyridoxal Phosphate Consumption
5. Conclusions
6. Acronyms
Author Contributions
Funding
Conflicts of Interest
References
- Bissell, D.M.; Anderson, K.E.; Bonkovsky, H.L. Porphyria. N. Eng. J. Med. 2017, 377, 862–872. [Google Scholar] [CrossRef]
- Simon, N.G.; Herkes, G.K. The neurologic manifestations of the acute porphyrias. J. Clin. Neurosci. 2011, 18, 1147–1153. [Google Scholar] [CrossRef]
- Bissell, D.M.; Wang, B. Acute Hepatic Porphyria. J. Clin. Transl. Hepatol. 2015, 3, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Bonkowsky, H.L.; Schady, W. Neurologic manifestations of acute porphyria. Semin. Liver Dis. 1982, 2, 108–124. [Google Scholar] [CrossRef]
- Meyer, U.A.; Schuurmans, M.M.; Lindberg, R.L. Acute porphyrias: Pathogenesis of neurological manifestations. Semin. Liver Dis. 1998, 18, 43–52. [Google Scholar] [CrossRef]
- Ventura, P.; Cuoghi, C.; Marcacci, M. The acute porphyric attack: A difficult diagnosis for a potential lethal event in emergency medicine. J. Emerg. Med. Trauma Surg. Care 2015, 2, 1–8. [Google Scholar] [CrossRef]
- Roveri, G.; Nascimbeni, F.; Rocchi, E.; Ventura, P. Drugs and acute porphyrias: Reasons for a hazardous relationship. Postgrad. Med. 2014, 126, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Pischik, E.; Kauppinen, R. Neurological Manifestations of Acute Intermittent Porphyria. Cell. Mol. Biol. 2015, 55, 72–83. [Google Scholar]
- Souza, P.V.S.; Badia, B.M.L.; Farias, I.B.; Gonçalves, E.A.; Pinto, W.B.V.R.; Oliveira, A.S.B. Acute hepatic porphyrias for the neurologist: Current concepts and perspectives. Arq. Neuropsiquiatr. 2021, 79, 68–80. [Google Scholar] [CrossRef]
- Jaramillo-Calle, D.A.; Solano, J.M.; Rabinstein, A.A.; Bonkovsky, H.L. Porphyria-induced posterior reversible encephalopathy syndrome and central nervous system dysfunction. Mol. Genet. Metab. 2019, 128, 242–253. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, X.; Wang, Y.; Zhao, R.; Qu, L.; Pei, H.; Tuo, M.; Zhang, Y.; Song, Y.; Ji, X.; et al. Acute intermittent porphyria presenting with seizures and posterior reversible encephalopathy syndrome: Two case reports and a literature review. Medicine 2018, 97, e11665. [Google Scholar] [CrossRef]
- Bylesjö, I.; Brekke, O.L.; Prytz, J.; Skjeflo, T.; Salvesen, R. Brain magnetic resonance imaging white-matter lesions and cerebrospinal fluid findings in patients with acute intermittent porphyria. Eur. Neurol. 2004, 51, 1–5. [Google Scholar] [CrossRef]
- Nordmann, Y.; Puy, H.; Da Silva, V.; Simonin, S.; Robreau, A.M.; Bonaiti, C.; Phung, L.N.; Deybach, J.C. Acute intermittent porphyria: Prevalence of mutations in the porphobilinogen deaminase gene in blood donors in france. J. Intern. Med. 1997, 242, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.; Harper, P.; Badminton, M.; Sandberg, S.; Deybach, J.C. The incidence of inherited porphyrias in europe. J. Inherit. Metab. Dis. 2013, 36, 849–857. [Google Scholar] [CrossRef]
- Chen, B.; Solis-Villa, C.; Hakenberg, J.; Qiao, W.; Srinivasan, R.R.; Yasuda, M.; Balwani, M.; Doheny, D.; Peter, I.; Chen, R.; et al. Acute intermittent porphyria: Predicted pathogenicity of HMBS variants indicates extremely low penetrance of the autosomal dominant disease. Hum. Mut. 2016, 37, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- McEwin, R.; Lawn, J.; Jonas, C.T. A survey of porphyria among psychiatric patients. Med. J. Aust. 1972, 2, 303–306. [Google Scholar] [CrossRef]
- Golechha, G.R.; Chatterjee, S.B.; Sethi, B.B.; Agarwal, S.S. Acute porphyria amongst psychiatric patients. Indian J. Psychiatry 1981, 23, 365–369. [Google Scholar]
- Tishler, P.V.; Woodward, B.; O’Connor, J.; Holbrook, D.A.; Seidman, L.J.; Hallett, M.; Knighton, D.J. High prevalence of intermittent acute porphyria in a psychiatric patient population. Am. J. Psychiatry 1985, 142, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.; Larochelle, J.; Lambert, M.; de Weerd, A.W.; Gianella-Borradori, A.; Michaud, J.; Grenier, A.; Ogier, H.; Gauthier, M.; Lacroix, J.; et al. Neurologic Crises in Hereditary Tyrosinemia. N. Engl. J. Med. 1990, 322, 432–437. [Google Scholar] [CrossRef]
- Bissell, D.M.; Lai, J.C.; Meister, R.K.; Blanc, P.D. Role of delta-aminolevulinic acid in the symptoms of acute porphyria. Am. J. Med. 2015, 128, 313–317. [Google Scholar] [CrossRef]
- Sassa, S.; Kappas, A. Hereditary tyrosinemia and the heme biosynthetic pathway. profound inhibition of δ-aminolevulinic acid de- hydratase activity by succinylacetone. J. Clin. Investig. 1983, 71, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Warren, M.J.; Cooper, J.B.; Wood, S.P.; Shoolingin-Jordan, P.M. Lead poisoning, haem synthesis and 5-aminolaevulinic acid dehydratase. Trends Biochem. Sci. 1998, 23, 217–221. [Google Scholar] [CrossRef]
- Foote, C.S. Definition of type I and type II photosensitized oxidation. Photochem. Photobiol. 1991, 54, 659. [Google Scholar] [CrossRef] [PubMed]
- Brun, A.; Sandberg, S. Mechanisms of photosensitivity in porphyric patients with special emphasis on erythropoietic protoporphyria. J. Photochem. Photobiol. B 1991, 10, 285–302. [Google Scholar] [CrossRef]
- Souza, P.V.S.; Badia, B.M.L.; Farias, I.B.; Pinto, W.B.V.R.; Oliveira, A.S.B. Acute hepatic porphyria: Pathophysiological basis of neuromuscular manifestations. Front. Neurosci. 2021, 15, 715523. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.S.; Levere, R.D.; Lieberman, J.S.; Cardinal, R.A.; Watson, C.J. Presynaptic neuromuscular inhibition by porphobilinogen and porphobilin. Proc. Natl. Acad. Sci. USA 1971, 68, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Wyss, P.A.; Boynton, S.; Chu, J.; Roth, A.S. Tissue distribution of succinylacetone in the rat in vivo: A possible basis for neurotoxicity in hereditary infantile tyrosinemia. Biochim. Biophys. Acta Mol. Basis Dis. 1993, 1182, 323–328. [Google Scholar] [CrossRef]
- Lelli, S.M.; de Viale, L.C.S.M.; Mazzetti, M.B. Response of glucose metabolism enzymes in an acute porphyria model: Role of reactive oxygen species. Toxicology 2005, 216, 49–58. [Google Scholar] [CrossRef]
- Handschin, C.; Lin, J.; Rhee, J.; Peyer, A.K.; Chin, S.; Wu, P.H.; Meyer, U.A.; Spiegelman, B.M. Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1α. Cell 2005, 122, 505–515. [Google Scholar] [CrossRef]
- De Matteis, F.; Rimington, C. Disturbance of porphyrin metabolism caused by griseofulvin in mice. Br. J. Dermatol. 1963, 75, 91–104. [Google Scholar] [CrossRef]
- Inafuku, K.; Takamiyagi, A.; Oshiro, M.; Kinjo, T.; Nakashima, Y.; Nonaka, S. Alteration of mRNA levels of δ-aminolevulinic acid synthase, ferrochelatase and heme oxygenase-1 in griseofulvin induced protoporphyria mice. J. Dermatol. Sci. 1999, 19, 189–198. [Google Scholar] [CrossRef]
- Juknat, A.A.; Kotler, M.L.; del Carmen Batlle, A.M. High δ-aminolevulinic acid uptake in rat cerebral cortex: Effect on porphyrin biosynthesis. Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 1995, 111, 143–150. [Google Scholar] [CrossRef]
- Princ, F.G.; Juknat, A.A.; Batlle, A.M.D.C. Porphyrinogenesis in rat cerebellum. Effect of high δ-aminolevulinic acid concentration. Gen. Pharmacol. Vasc. Syst. 1994, 25, 761–766. [Google Scholar] [CrossRef]
- Lavandera, J.; Rodr’ıguez, J.; Ruspini, S.; Meiss, R.; Zuccoli, J.R.; Mart’ınez, M.D.C.; Gerez, E.; Batlle, A.; Buzaleh, A.M. Pleiotropic effects of 5-aminolevulinic acid in mouse brain. Biochem. Cell Biol. 2016, 94, 297–305. [Google Scholar] [CrossRef]
- Felitsyn, N.; McLeod, C.; Shroads, A.L.; Stacpoole, P.W.; Notterpek, L. The heme precursor delta-aminolevulinate blocks peripheral myelin formation. J. Neurochem. 2008, 106, 2068–2079. [Google Scholar] [CrossRef]
- Yasuda, M.; Desnick, R.J. Murine models of the human porphyrias: Contributions toward understanding disease pathogenesis and the development of new therapies. Mol. Genet. Metab. 2019, 128, 332–341. [Google Scholar] [CrossRef]
- Lindberg, R.L.; Porcher, C.; Grandchamp, B.; Ledermann, B.; Bu¨rki, K.; Brandner, S.; Aguzzi, A.; Meyer, U.A. Porphobilinogen deaminase deficiency in mice causes a neuropathy resembling that of human hepatic porphyria. Nat. Genet. 1996, 12, 195–199. [Google Scholar] [CrossRef]
- Lindberg, R.L.; Martini, R.; Baumgartner, M.; Erne, B.; Borg, J.; Zielasek, J.; Ricker, K.; Steck, A.; Toyka, K.V.; Meyer, U.A. Motor neuropathy in porphobilinogen deaminase-deficient mice imitates the peripheral neuropathy of human acute porphyria. J. Clin. Investig. 1999, 103, 1127–1134. [Google Scholar] [CrossRef]
- Yasuda, M.; Gan, L.; Chen, B.; Yu, C.; Zhang, J.; Gama-Sosa, M.A.; Pollak, D.D.; Berger, S.; Phillips, J.D.; Edelmann, W.; et al. Homozygous hydroxymethylbilane synthase knock-in mice provide pathogenic insights into the severe neurological impairments present in human homozygous dominant acute intermittent porphyria. Hum. Mol. Genet. 2019, 28, 1755–1767. [Google Scholar] [CrossRef]
- Ennis, S.R.; Novotny, A.; Xiang, J.; Shakui, P.; Masada, T.; Stummer, W.; Smith, D.E.; Keep, R.F. Transport of 5-aminolevulinic acid between blood and brain. Brain Res. 2003, 959, 226–234. [Google Scholar] [CrossRef]
- Novotny, A.; Xiang, J.; Stummer, W.; Teuscher, N.S.; Smith, D.E.; Keep, R.F. Mechanisms of 5-aminolevulinic acid uptake at the choroid plexus. J. Neurochem. 2000, 75, 321–328. [Google Scholar] [CrossRef]
- Shu, C.; Shen, H.; Teuscher, N.S.; Lorenzi, P.J.; Keep, R.F.; Smith, D.E. Role of PEPT2 in peptide/mimetic trafficking at the blood-cerebrospinal fluid barrier: Studies in rat choroid plexus epithelial cells in primary culture. J. Pharmacol. Exp. Ther. 2002, 301, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Berger, U.V.; Hediger, M.A. Distribution of peptide transporter PEPT2 mRNA in the rat nervous system. Anat. Embryol. 1999, 199, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Groneberg, D.A.; D¨oring, F.; Nickolaus, M.; Daniel, H.; Fischer, A. Expression of PEPT2 peptide transporter mRNA and protein in glial cells of rat dorsal root ganglia. Neurosci. Lett. 2001, 304, 181–184. [Google Scholar] [CrossRef]
- Whetsell, W.O. Porphyrin-heme biosynthesis in organotypic cultures of mouse dorsal root ganglia. Effects of heme and lead on porphyrin synthesis and peripheral myelin. J. Clin. Investig. 1984, 74, 600–607. [Google Scholar] [CrossRef]
- Xiang, J.; Hu, Y.; Smith, D.E.; Keep, R.F. PEPT2-mediated transport of 5-aminolevulinic acid and carnosine in astrocytes. Brain Res. 2006, 1122, 18–23. [Google Scholar] [CrossRef]
- Shen, H.; Smith, D.E.; Keep, R.F.; Brosius, F.C. Immunolocalization of the proton-coupled oligopeptide transporter PEPT2 in developing rat brain. Mol. Pharm. 2004, 1, 248–256. [Google Scholar] [CrossRef]
- Hu, Y.; Shen, H.; Keep, R.F.; Smith, D.E. Peptide transporter 2 (PEPT2) expression in brain protects against 5-aminolevulinic acid neurotoxicity. J. Neurochem. 2007, 103, 2058–2065. [Google Scholar] [CrossRef]
- Tchernitchko, D.; Tavernier, Q.; Lamoril, J.; Schmitt, C.; Talbi, N.; Lyoumi, S.; Robreau, A.M.; Karim, Z.; Gouya, L.; Thervet, E.; et al. A Variant of Peptide Transporter 2 Predicts the Severity of Porphyria-Associated Kidney Disease. J. Am. Soc. Nephrol. 2017, 28, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Sobin, C.; Flores-Montoya, M.G.; Gutierrez, M.; Parisi, N.; Schaub, T. δ-Aminolevulinic acid dehydratase single nucleotide polymorphism 2 (ALAD2) and peptide transporter 2*2 haplotype (hPEPT2*2) differently influence neurobehavior in low-level lead exposed children. Neurotoxicol. Teratol. 2015, 47, 137–145. [Google Scholar] [CrossRef]
- Chen, N.H.; Reith, M.E.; Quick, M.W. Synaptic uptake and beyond: The sodium-and chloride-dependent neurotransmitter transporter family SLC6. Pflüger’s Gers Arch. 2004, 447, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Baglo, Y.; Gabrielsen, M.; Sylte, I.; Gederaas, O.A. Homology Modeling of Human γ-Butyric Acid Transporters and the Binding of Pro-Drugs 5-Aminolevulinic Acid and Methyl Aminolevulinic Acid Used in Photodynamic Therapy. PLoS ONE 2013, 8, e65200. [Google Scholar] [CrossRef]
- Tran, T.T.; Mu, A.; Adachi, Y.; Adachi, Y.; Taketani, S. Neurotransmitter transporter family including SLC6A6 and SLC6A13 contributes to the 5-aminolevulinic acid (ALA)-induced accumulation of protoporphyrin IX and photodamage, through uptake of ALA by cancerous cells. Photochem. Photobiol. 2014, 90, 1136–1143. [Google Scholar] [CrossRef]
- Frølund, S.; Marquez, O.C.; Larsen, M.; Brodin, B.; Nielsen, C.U. δ-aminolevulinic acid is a substrate for the amino acid transporter SLC36A1 (HPAT1). Br. J. Pharmacol. 2010, 159, 1339–1353. [Google Scholar] [CrossRef]
- Palladino, S.P.; Helton, E.S.; Jain, P.; Dong, C.; Crowley, M.R.; Crossman, D.K.; Ubogu, E.E. The human blood-nerve barrier transcriptome. Sci. Rep. 2017, 7, 17477. [Google Scholar] [CrossRef] [PubMed]
- Kazamel, M.; Desnick, R.J.; Quigley, J.G. Porphyric neuropathy: Pathophysiology, diagnosis, and updated management. Curr. Neurol. Neurosci. Rep. 2020, 20, 56. [Google Scholar] [CrossRef]
- Cutler, M.G.; Turner, J.M.; Moore, M.R. A comparative study of the effects of δ-aminolaevulinic acid and the GABAA agonist, muscimol, in rat jejunal preparations. Pharmacol. Toxicol. 1991, 69, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Sylantiev, C.; Schoenfeld, N.; Mamet, R.; Groozman, G.B.; Drory, V.E. Acute neuropathy mimicking porphyria induced by aminolevulinic acid during photodynamic therapy. Muscle Nerve 2005, 31, 390–393. [Google Scholar] [CrossRef]
- Lombardo, M.E.; Araujo, L.S.; Batlle, A. 5-Aminolevulinic acid synthesis in epimastigotes of Trypanosoma cruzi. Int. J. Biochem. Cell Biol. 2003, 35, 1263–1271. [Google Scholar] [CrossRef]
- Alves, J.M.; Voegtly, L.; Matveyev, A.V.; Lara, A.M.; da Silva, F.M.; Serrano, M.G.; Buck, G.A.; Teixeira, M.M.; Camargo, E.P. Identification and phylogenetic analysis of heme synthesis genes in trypanosomatids and their bacterial endosymbionts. PLoS ONE 2011, 6, e23518. [Google Scholar] [CrossRef]
- Sica, R.; Gonzalez, S.C.; Sanz, O.; Mirkin, G. Peripheral nervous system involvement in human and experimental chronic american trypanosomiasis. Bull Soc. Pathol. Exot. 1995, 88, 156–163. [Google Scholar] [PubMed]
- Lopes, E.R.; Tafuri, W.L. Involvement of the autonomic nervous system in Chagas heart disease. Rev. Soc. Bras. Med. Trop. 1983, 16, 206–212. [Google Scholar] [CrossRef][Green Version]
- P´erez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Lissing, M.; Nowak, G.; Adam, R.; Karam, V.; Boyd, A.; Gouya, L.; Meersseman, W.; Melum, E.; O-ldakowska-Jedynak, U.; Reiter, F.P.; et al. Liver transplantation for acute intermittent porphyria. Liver Transplant. 2021, 27, 491–501. [Google Scholar] [CrossRef]
- Dowman, J.K.; Gunson, B.K.; Bramhall, S.; Newsome, P.N.; Badminton, M.N. Liver transplantation from donors with acute intermittent porphyria. Ann. Intern. Med. 2011, 154, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Paterniti, J.R.; Simone, J.J.; Beattie, D.S. Detection and regulation of δ-aminolevulinic acid synthetase activity in the rat brain. Arch. Biochem. Biophys. 1978, 189, 86–91. [Google Scholar] [CrossRef]
- De Matteis, F.; Zetterlund, P.; Wetterberg, L. Brain 5-aminolaevulinate synthase. Developmental aspects and evidence for regulatory role. Biochem. J. 1981, 196, 811–817. [Google Scholar] [CrossRef]
- De Matteis, F.; Ray, D.E. Studies on Cerebellar Haem Metabolism in the Rat In Vivo. J. Neurochem. 1982, 39, 551–556. [Google Scholar] [CrossRef]
- Ruspini, S.F.; Zuccoli, J.R.; Lavandera, J.V.; Mart´ınez, M.D.C.; Oliveri, L.M.; Gerez, E.N.; Batlle, A.M.D.C.; Buzaleh, A.M. Effects of volatile anaesthetics on heme metabolism in a murine genetic model of Acute Intermittent Porphyria. A comparative study with other por-phyrinogenic drugs. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1296–1305. [Google Scholar] [CrossRef]
- Kang, K.; Anderson-Burham, L.A.; Bloomer, J.R. Effect of succinylacetone administration on brain heme metabolism and behavior in mice. Biochem. Pharmacol. 1987, 36, 3084–3086. [Google Scholar] [CrossRef]
- Chernova, T.; Steinert, J.R.; Guerin, C.J.; Nicotera, P.; Forsythe, I.D.; Smith, A.G. Neurite degeneration induced by heme deficiency mediated via inhibition of NMDA receptor-dependent extracellular signal- regulated kinase 1/2 activation. J. Neurosci. 2007, 27, 8475–8485. [Google Scholar] [CrossRef]
- Rocha, M.E.M.; Dutra, F.; Bandy, B.; Baldini, R.L.; Gomes, S.L.; Faljoni-Al´ario, A.; Liria, C.W.; Miranda, M.T.M.; Bechara, E.J.H. Oxidative damage to ferritin by 5-aminolevulinic acid. Arch. Biochem. Biophys. 2003, 409, 349–356. [Google Scholar] [CrossRef]
- Oteiza, P.I.; Bechara, E.J. 5-Aminolevulinic acid induces lipid peroxidation in cardiolipin-rich liposomes. Arch. Biochem. Biophys. 1993, 305, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Hermes-Lima, M.; Castilho, R.F.; Valle, V.G.; Bechara, E.J.; Vercesi, A.E. Calcium-dependent mitochondrial oxidative damage pro moted by 5-aminolevulinic acid. Biochim. Biophys. Acta Mol. Basis Dis. 1992, 1180, 201–206. [Google Scholar] [CrossRef]
- Vercesi, A.E.; Castilho, R.F.; Meinicke, A.R.; Valle, V.G.; Hermes-Lima, M.; Bechara, E.J. Oxidative damage of mitochondria induced by 5-aminolevulinic acid: Role of Ca2+ and membrane protein thiols. Biochim. Biophys. Acta (BBA)—Bioenerg. 1994, 1188, 86–92. [Google Scholar] [CrossRef]
- Laafi, J.; Homedan, C.; Jacques, C.; Gueguen, N.; Schmitt, C.; Puy, H.; Reynier, P.; Carmen Martinez, M.; Malthi‘ery, Y. Pro-oxidant effect of ALA is implicated in mitochondrial dysfunction of HepG2 cells. Biochimie 2014, 106, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Emanuelli, T.; Pagel, F.W.; Porciu´ncula, L.O.; Souza, D.O. Effects of 5-aminolevulinic acid on the glutamatergic neurotransmission. Neurochem. Int. 2003, 42, 115–121. [Google Scholar] [CrossRef]
- Oteiza, P.I.; Kleinman, C.G.; Demasi, M.; Bechara, E.J. 5-aminolevulinic acid induces iron release from ferritin. Arch. Biochem. Biophys. 1995, 316, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.E.; Bandy, B.; Costa, C.A.; de Barros, M.P.; Pinto, A.M.; Bechara, E.J. Iron mobilization by succinylacetone methyl ester in rats. A model study for hereditary tyrosinemia and porphyrias charac- terized by 5-aminolevulinic acid overload. Free Radic. Res. 2000, 32, 343–353. [Google Scholar] [CrossRef]
- Demasi, M.; Penatti, C.A.; DeLucia, R.; Bechara, E.J. The prooxidant effect of 5-aminolevulinic acid in the brain tissue of rats: Impli- cations in neuropsychiatric manifestations in porphyrias. Free Radic. Biol. Med. 1996, 20, 291–299. [Google Scholar] [CrossRef]
- Carvalho, H.; Bechara, E.J.; Meneghini, R.; Demasi, M. Haem precursor delta-aminolaevulinic acid induces activation of the cytosolic iron regulatory protein 1. Biochem. J. 1997, 328 Pt 3, 827–832. [Google Scholar] [CrossRef]
- Brennan, M.J.; Cantrill, R.C. δ-Aminolaevulinic acid is a potent agonist for GABA autoreceptors. Nature 1979, 280, 514–515. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.J.; Cantrill, R.C.; Kramer, S. Effect of delta- aminolaevulinic acid on GABA receptor binding in synaptic plasma membranes. Int. J. Biochem. 1980, 12, 833–835. [Google Scholar] [CrossRef]
- Adhikari, A.; Penatti, C.A.A.; Resende, R.R.; Ulrich, H.; Britto, L.R.G.; Bechara, E.J.H. 5-Aminolevulinate and 4, 5-dioxovalerate ions decrease GABA(A) receptor density in neuronal cells, synaptosomes and rat brain. Brain Res. 2006, 1093, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Puy, H.; Deybach, J.C.; Bogdan, A.; Callebert, J.; Baumgartner, M.; Voisin, P.; Nordmann, Y.; Touitou, Y. Increased δ aminolevulinic acid and decreased pineal melatonin production: A common event in acute porphyria studies in the rat. J. Clin. Investig. 1996, 97, 104–110. [Google Scholar] [CrossRef]
- Lin, T.C.; Lai, S.L.; Hsu, S.P.; Ro, L.S. Treatment of neuropathic pain in acute intermittent porphyria with gabapentin. J. Formos Med. Assoc. 2013, 112, 578–579. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Emanuelli, T.; Prauchner, C.A.; Dacanal, J.; Zeni, A.; Reis, E.C.; De Mello, C.F.; De Souza, D.O. Intrastriatal administration of 5-aminolevulinic acid induces convulsions and body asymmetry through glutamatergic mechanisms. Brain Res. 2000, 868, 88–94. [Google Scholar] [CrossRef]
- Becker, D.; Viljoen, D.; Kramer, S. The inhibition of red cell and brain atpase by δ-aminolaevulinic acid. Biochim. Biophys. Acta Biomembr. 1971, 225, 26–34. [Google Scholar] [CrossRef]
- Russell, V.A.; Lamm, M.C.; Taljaard, J.J. Inhibition of Na+, K+-ATPase activity by delta-aminolevulinic acid. Neurochem. Res. 1983, 8, 1407–1415. [Google Scholar] [CrossRef]
- Emanuelli, T.; Pagel, F.W.; Alves, L.B.; Regner, A.; Souza, D.O. Inhibition of adenylate cyclase activity by 5-aminolevulinic acid in rat and human brain. Neurochem. Int. 2001, 38, 213–218. [Google Scholar] [CrossRef]
- Zhu, Y.; Hon, T.; Ye, W.; Zhang, L. Heme deficiency interferes with the Ras-mitogen-activated protein kinase signaling pathway and expression of a subset of neuronal genes. Cell Growth Differ. 2002, 13, 431–439. [Google Scholar] [PubMed]
- Zhu, Y.; Lee, H.C.; Zhang, L. An Examination of Heme Action in Gene Expression: Heme and Heme Deficiency Affect the Expression of Diverse Genes in Erythroid K562 and Neuronal PC12 Cells. DNA Cell Biol. 2002, 21, 333–346. [Google Scholar] [CrossRef]
- Sengupta, A.; Hon, T.; Zhang, L. Heme deficiency suppresses the expression of key neuronal genes and causes neuronal cell death. Brain Res. Mol. Brain Res. 2005, 137, 23–30. [Google Scholar] [CrossRef]
- Weinshenker, D.; Szot, P.; Miller, N.S.; Rust, N.C.; Hohmann, J.G.; Pyati, U.; White, S.S.; Palmiter, R.D. Genetic comparison of seizure control by norepinephrine and neuropeptide Y. J. Neurosci. 2001, 21, 7764–7769. [Google Scholar] [CrossRef]
- Chernova, T.; Nicotera, P.; Smith, A.G. Heme deficiency is associated with senescence and causes suppression of N-methyl-D-aspartate receptor subunits expression in primary cortical neurons. Mol. Phar. Macol. 2006, 69, 697–705. [Google Scholar] [CrossRef]
- Song, G.; Zhang, S.; Tian, M.; Zhang, L.; Guo, R.; Zhuo, W.; Yang, M. Molecular insights into the human ABCB6 transporter. Cell Discovery 2021, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Homedan, C.; Schmitt, C.; Laafi, J.; Gueguen, N.; Desquiret-Dumas, V.; Lenglet, H.; Karim, Z.; Gouya, L.; Deybach, J.C.; Simard, G.; et al. Mitochondrial energetic defects in muscle and brain of a Hmbs-/- mouse model of acute intermittent porphyria. Hum. Mol. Genet. 2015, 24, 5015–5023. [Google Scholar] [CrossRef]
- Dixon, N.; Li, T.; Marion, B.; Faust, D.; Dozier, S.; Molina, A.; Rud- Nick, S.; Bonkovsky, H.L. Pilot study of mitochondrial bioenergetics in subjects with acute porphyrias. Mol. Genet. Metab. 2019, 128, 228–235. [Google Scholar] [CrossRef]
- Jover, R.; Hoffmann, F.; Scheffler-Koch, V.; Lindberg, R.L. Limited heme synthesis in porphobilinogen deaminase-deficient mice impairs transcriptional activation of specific cytochrome P450 genes by phenobarbital. Eur. J. Biochem. 2000, 267, 7128–7137. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.B. The functions and regulation of tryptophan pyrrolase. Life Sci. 1977, 21, 755–767. [Google Scholar] [CrossRef]
- Salter, M.; Hazelwood, R.; Pogson, C.I.; Iyer, R.; Madge, D.J. The effects of a novel and selective inhibitor of tryptophan 2,3-dioxygenase on tryptophan and serotonin metabolism in the rat. Biochem. Pharmacol. 1995, 49, 1435–1442. [Google Scholar] [CrossRef]
- Litman, D.A.; Correia, M.A. L-tryptophan: A common denominator of biochemical and neurological events of acute hepatic porphyria? Science 1983, 222, 1031–1033. [Google Scholar] [CrossRef]
- Lelli, S.M.; Mazzetti, M.B.; San Mart´ın de Viale, L.C. Hepatic alteration of tryptophan metabolism in an acute porphyria model Its relation with gluconeogenic blockage. Biochem. Pharmacol. 2008, 75, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gomez, A.; Marcos, J.; Aguilera, P.; To-Figueras, J.; Pozo, O.J. Comprehensive analysis of the tryptophan metabolome in urine of patients with acute intermittent porphyria. J. Chromatogr. B 2017, 1060, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Correia, M.; Lunetta, J. Acute hepatic heme depletion: Impaired gluconeogenesis in rats. Semin. Hematol. 1989, 26, 120–127. [Google Scholar] [PubMed]
- Collantes, M.; Serrano-Mendioroz, I.; Benito, M.; Molinet-Dronda, F.; Delgado, M.; Vinaixa, M.; Sampedro, A.; Enr´ıquez de Salamanca, R.; Prieto, E.; Pozo, M.A.; et al. Glucose metabolism during fasting is altered in experimental porphobilinogen deaminase deficiency. Hum. Mol. Genet. 2016, 25, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Haruki, H.; Hovius, R.; Pedersen, M.G.; Johnsson, K. Tetrahydro- biopterin biosynthesis as a potential target of the kynurenine pathway metabolite xanthurenic acid. J. Biol. Chem. 2016, 291, 652–657. [Google Scholar] [CrossRef]
- Staats Pires, A.; Tan, V.X.; Heng, B.; Guillemin, G.J.; Latini, A. Kynurenine and tetrahydrobiopterin pathways crosstalk in pain hypersensitivity. Front. Neurosci. 2020, 14, 620. [Google Scholar] [CrossRef]
- Hamfelt, A.; Wetterberg, L. Pyridoxal phosphate in acute intermittent porphyria. Ann. N. Y. Acad. Sci. 1969, 166, 361–364. [Google Scholar] [CrossRef]
- Haugen, V.E.; Storjord, E.; Bjørke Monsen, A.; Brekke, O.; Sandberg, S.; Dahl, J.; Landsem, A.; Mollnes, T.; Waage Nielsen, E.; Ueland, P.; et al. Impaired vitamin b6 status in patients with acute intermittent porphyria. In Proceedings of the International Congress on Porphyrins and Porphyrias, Bordeaux, France, 3 July 2017. [Google Scholar]
- Alkaitis, M.S.; Crabtree, M.J. Recoupling the cardiac nitric oxide synthases: Tetrahydrobiopterin synthesis and recycling. Curr. Heart Fail. Rep. 2012, 9, 200–210. [Google Scholar] [CrossRef]
- Lelli, S.M.; Mazzetti, M.B.; de Viale, L.C.S.M. Melatonin modulates drug-induced acute porphyria. Toxicol. Rep. 2016, 3, 141–147. [Google Scholar] [CrossRef]
- Puy, H.; Deybach, J.C.; Baudry, P.; Callebert, J.; Touitou, Y.; Nordmann, Y. Decreased nocturnal plasma melatonin levels in patients with recurrent acute intermittent porphyria attacks. Life Sci. 1993, 53, 621–627. [Google Scholar] [CrossRef]
- Lukat-Rodgers, G.S.; Correia, C.; Botuyan, M.V.; Mer, G.; Rodgers, K.R. Heme-based sensing by the mammalian circadian protein CLOCK. Inorg. Chem. 2010, 49, 6349–6365. [Google Scholar] [CrossRef][Green Version]
- Yang, J.; Kim, K.D.; Lucas, A.; Drahos, K.E.; Santos, C.S.; Mury, S.P.; Capelluto, D.G.S.; Finkielstein, C.V. A novel heme-regulatory motif mediates heme-dependent degradation of the circadian factor Period 2. Mol. Cell. Biol. 2008, 28, 4697–4711. [Google Scholar] [CrossRef]
- Burris, T.P. Nuclear Hormone Receptors for Heme: REV-ERBα and REV-ERBβ Are Ligand-Regulated Components of the Mammalian Clock. Mol. Endocrinol. 2008, 22, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Wu, N.; Curtin, J.C.; Qatanani, M.; Szwergold, N.R.; Reid, R.A.; Waitt, G.M.; Parks, D.J.; Pearce, K.H.; Wisely, G.B.; et al. Rev-erbα, a heme sensor that coordinates metabolic and circadian pathways. Science 2007, 318, 1786–1789. [Google Scholar] [CrossRef]
- Rogers, P.M.; Ying, L.; Burris, T.P. Relationship between circadian oscillations of REV-ERBα expression and intracellular levels of its ligand, heme. Biochem. Biophys. Res. Commun. 2008, 368, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Iwadate, R.; Satoh, Y.; Watanabe, Y.; Kawai, H.; Kudo, N.; Kawashima, Y.; Mashino, T.; Mitsumoto, A. Impairment of heme biosynthesis induces short circadian period in body temperature rhythms in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R8–R18. [Google Scholar] [CrossRef] [PubMed]
- Thachil, J. L-Asparaginase, nitric oxide and posterior reversible encephalopathy syndrome. Ann. Hematol. 2013, 92, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Klatt, P.; Pfeiffer, S.; List, B.M.; Lehner, D.; Glatter, O.; B¨achinger, H.P.; Werner, E.R.; Schmidt, K.; Mayer, B. Characterization of heme-deficient neuronal nitric-oxide synthase reveals a role for heme in subunit dimerization and binding of the amino acid substrate and tetrahydrobiopterin. J. Biol. Chem. 1996, 271, 7336–7342. [Google Scholar] [CrossRef]
- Bredt, D.S.; Hwang, P.M.; Snyder, S.H. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 1990, 347, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O.; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; Wang, X.; Sapsford, R.J.; Russell, R.P.; Farrell, A.L.; Jessop, H.A.; McGawley, G.M.; Oxborough, D.L.; Pleasants, P.; Richards, S.J.; et al. Nitric oxide consumption and pulmonary hypertension in patients with paroxysmal nocturnal hemoglobinuria. Blood 2005, 106, 1046. [Google Scholar] [CrossRef]
- Thachil, J. Nitric oxide and the clinical manifestations of acute porphyria. Intern. Med. J. 2008, 38, 732–735. [Google Scholar] [CrossRef]
- Buzaleh, A.; Meiss, R.; Lavandera, J.; Vallecorsa, P.; Ruspini, S.; Batlle, A. Óxido nítrico sintasa y hemo oxigenasa en encéfalo de ratones tratados con anestésicos volatiles y otros agentes porfirinogénicos: Estudio inmunohistoquímico de la expresión proteica. Medicina 2012, 72, 121. [Google Scholar]
- Bourque, S.L.; Benjamin, C.D.; Adams, M.A.; Nakatsu, K. Lack of hemodynamic effects after extended heme synthesis inhibition by succinylacetone in rats. J. Pharmacol. Exp. Ther. 2010, 333, 290–296. [Google Scholar] [CrossRef]
- Soong, J.; Adams, M.A.; Nakatsu, K. Acute depletion of heme by succinylacetone alters vascular responses but does not induce hyperten-sion. Can. J. Physiol. Pharmacol. 2008, 86, 613–619. [Google Scholar] [CrossRef]
- Homedan, C.; Laafi, J.; Schmitt, C.; Gueguen, N.; Lefebvre, T.; Karim, Z.; Desquiret-Dumas, V.; Wetterwald, C.; Deybach, J.C.; Gouya, L.; et al. Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model. Int. J. Biochem. Cell Biol. 2014, 51, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Luck, M.; Schmitt, C.; Talbi, N.; Gouya, L.; Caradeuc, C.; Puy, H.; Bertho, G.; Pallet, N. Urinary metabolic profiling of asymptomatic acute intermittent porphyria using a rule-mining-based algorithm. Metabolomics 2018, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Elder, T.; Mengel, C.E. Effect of pyridoxine deficiency on porphyrin precursor excretion in acute intermittent porphyria. Am. J. Med. 1966, 41, 369–374. [Google Scholar] [CrossRef]
- Ventura, P.; Marcacci, M.; Marchini, S.; Cuoghi, C.; Vaccari, D.; Pietrangelo, A. Is poor vitamin status a reliable target for treatment of symptomatic patients with hepatic acute porphyrias? Dig. Liver Dis. 2019, 51, e23–e24. [Google Scholar] [CrossRef]
- Mills, P.B.; Footitt, E.J.; Mills, K.A.; Tuschl, K.; Aylett, S.; Varadkar, S.; Hemingway, C.; Marlow, N.; Rennie, J.; Baxter, P.; et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (aldh7a1 deficiency). Brain 2010, 133, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Bagci, S.; Zschocke, J.; Hoffmann, G.; Bast, T.; Klepper, J.; Mu¨ller, A.; Heep, A.; Bartmann, P.; Franz, A. Pyridoxal phosphate-dependent neonatal epileptic encephalopathy. BMJ Case Rep. 2009, 2009, bcr1120081247. [Google Scholar] [CrossRef]
- Plecko, B.; St¨ockler, S. Vitamin B6 dependent seizures. Can. J. Neurol. Sci. 2009, 36 (Suppl. 2), S73–S77. [Google Scholar]
- Romero, J.A.; Kuczler, F.J., Jr. Isoniazid overdose: Recognition and treatment. Am. Fam. Physician 1998, 57, 749. [Google Scholar] [PubMed]
- Cavanagh, J. On the pattern of change in peripheral nerves produced by isoniazid intoxication in rats. J. Neurol. Neurosurg. Psychiatry 1967, 30, 26. [Google Scholar] [CrossRef]
- Gouya, L.; Ventura, P.; Balwani, M.; Bissell, D.M.; Rees, D.C.; St¨olzel, U.; Phillips, J.D.; Kauppinen, R.; Langendonk, J.G.; Desnick, R.J.; et al. Explore: A prospective, multinational, natural history study of patients with acute hepatic porphyria with recurrent attacks. Hepatology 2020, 71, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Ventura, P.; Cappellini, M.D.; Rocchi, E. The acute porphyrias: A diagnostic and therapeutic challenge in internal and emergency medicine. Intern. Emerg. Med. 2009, 4, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Ventura, P.; Cappellini, M.D.; Biolcati, G.; Guida, C.C.; Rocchi, E.; Porfiria, G.I. A challenging diagnosis for potential fatal diseases: Recommendations for diagnosing acute porphyrias. Eur. J. Intern. Med. 2014, 25, 497–505. [Google Scholar] [CrossRef]
- Soonawalla, Z.F.; Orug, T.; Badminton, M.N.; Elder, G.H.; Rhodes, J.M.; Bramhall, S.R.; Elias, E. Liver transplantation as a cure for acute intermittent porphyria. Lancet 2004, 363, 705–706. [Google Scholar] [CrossRef]
- Singal, A.K.; Parker, C.; Bowden, C.; Thapar, M.; Liu, L.; McGuire, B.M. Liver transplantation in the management of porphyria. Hepatology 2014, 60, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Sardh, E.; Ventura, P.; Peir´o, P.A.; Rees, D.C.; St¨olzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N. Eng. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef]
- Lazareth, H.; Poli, A.; Bignon, Y.; Mirmiran, A.; Rabant, M.; Cohen, R.; Schmitt, C.; Puy, H.; Karras, A.; Gouya, L.; et al. Renal function decline under therapy with small interfering RNA silencing ALAS1 for acute intermittent porphyria. Kidney Int. Rep. 2021, 6, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Petrides, P.E.; Klein, M.; Schuhmann, E.; Torkler, H.; Molitor, B.; Loehr, C.; Obermeier, Z.; Beykirch, M.K. Severe homocysteinemia in two givosiran treated porphyria patients: Is free heme deficiency the culprit? Ann. Hematol. 2021, 100, 1685–1693. [Google Scholar] [CrossRef]
- To-Figueras, J.; Wijngaard, R.; García-Villoria, J.; Aarsand, A.K.; Aguilera, P.; Deulofeu, R.; Brunet, M.; Gómez-Gómez, À.; Pozo, O.J.; Sandberg, S. Dysregulation of homocysteine homeostasis in acute intermittent porphyria patients receiving heme arginate or Givosiran. J. Inherit. Metab. Dis. 2021, 44, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.; Marcacci, M.; Cuoghi, C.; Pietrangelo, A.; Ventura, P. Hyperhomocysteinemia in patients with acute porphyrias: A possible effect of ALAS1 modulation by siRNAm therapy and its control by vitamin supplementation. Eur. J. Intern. Med. 2021, 92, 121–123. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricci, A.; Di Pierro, E.; Marcacci, M.; Ventura, P. Mechanisms of Neuronal Damage in Acute Hepatic Porphyrias. Diagnostics 2021, 11, 2205. https://doi.org/10.3390/diagnostics11122205
Ricci A, Di Pierro E, Marcacci M, Ventura P. Mechanisms of Neuronal Damage in Acute Hepatic Porphyrias. Diagnostics. 2021; 11(12):2205. https://doi.org/10.3390/diagnostics11122205
Chicago/Turabian StyleRicci, Andrea, Elena Di Pierro, Matteo Marcacci, and Paolo Ventura. 2021. "Mechanisms of Neuronal Damage in Acute Hepatic Porphyrias" Diagnostics 11, no. 12: 2205. https://doi.org/10.3390/diagnostics11122205
APA StyleRicci, A., Di Pierro, E., Marcacci, M., & Ventura, P. (2021). Mechanisms of Neuronal Damage in Acute Hepatic Porphyrias. Diagnostics, 11(12), 2205. https://doi.org/10.3390/diagnostics11122205