Abstract
This study aimed to compare and assess the genetic diversity and trends among the introduced family provenance, first-cycle superior trees breeding provenance, and improved-generation superior trees breeding provenance of Pinus elliottii using EST-SSR markers. The goal was to provide a foundation for advanced genetic improvement and sustainable utilization of P. elliottii in Jiangxi Province. A total of 417 individuals were analyzed for their genetic diversity and population structure using 19 pairs of SSR markers. The analysis identified 103 alleles across all the samples, with an average of 5.421 alleles per locus. Compared to other coniferous species, P. elliottii exhibited a moderate to high level of genetic diversity (I = 0.862, He = 0.457). Analysis of the molecular variance (AMOVA) revealed that 97.90% of the genetic variation occurred within provenances, consistent with a low genetic differentiation coefficient (Fst = 0.016 < 0.05) and high gene flow (Nm = 15.715) among provenances. In addition, analysis using STRUCTURE v. 2.3.4 software divided the 417 germplasm samples into two distinct groups, corroborating the results of the principal coordinates analysis (PCoA) and the unweighted pair group method with arithmetic (UPGMA) clustering analysis. Overall, the germplasm resources of P. elliottii exhibited rich genetic diversity, with the majority of the genetic variation occurring within provenances. For the genetic improvement of high-resin-yielding slash pines, breeding programs should prioritize populations with high genetic diversity while carefully selecting superior individuals from within those populations. These findings provide a solid foundation for breeding high-resin-yielding varieties and for future research on the sustainable utilization of these valuable resources.
1. Introduction
Pinus elliottii, native to the southeastern United States, is a valuable species for timber and pulp production, as well as a high-quality source of resin [1,2]. The resin from P. elliottii is renowned for its high yield, excellent quality, resistance to crystallization, slow coagulation, and low impurity content. Since its introduction into Jiangxi Province in 1948, P. elliottii has become a key afforestation species in the hilly areas of the Jitai Plain, primarily due to its strong adaptability, rapid growth, and high resin yield [3]. To accelerate the selection and breeding of high-resin-yielding P. elliottii varieties, genetic improvement efforts began in China in the 1980s. These initiatives have focused on genetic variation in resin-related traits, resin composition, and molecular breeding for resin production, laying the groundwork for advanced genetic improvement of P. elliottii [4,5,6]. Interspecific hybridization is a vital approach for germplasm innovation in pine trees, expanding genetic resources and incorporating favorable traits from both closely related and distantly related species. This strategy addresses the limitations of close-relative hybridization by broadening the gene pool [7]. Previous research has explored the cross-compatibility of P. elliottii with native Chinese pine species (Pinus massoniana) and exotic species (Pinus taeda and Pinus caribaea). The results indicated that three hybrid combinations—Pinus caribaea × P. elliottii, P. elliottii × Pinus caribaea, and P. elliottii × Pinus taeda—exhibited fertility levels equal to or higher than their corresponding half-sib controls. In contrast, the Pinus massoniana × P. elliottii and Pinus taeda × P. elliottii hybrids showed significantly lower fertility than the half-sib controls, with some exhibiting little to no fertility [8]. Furthermore, P. elliottii is typically characterized by dense foliage and large lateral branches during its juvenile phase, contributing to the severe damage during the 2008 snow disaster in southern China. Therefore, hybridization to improve the needle size and lateral branch traits holds significant potential for enhancing its resilience to such events [9,10].
However, due to the lack of natural P. elliottii germplasm resources in China, the genetic foundation for breeding improvements depends heavily on the diversity of the introduced germplasm. Early introductions were often driven by specific objectives, with limited consideration of population genetic diversity, which has limited the efficiency and effectiveness of P. elliottii breeding programs. As a result, a comprehensive study of the genetic base of breeding resources in Jiangxi Province is crucial to better understand the genetic information and background of the germplasm. This will facilitate the development of breeding populations with enhanced genetic quality and provide essential theoretical guidance for future genetic improvement and the creation of P. elliottii germplasm. Foundational studies on the genetic diversity of breeding populations have been conducted for many conifer species, both domestically and internationally, including Masson pine (Pinus massoniana) [11], Scots pine (Pinus sylvestris) [12], loblolly pine (Pinus taeda) [13], Korean pine (Pinus koraiensis) [14], and European larch (Larix decidua) [15].
Genetic diversity can be assessed through a variety of approaches, including morphological, cytological, physiological, biochemical, and molecular markers [16]. Early studies primarily used observable morphological traits to analyze genetic variation, focusing on phenotypic evaluation. However, morphological markers are limited in number, lack standardized observation criteria, and are highly influenced by environmental factors, making them less suitable for genetic variation studies [17]. In contrast, molecular markers represent the genetic diversity at the DNA level, offering greater precision as they are unaffected by environmental conditions [18]. Among the available molecular markers, simple sequence repeat (SSR) markers are particularly favored due to their abundance, high reliability, and convenient operation [19,20]. SSR markers have been widely applied in studies of genetic diversity, population structure, and phylogenetic relationships in forest tree germplasm, such as Masson pine [11], Scots pine [21], Chinese pine (Pinus tabuliformis) [22], and Korean pine [14]. Although SSR analysis of P. elliottii has been employed in studies such as transcriptomes [6,23], SSR marker development [24], genetic diversity studies using RAPD molecular markers in seed orchards [25], exome resequencing [26], and genetic map construction [27], comprehensive studies on the genetic diversity of P. elliottii germplasm resources remain limited.
Although our research group has collected a substantial amount of P. elliottii germplasm from both the United States and various regions across China, the genetic background of these diverse sources remains unclear. This has led to issues, such as germplasm admixture, genetic background ambiguity, and a lack of systematic integrated research, making it difficult to select suitable breeding materials. Therefore, assessing the genetic background of these populations is crucial. To ensure a robust genetic foundation for the sustainable improvement of P. elliottii, this study focused on the analysis of the genetic diversity and population structure across breeding provenances with different levels of improvement, using SSR molecular markers. The materials included the introduced family provenance, first-cycle superior trees breeding provenance, and improved-generation superior trees breeding provenance collected from Jiangxi Province. The aim was to elucidate the complexity of their genetic backgrounds, the relationships among materials, and the degree of genetic variation. This research provides a theoretical basis and material foundation for parent selection in hybrid breeding, as well as for constructing core germplasm and fingerprinting profiles. Moreover, it is expected to improve the efficiency of breeding high-resin-yielding P. elliottii varieties while laying the groundwork for further exploration and utilization of high-resin-yielding germplasm in the future.
2. Materials and Methods
2.1. Experimental Materials
The experimental materials for this study consisted of 417 P. elliottii germplasm resources currently preserved in Jiangxi Province. Samples were collected from young buds or leaves, which were immediately flash-frozen in liquid nitrogen and stored at −80 °C until further use. The primary sources of these samples are detailed below (Table 1).

Table 1.
Information on the P. elliottii germplasm resources.
Introduced family provenance (IP): This group includes 113 families from the P. elliottii provenance/family trial forest established in 1990 at Baiyunshan Forest Farm (27.22° N, 115.13° E), Qingyuan District, Ji’an City. The seeds were sourced from seed orchards in Georgia, Mississippi, and Florida, USA, with a total of 112 samples successfully amplified. Additionally, a family trial forest established in 2011 and 2018 at Fengshushan Forest Farm (29.37° N, 117.25° E), Jingdezhen, includes 30 and 10 families, respectively, also sourced from seed orchards in Florida, USA, with 18 samples successfully amplified. Furthermore, the superior family trial forest established in 2018 by the Ganzhou Forestry Research Institute (25.38° N, 114.93° E) includes 40 families sourced from seed orchards in Arkansas, USA, with all 40 samples successfully amplified.
First-cycle superior trees breeding provenance (FP): This group primarily consists of slash pine plantation stands from early introductions in Jiangxi Province. A total of 44 samples were collected from the first-generation seed orchard established in May 1992 at Baiyunshan Forest Farm, Qingyuan District, Ji’an City. An additional 77 samples were collected from the first-generation seed orchard established in 1980 at Xiajiang County Superior Tree Breeding Farm (27.58° N, 115.32° E).
Improved-generation superior trees breeding provenance (IGP): This group includes 126 samples from experimental forests planted with selected superior trees from across the province, with 79 samples from Baiyunshan Forest Farm, Qingyuan District, Ji’an City, and 47 samples from Xiajiang County Superior Tree Breeding Farm.
2.2. Research Methods
2.2.1. Genomic DNA Extraction and Detection
Genomic DNA was extracted using the DNAsecure Plant Kit (DP320) from Tiangen Biotech Co., Ltd. (Beijing, China). The concentration and quality of the extracted DNA were assessed via 1% agarose gel electrophoresis. The DNA samples were then diluted to a uniform concentration of 20–50 ng/μL and stored at −20 °C for further analysis.
2.2.2. SSR Primer Screening
A total of 24 pairs of EST-SSR primers were selected from the published literature [24]. Additionally, SSR loci were identified within unigenes obtained from the P. elliottii transcriptome using MISA software (http://misaweb.ipk-gatersleben.de/, accessed on 25 October 2024), with the following criteria: dinucleotide repeats > 6 times, and trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide repeats > 5 times. Based on the conserved regions flanking the SSR loci, 103 pairs of SSR primers were designed using Primer 3.0 software [28] and synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). The primers were validated for polymorphism using 11 slash pine samples.
2.2.3. PCR Amplification and Capillary Electrophoresis
The PCR reaction system and program are detailed in Table 2. Primers that produced clear and polymorphic amplification bands were labeled with one of four fluorescent dyes (FAM, Rox, Tem, or Hex). The PCR products were then analyzed using capillary electrophoresis on an ABI 3730XL DNA sequencer in collaboration with Sangon Biotech Co., Ltd. (Shanghai, China).
Table 2.
Reaction system and SSR-PCR procedure for P. elliottii.
2.2.4. Statistical Analysis
Microsatellite allele data for each P. elliottii sample, obtained from capillary electrophoresis, were analyzed using GeneMarker v.2.2.0 software [29]. DataFormater v.2.7 [30] and GenAlEx v.6.5 software [31] were then used to convert the resulting data into the necessary formats for further analysis.
The genetic diversity parameters, including the number of alleles (Na), the effective number of alleles (Ne), Shannon’s information index (I), observed (Ho) and expected heterozygosity (He), F-statistics (Fis, Fit and Fst) and gene flow (Nm), were calculated using POPGENE v.1.3.1 software [32]. The allelic frequency variation and polymorphic information content (PIC) were assessed and calculated using PowerMarker v.3.25 software [33].
To assess the degree of genetic variation within and between the three P. elliottii provenances, an analysis of molecular variance (AMOVA) was performed using ARLEQUIN v.3.5.2.2 software [34].
The genetic structure of the 417 individuals was analyzed using STRUCTURE v.2.3.4 software [35]. The number of clusters (K) was set from 1 to 10, with a burn-in period of 100,000 iterations and 10,000 MCMC repetitions. Each K value was run three times, and the optimal number of clusters was determined by plotting K against ∆K and selecting the peak value.
The genetic distances between samples or provenances were calculated using PowerMarker v.3.25 software, and a UPGMA dendrogram was constructed based on these distances using MEGA v.11.0 software [36]. In addition, principal coordinate analysis (PCoA) was conducted using GenAlEx v.6.5 software to further explore the genetic relationships between provenances based on the microsatellite allele data.
3. Results
3.1. Polymorphism Analysis of EST-SSR Markers
The capillary electrophoresis results showed that out of the 127 primer pairs tested, 112 successfully amplified bands, yielding an amplification success rate of 88.19%. Among these, 19 primer pairs were identified as highly polymorphic SSR markers, with a polymorphic loci ratio of 14.96% (Figure 1; Table 3).
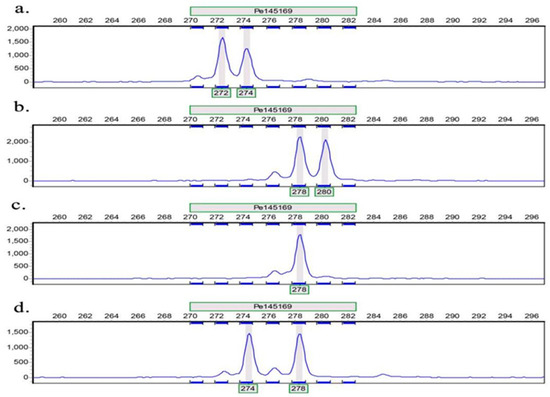
Figure 1.
Amplification of some samples on primer Pe145169. The horizontal axis represents the size of the product (bp); The vertical axis represents the relative fluorescence signal; (a–d) represent four different samples.
Table 3.
Sequence information of 19 pairs of polymorphic EST-SSR primers.
3.2. Cross-Species Applicability of EST-SSR Markers
To evaluate the cross-species applicability of the 19 polymorphic SSR markers developed for P. elliottii, 15 samples each of Pinus massoniana and Pinus taeda were selected for validation. The results (Table 4) demonstrated that all 19 SSR markers successfully amplified bands in both species, achieving a cross-species amplification rate of 100%.
Table 4.
Transferability and percentage of the polymorphic loci for the tested SSR markers.
3.3. Genetic Diversity Analysis of Loci
A total of 103 alleles were detected across the 417 P. elliottii samples using the 19 pairs of SSR primers, with an average of 5.421 alleles per locus (Table 5). Primer Pe103802 exhibited the highest number of alleles (13), with an effective number of alleles of 1.777, followed by primer Pe113019, which had 8 alleles and an effective allele number of 1.263. Primers Pe123077 and Pe139538 had the fewest alleles, with two each, and effective allele numbers of 1.964 and 1.352, respectively. Shannon’s information index (I) ranged from 0.370 (Pe114582) to 1.638 (Pe145169), with an average value of 0.862. The observed heterozygosity (Ho) ranged from 0.138 (Pe114582) to 0.795 (Pe123077), while the expected heterozygosity (He) ranged from 0.149 (Pe114582) to 0.777 (Pe145169), with average values of 0.402 and 0.457, respectively. For most loci, He was greater than Ho, except for primers Pe119033, Pe106732, and Pe123077, indicating varying degrees of heterozygote deficiency. The polymorphism information content (PIC) values ranged from 0.146 (Pe114582) to 0.743 (Pe145169), with an average of 0.411. Among the loci, 3 had PIC values below 0.25, indicating low polymorphism; 10 loci had PIC values between 0.25 and 0.5, indicating moderate polymorphism; and 6 loci had PIC values greater than 0.5, indicating high polymorphism. Overall, these 19 SSR markers proved highly effective for analyzing the genetic diversity of the P. elliottii provenances.
Table 5.
Diversity parameters of 19 microsatellite loci in P. elliottii.
3.4. Comparison of Genetic Diversity Among Provenances
A total of 83 and 95 alleles were detected in the FP and IGP of P. elliottii. In contrast, only 82 alleles were found in the introduced family provenance (Table 6). The ratios of the effective number of alleles (Ne) to the total number of alleles (Na) were higher in both the FP and IGP compared to the IP. Notably, the IP had a higher number of alleles at locus 135178 than the other two provenances. The FP exhibited the highest number of alleles at loci 144426 and 131259, while the IGP showed the highest number of alleles at seven loci: 145380, 119033, 114582, 138370, 134815, 113019, and 103802.
Table 6.
Polymorphism analysis of 19 pairs of primers in three provenances.
The Shannon’s information index (I) values for the IP, FP, and IGP were 0.734, 0.839, and 0.964, respectively. The PIC values were 0.356, 0.407, and 0.460, respectively. The average Ho values were 0.371, 0.421, and 0.424, while the average He values were 0.398, 0.457, and 0.512, respectively. Overall, the genetic diversity of the FP and IGP was higher than that of the IP. These findings suggest that the P. elliottii provenances have maintained a relatively high level of genetic diversity following selective breeding and improvement efforts.
3.5. Genetic Differentiation and Gene Flow Among Provenances
As shown in Table 7, the Fis values across the 19 SSR loci ranged from −0.655 (Pe123077) to −0.401 (Pe139538), with an average of 0.110, indicating the presence of heterozygote deficiency within the provenances. The average Ho (0.402) was lower than the average He (0.457), further confirming the occurrence of heterozygote deficiency. The Fit values ranged from −0.634 (Pe123077) to −0.451 (Pe139538), with an average of 0.124. The overall Fst values ranged from 0.005 (Pe145380) to 0.084 (Pe139538), with an average of 0.016, indicating low genetic differentiation among the P. elliottii provenances. The gene flow (Nm) values ranged from 2.710 (Pe139538) to 53.260 (Pe145380), with an average of 15.715, suggesting substantial gene flow between the provenances, which likely reduces the genetic drift and contributes to the observed low genetic differentiation.
Table 7.
Genetic differentiation and gene flow in the provenances of P. elliottii.
The AMOVA analysis based on the 19 loci (Table 8) revealed that the genetic variation among the provenances accounted for only 2.10% of the total variation, while 97.90% of the genetic variation was attributable to differences within individuals and among individuals within provenances. This indicated that most of the genetic variation in P. elliottii occurred within individuals, rather than between the IP, FP, and IGP.
Table 8.
Analysis of the molecular variance (AMOVA) of P. elliottii.
The genetic similarity coefficients and genetic distances among the three provenances were calculated using Nei’s method (Table 9). The genetic distance between the IP and the FP was 0.014, with a genetic similarity coefficient of 0.986. The genetic distance between the IP and the IGP was 0.020, with a genetic similarity coefficient of 0.980. The genetic distance between the FP and the IGP was 0.019, with a genetic similarity coefficient of 0.981. These results indicate that the IP is more closely related to the FP than to the IGP.
Table 9.
Genetic distance (lowerleft) and genetic similarity (upperright) between three provenances.
3.6. Population Genetic Structure Analysis
To further investigate the population genetic structure and relationships among the 417 P. elliottii samples, STRUCTURE software was used to analyze the data based on 19 pairs of SSR primers. The results (Figure 2) showed that as the K value increased, the lnP(D) also increased continuously. The most significant change in the lnP(D) occurred when the K value increased from 1 to 2, with the ∆K reaching its maximum peak at K = 2, indicating that the 417 P. elliottii samples were divided into two distinct subgroups (Figure 3).
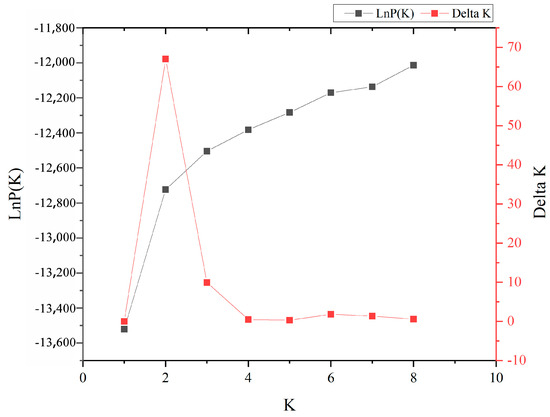
Figure 2.
Diagnostic plots of the LnP(D) and ΔK from the STRUCTURE analysis of the P. elliottii provenances.

Figure 3.
Results of the structure analysis of the P. elliottii provenances when K = 2. Red represents Subgroup I, blue represents Subgroup II.
To better characterize the genetic background of the materials, a Q value > 0.6 was used as the threshold for assigning the 417 P. elliottii samples into different subgroups. Based on this criterion, 410 samples were assigned to either Subgroup I or Subgroup II, indicating the relatively homogeneous genetic backgrounds among these samples. The 417 samples originated from the IP, FP and IGP. However, they were not strictly grouped according to their original provenances. Thus, the origins of the materials within each subgroup were further analyzed (Table 10). Subgroup I comprised 84.41% of the total samples, making it the most abundant group, with 352 samples derived from the introduced family trial forest (159 samples), the first-generation seed orchard (105 samples), and the advanced-generation seed orchard (88 samples). Subgroup II included 58 samples, with 4 from the introduced family trial forest, 16 from the first-generation seed orchard, and 38 from the advanced-generation seed orchard. Additionally, seven samples from the introduced family trial forest were placed in a mixed group, as they displayed mixed ancestry and complex genetic structures. These findings suggest that caution should be exercised when selecting these samples as breeding parents.
Table 10.
Statistics of the population structure for 417 P. elliottii.
3.7. PCoA and Genetic Clustering
To further analyze the cluster patterns, PCoA of P. elliottii was performed based on the pairwise genetic distance matrix of 19 EST-SSRs, with the results shown in Figure 4. The analysis revealed that the first principal coordinate (Coord. 1) and second principal coordinate (Coord. 2) accounted for 11.51% and 17.69% of the genetic variation, respectively. The individuals from the IP, FP, and IGP could not be completely separated, suggesting relatively close genetic relationships among these provenances. The entire P. elliottii germplasm could be roughly divided into two groups, which is consistent with the results of the STRUCTURE analysis at K = 2.
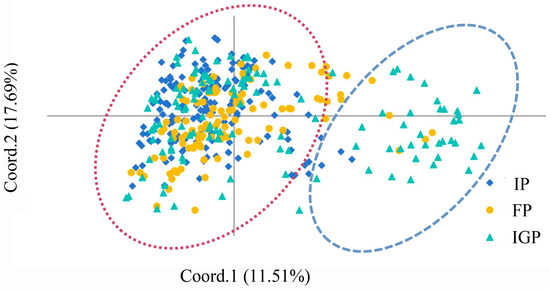
Figure 4.
Principal coordinates analysis based on Nei’s genetic distance of P. elliottii.
A clustering dendrogram of the 417 P. elliottii germplasm samples was constructed using the unweighted pair group method with arithmetic mean (UPGMA) in MEGA software. As indicated by the clustering results (Figure 5), the P. elliottii germplasm did not cluster strictly according to the IP, FP, and IGP. Instead, the samples were broadly divided into two groups. Except for a small number of samples with discrepancies, the clustering results were largely consistent with the findings from the PCoA and STRUCTURE analyses.
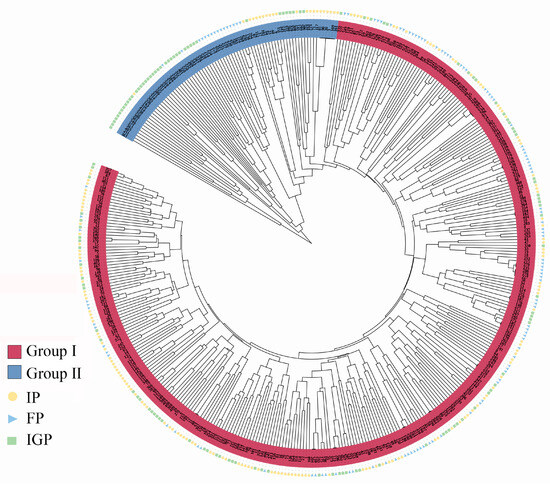
Figure 5.
UPGMA cluster analysis of the P. elliottii provenances based on the genetic distance.
4. Discussion
4.1. Development and Cross-Species Applicability of P. elliottii Primers
The limited availability of SSR primers has hindered molecular-level research on P. elliottii populations. In this study, we developed 19 polymorphic SSR loci based on the P. elliottii transcriptome and conducted cross-species applicability analyses across three pine species. The results demonstrated that the SSR markers developed for P. elliottii exhibited high cross-species applicability in other Pinus species, such as P. taeda and P. massoniana, with a 100% amplification rate and a high proportion of polymorphic loci—89.47% in P. taeda and 100% in P. massoniana. This high level of cross-species applicability is likely due to the conserved nature of the EST sequences compared to the non-coding DNA, along with the low molecular evolution rates within the Pinus genus, which facilitate the cross-transferability of SSRs and other molecular markers [37,38]. For instance, 14 EST-SSR markers developed for Aleppo pine (Pinus halepensis) showed high applicability in Mediterranean pines (Pinus halepensis) [39], and 184 EST-SSR markers developed from the lodgepole pine transcriptome were effectively amplified across different Pinus species, with amplification rates ranging from 54% to 73%, including a 65% amplification rate in P. elliottii [40]. Similarly, 20 EST-SSR markers developed for Larix principis-rupprechtii produced clear and stable bands across three related Larix species [41]. In this study, the SSR primers developed for P. elliottii demonstrated 100% cross-species applicability and a high proportion of polymorphic loci in P. massoniana, consistent with the findings from previous studies. These EST-SSR markers can also be employed in the future for interspecific hybrid identification, comparative mapping, and phylogenetic relationship evaluation among P. elliottii and its related species.
The PIC is a crucial genetic parameter often used to assess the discriminatory power of primers and the reliability of the information they provide in genetic diversity studies [42]. According to the definition of polymorphism by Botstein et al., the SSR primers selected in this study had an average PIC value of 0.411, with six loci exhibiting PIC values greater than 0.5, indicating a high level of polymorphism [43]. This suggests that the 19 SSR primers exhibit good polymorphism, effectively reflecting the genetic diversity of the materials. Therefore, these primers can provide technical support for the identification of P. elliottii germplasm, variety breeding, and the construction of genetic fingerprints. In comparison to previously developed SSR markers from our research group (average PIC = 0.349), the polymorphism of the primers used in this study was improved and the number of analyzed populations was expanded.
4.2. Genetic Diversity of P. elliottii Populations
Genetic diversity is a crucial factor in assessing a species’ ability to adapt to environmental changes. Enhancing the genetic base of a population can increase its adaptability and resilience, thereby improving its overall survival potential [17]. The level of genetic diversity within populations can be reflected by the He and the I [44].
In this study, 19 pairs of SSR primers were used to amplify three P. elliottii provenances, resulting in I = 0.862 and He = 0.457. These values were higher than those reported for Scots pine (He = 0.167; I = 0.253) [45], P. yunnanensis var. tenuifolia (He = 0.342; I = 0.488) [46], and P. taiwanensis Hayata var. damingshanensis (He = 0.40; I = 0.62) [47], but lower than those for P. massoniana (He = 0.5438; I = 0.8696) [11], Chinese pine (He = 0.604; I = 1.230) [48], and Korean pine (He = 0.521; I = 1.019) [49]. These results indicate that P. elliottii exhibits a moderate to high level of genetic diversity compared to other conifer species.
Some studies suggest that while breeding practices can effectively increase the genetic gains of improved populations, they may also reduce the genetic diversity of these populations due to intensified human selection pressure [50]. However, empirical research has shown that the impact of artificial selection on genetic diversity is not significant. For instance, comparative analyses of the genetic diversity between improved populations obtained through phenotypic selection and their natural source populations in white spruce (Picea glauca) found only a slight, non-significant reduction in diversity in the improved populations [51]. Similarly, genetic diversity analyses of first- and second-generation improved seed orchards of Pinus taeda and first- to third-generation improved seed orchards of Larix principis-rupprechtii revealed that the genetic diversity remained relatively stable across different breeding generations, with only minor population differentiation [52,53]. Even in the fourth-generation breeding population of Cunninghamia lanceolata, high genetic diversity was maintained [54], and studies on Picea sitchensis [55], Pinus taeda [56], and Larix kaempferi [50] have found that the genetic diversity of improved populations can sometimes exceed that of their source populations.
In this study, no significant differences were found in the I, Ho, and He among the three provenances of slash pine. However, the genetic diversity of the FP and IGP was slightly higher than that of the IP. These findings align with the results observed in Larix kaempferi [50], Picea sitchensis [55], and Pinus taeda [56], suggesting that the breeding strategies and measures applied during the genetic improvement of slash pine in Jiangxi have been relatively effective. As an introduced species without natural germplasm resources in China, the genetic diversity of slash pine relies on the diversity of the introduced germplasm. To further enhance the genetic diversity of breeding populations, it may be advantageous to introduce superior individuals from native populations with distinct genetic backgrounds or to select elite offspring from already introduced families. This approach would help to further broaden the genetic base of the breeding populations.
4.3. Genetic Differentiation and Gene Flow in P. elliottii Populations
Understanding the genetic differentiation and structure of populations is fundamental for developing species conservation strategies and constructing plant breeding populations. Research suggests that a Fst value of less than 0.05 indicates low genetic differentiation, 0.05 < Fst < 0.15 indicates moderate differentiation, 0.15 < Fst < 0.25 indicates relatively high differentiation, and Fst > 0.25 indicates high genetic differentiation [57]. In this study, the Fst value for P. elliottii was 0.016, which is less than 0.05, indicating low genetic differentiation among the populations. Additionally, the AMOVA analysis showed that the within-provenance variation (97.9%) was much greater than the between-provenance variation (2.10%). This, combined with the genetic differentiation analysis, demonstrates that P. elliottii has significantly higher genetic diversity within populations than between them, suggesting that selection efforts should focus more on individual trees within populations. This phenomenon has also been observed in other conifer species, such as Korean pine [14], P. massoniana [11], Norway spruce [58], Pinus yunnanensis var. tenuifolia [46], and Cunninghamia lanceolata [59]. The likely cause of this pattern is the increased frequency of gene exchange between different population germplasm resources [60].
The gene flow (Nm) is a critical factor influencing genetic differentiation among populations. Hamrick et al. suggested that when Nm > 4, the gene exchange between populations was relatively sufficient to counteract genetic drift and prevent genetic differentiation among populations [61]. In this study, the overall gene flow among provenances was 15.715, which aligns with the observed low level of genetic differentiation (Fst = 0.016). This high level of Nm has inhibited population differentiation and inbreeding, promoting individual variation and leading to a relatively uniform genetic background across populations. These findings are consistent with the results of the genetic structure analysis and variance analysis. Two primary factors may contribute to this pattern. First, it is related to the biological characteristics of P. elliottii. The species produces winged seeds and is wind-pollinated, with pollen-containing air sacs that allow it to travel relatively long distances [62,63]. Second, there has been frequent human-mediated exchange of germplasm materials between different populations over a long period, with most population materials being confined to Jiangxi Province [54]. This has resulted in low genetic diversity and a lack of significant genetic differentiation among populations. In the future selection of P. elliottii breeding materials, the focus should be on selecting and preserving superior individuals within each population. Additionally, selecting elite trees from major P. elliottii introduction areas could help expand the range of resource collection, enrich the diversity of genetic resources, and improve the breeding efficiency.
4.4. Genetic Structure and Clustering Analysis of P. elliottii Populations
Population structure analysis in plants primarily utilizes three different methods: STRUCTURE analysis, UPGMA clustering, and PCoA analysis. PCoA and UPGMA offer a visual representation of the distribution relationships among materials, while STRUCTURE analysis provides insights into the genetic background and gene flow within populations [64,65]. These three methods complement and corroborate each other, facilitating a comprehensive understanding of the genetic differences among materials. In this study, STRUCTURE analysis revealed that the 417 P. elliottii samples could be divided into two subgroups. Individuals from different sources clustered together within the same genetic structure, indicating that most samples had relatively homogeneous genetic backgrounds. Using a Q value threshold of 0.6 for classification, only seven samples had Q values below 0.6, suggesting that these samples have a more complex genetic structure and evidence of gene flow. Therefore, caution should be exercised when selecting these samples as breeding parents. Analysis using the UPGMA clustering also grouped the samples into two main clusters, and with few exceptions, the clustering patterns were consistent with the results of both the PCoA and STRUCTURE analyses.
5. Conclusions
This study examined the genetic diversity and population structure of 417 P. elliottii samples from three provenances using 19 polymorphic SSR markers. The results showed that P. elliottii exhibited moderate to high genetic diversity compared to other conifer species. The gene flow and genetic differentiation analyses indicated that the gene exchange among the provenances was relatively extensive, limiting genetic differentiation and resulting in low overall genetic differentiation. The molecular variance analysis revealed that most genetic variation occurred within populations. Therefore, breeding efforts should focus on selecting superior individuals from genetically diverse populations. The population structure analysis revealed that individuals from different sources were not strictly clustered according to their origins but were distributed across two subgroups, with seven samples classified into a mixed group, indicating complex genetic relationships. The presence of redundant genetic diversity among the 417 germplasm samples suggests that future work should aim to remove genetic redundancies. Overall, these findings provide valuable guidance for the efficient utilization of high-resin-yielding P. elliottii germplasm, the development of core germplasm, and the selection of hybrid parents for breeding new germplasm.
Author Contributions
Conceptualization, M.Y. and M.L.; methodology, M.Y.; software, R.H.; validation, M.Y., R.H. and W.H.; formal analysis, H.X. and W.H.; investigation, H.X., W.X. and X.L.; resources, M.Y. and M.L.; data curation, R.H., W.X. and T.C.; writing—original draft preparation, M.Y.; writing—review and editing, M.L.; supervision, M.L.; project administration, M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by several programs, including the Jiangxi Province Science Foundation for Youths (No. 20212BAB215013), the Science and Technology Research Project of Education Department of Jiangxi Province (No. GJJ200407), the National Natural Science Foundation of China (No. 32160385 and 32460385), and the Jiangxi Forestry Science and Technology Innovation Project (No. 202311).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Acknowledgments
We thank the editor and reviewers for their constructive comments and advice on our manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Susaeta, A.; Peter, G.F.; Hodges, A.W.; Carter, D.R. Oleoresin tapping of planted slash pine (Pinus elliottii Engelm. var. elliottii) adds value and management flexibility to landowners in the southern United States. Biomass Bioenergy 2014, 68, 55–61. [Google Scholar]
- Neis, F.A.; de Costa, F.; de Almeida, M.R.; Colling, L.C.; de Oliveira Junkes, C.F.; Fett, J.P.; Fett-Neto, A.G. Resin exudation profile, chemical composition, and secretory canal characterization in contrasting yield phenotypes of Pinus elliottii Engelm. Ind. Crop. Prod. 2019, 132, 76–83. [Google Scholar] [CrossRef]
- Lai, M.; Dong, L.M.; Su, R.F.; Zhang, L.; Jia, T.; Chen, T.X.; Yi, M. Needle functional features in contrasting yield phenotypes of slash pine at three locations in southern China. Ind. Crop. Prod. 2023, 206, 117613. [Google Scholar] [CrossRef]
- Ding, X.; Li, Y.; Zhang, Y.; Diao, S.; Luan, Q.; Jiang, J. Genetic analysis and elite tree selection of the main resin components of slash pine. Front. Plant Sci. 2023, 14, 1079952. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Zhang, L.; Lei, L.; Liu, S.; Jia, T.; Yi, M. Inheritance of resin yield and main resin components in Pinus elliottii Engelm. at three locations in southern China. Ind. Crop. Prod. 2020, 144, 112065. [Google Scholar] [CrossRef]
- Yi, M.; Zhang, L.; Cheng, Z.; Hu, R.; Gao, Y.; Jin, C.; Yuan, S.; Sun, S.; Lai, M. Identification of key genes for oleoresin biosynthesis in high and low oleoresin-yielding slash pine based on transcriptome analysis. Forests 2022, 13, 1337. [Google Scholar] [CrossRef]
- Dungey, H.S. Pine hybrids—A review of their use performance and genetics. For. Ecol. Manage. 2001, 148, 243–258. [Google Scholar] [CrossRef]
- Sun, M.S.; Feng, Y.H.; Jia, J.; Yang, Z.Q. Fertility of different interspecific hybrid types of pines. Guihaia 2021, 41, 1270–1279. [Google Scholar]
- Luan, Q.; Lu, P.; Xiao, F.; Jiang, J.; Yu, M. Investigation on the damage of Pinus elliottii in the freezing rain and snow area and the analysis on the reason. Sci. Silv. Sin. 2008, 44, 50–55. [Google Scholar]
- Zhou, B.; Gu, L.; Ding, Y.; Shao, L.; Wu, Z.; Yang, X.; Li, C.; Li, Z.; Wang, X.; Cao, Y.; et al. The great 2008 Chinese ice storm: Its socioeconomic-ecological impact and sustainability lessons learned. Bull. Am. Meteorol. Soc. 2011, 92, 47–60. [Google Scholar] [CrossRef]
- Mei, L.; Wen, X.; Fan, F.; Yang, Z.; Xie, W.; Hong, Y. Genetic diversity and population structure of masson pine (Pinus massoniana Lamb.) superior clones in South China as revealed by EST-SSR markers. Genet. Resour. Crop. Evol. 2021, 68, 1987–2002. [Google Scholar] [CrossRef]
- Sheller, M.; Tóth, E.G.; Ciocîrlan, E.; Mikhaylov, P.; Kulakov, S.; Kulakova, N.; Melnichenko, N.; Ibe, A.; Sukhikh, T.; Curtu, A.L. Genetic diversity and population structure of scots pine (Pinus sylvestris L.) in Middle Siberia. Forests 2023, 14, 119. [Google Scholar] [CrossRef]
- Lu, M.M.; Krutovsky, K.V.; Nelson, C.D.; Koralewski, T.E.; Byram, T.D.; Loopstra, C.A. Exome genotyping, linkage disequilibrium and population structure in loblolly pine (Pinus taeda L.). BMC Genom. 2016, 17, 730. [Google Scholar] [CrossRef]
- Yan, P.; Xie, Z.; Feng, K.; Qiu, X.; Zhang, L.; Zhang, H. Genetic diversity analysis and fingerprint construction of Korean pine (Pinus koraiensis) clonal seed orchard. Front. Plant Sci. 2023, 13, 1079571. [Google Scholar] [CrossRef]
- Gramazio, P.; Plesa, I.M.; Truta, A.M.; Sestras, A.F.; Vilanova, S.; Plazas, M.; Vicente, O.; Boscaiu, M.; Prohens, J.; Sestras, R.E. Highly informative SSR genotyping reveals large genetic diversity and limited differentiation in European larch (Larix decidua) populations from Romania. Turk. J. Agric. For. 2018, 42, 165–175. [Google Scholar] [CrossRef]
- Rana, J.C.; Chahota, R.K.; Sharma, V.; Rana, M.; Verma, N.; Verma, B.; Sharma, T.R. Genetic diversity and structure of Pyrus accessions of Indian Himalayan region based on morphological and SSR markers. Tree Genet. Genomes 2015, 11, 821. [Google Scholar] [CrossRef]
- Porth, I.; El-Kassaby, Y.A. Assessment of the genetic diversity in forest tree populations using molecular markers. Diversity 2014, 6, 283–295. [Google Scholar] [CrossRef]
- Grover, A.; Sharma, P.C. Development and use of molecular markers: Past and present. Crit. Rev. Biotechnol. 2016, 36, 290–302. [Google Scholar] [CrossRef]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Carletti, G.; Cattivelli, L.; Vietto, L.; Nervo, G. Multiallelic and multilocus simple sequence repeats (SSRs) to assess the genetic diversity of a Salix spp. germplasm collection. J. For. Res. 2021, 32, 263–271. [Google Scholar] [CrossRef]
- Kavaliauskas, D.; Danusevičius, D.; Baliuckas, V. New insight into genetic structure and diversity of Scots pine (Pinus sylvestris L.) populations in Lithuania based on nuclear, chloroplast and mitochondrial DNA markers. Forests 2022, 13, 1179. [Google Scholar] [CrossRef]
- Zhou, B.; Zhang, Z.; Li, Y.; Ma, Y.; Zhang, S.; Niu, S.; Li, Y. Genetic diversity, genetic structure, and germplasm source of Chinese pine in North China. Eur. J. For. Res. 2023, 142, 183–195. [Google Scholar] [CrossRef]
- de Oliveira Junkes, C.F.; de Araújo Júnior, A.T.; de Lima, J.C.; de Costa, F.; Füller, T.; de Almeida, M.R.; Neis, F.A.; da Silva Rodrigues-Correa, K.C.; Fett, J.P.; Fett-Neto, A.G. Resin tapping transcriptome in adult slash pine (Pinus elliottii var. elliottii). Ind. Crop. Prod. 2019, 139, 111545. [Google Scholar] [CrossRef]
- Yi, M.; Zhang, L.; Lei, L.; Cheng, Z.; Sun, S.; Lai, M. Analysis of SSR information in transcriptome and development of EST-SSR molecular markers in Pinus elliottii Engelm. J. Nanjing For. Univ. 2020, 44, 75. [Google Scholar]
- Nelson, C.D.; Nance, W.L.; Doudrick, R.L. A partial genetic linkage map of slash pine (Pinus elliottii Engelm. var. elliottii) based on random amplified polymorphic DNAs. Theor. Appl. Genet. 1993, 87, 145–151. [Google Scholar]
- Acosta, J.J.; Fahrenkrog, A.M.; Neves, L.G.; Resende, M.F.; Dervinis, C.; Davis, J.M.; Holliday, J.A.; Kirst, M. Exome resequencing reveals evolutionary history, genomic diversity, and targets of selection in the conifers Pinus taeda and Pinus elliottii. Genome Biol. Evol. 2019, 11, 508–520. [Google Scholar] [CrossRef]
- Westbrook, J.W.; Chhatre, V.E.; Wu, L.S.; Chamala, S.; Neves, L.G.; Muñoz, P.; Martínez-García, P.J.; Neale, D.B.; Kirst, M.; Mockaitis, K.; et al. A consensus genetic map for Pinus taeda and Pinus elliottii and extent of linkage disequilibrium in two genotype-phenotype discovery populations of Pinus taeda. G3 Genes Genomes Genet. 2015, 5, 1685–1694. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.; Remm, M.; Rozen, S. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, 115. [Google Scholar] [CrossRef]
- Holland, M.; Parson, W. GeneMarker® HID: A reliable software tool for the analysis of forensic STR data. J. Forensic Sci. 2011, 56, 29–35. [Google Scholar] [CrossRef]
- Fan, W.; Gai, H.; Sun, X.; Yang, A.; Zhang, Z.; Ren, M. DataFormater, a software for SSR data formatting to develop population genetics analysis. Mol. Plant Breed. 2016, 14, 265–270. [Google Scholar]
- Peakall, R.O.D.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Yeh, F.C.; Yang, R.C.; Boyle, T.B.J.; Ye, Z.H.; Mao, J.X. Popgene, the user friendly shareware for population genetic analysis. In Molecular Biology and Biotechnology Centre; University of Alberta: Edmonton, AB, Canada, 1997. [Google Scholar]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Neale, D.B. Genomics to tree breeding and forest health. Curr. Opin. Genet. Dev. 2007, 17, 539–544. [Google Scholar] [CrossRef]
- Neale, D.B.; Ingvarsson, P.K. Population, quantitative and comparative genomics of adaptation in forest trees. Curr. Opin. Plant Biol. 2008, 11, 149–155. [Google Scholar] [CrossRef]
- Leonarduzzi, C.; Spanu, I.; Labriola, M.; González-Martínez, S.C.; Piotti, A.; Vendramin, G.G. Development and characterization of three highly informative EST-SSR multiplexes for Pinus halepensis Mill. and their transferability to other Mediterranean pines. Plant Mol. Biol. Rep. 2016, 34, 993–1002. [Google Scholar] [CrossRef]
- Lesser, M.R.; Parchman, T.L.; Buerkle, C.A. Cross-species transferability of SSR loci developed from transciptome sequencing in lodgepole pine. Mol. Ecol. Resour. 2012, 12, 448–455. [Google Scholar] [CrossRef]
- Dong, M.; Wang, Z.; He, Q.; Zhao, J.; Fan, Z.; Zhang, J. Development of EST-SSR markers in Larix principis-rupprechtii Mayr and evaluation of their polymorphism and cross-species amplification. Trees 2018, 32, 1559–1571. [Google Scholar] [CrossRef]
- Uzun, A.; Yesiloglu, T.; Polat, I.; Aka-Kacar, Y.; Gulsen, O.; Yildirim, B.; Tuzcu, O.; Tepe, S.; Canan, I.; Anil, S. Evaluation of genetic diversity in lemons and some of their relatives based on SRAP and SSR markers. Plant Mol. Biol. Rep. 2011, 29, 693–701. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314. [Google Scholar] [PubMed]
- Schmidt, T.L.; Jasper, M.E.; Weeks, A.R.; Hoffmann, A.A. Unbiased population heterozygosity estimates from genome-wide sequence data. Methods Ecol. Evol. 2021, 12, 1888–1898. [Google Scholar] [CrossRef]
- Vasilyeva, Y.; Chertov, N.; Nechaeva, Y.; Sboeva, Y.; Pystogova, N.; Boronnikova, S.; Kalendar, R. Genetic structure, differentiation and originality of Pinus sylvestris L. populations in the east of the East European Plain. Forests 2021, 12, 999. [Google Scholar] [CrossRef]
- Yang, Z.; Feng, Y.; Wu, D. Analysis of genetic diversity of Pinus yunnanensis var. tenuifolia nature populations by SSR marker. Guihaia 2014, 34, 10–14. [Google Scholar]
- Luo, Q.; Feng, Y.; Wu, D.; Yang, Z. Genetic diversity of Pinus taiwanensis var. damingshanensis natural populations by SSR markers. Guihaia 2022, 42, 1367–1373. [Google Scholar]
- Yang, B.; Niu, S.; El-Kassaby, Y.A.; Li, W. Monitoring genetic diversity across Pinus tabuliformis seed orchard generations using SSR markers. Can. J. For. Res. 2021, 51, 1534–1540. [Google Scholar] [CrossRef]
- Li, X.; Zhao, M.; Xu, Y.; Li, Y.; Tigabu, M.; Zhao, X. Genetic diversity and population differentiation of Pinus koraiensis in China. Horticulturae 2021, 7, 104. [Google Scholar] [CrossRef]
- Du, C.; Sun, X.; Xie, Y.; Hou, Y. Genetic diversity of Larix kaempferi populations with different levels of improvement in northern subtropical region. Sci. Silvae Sin. 2021, 57, 68–76. [Google Scholar]
- Fageria, M.S.; Rajora, O.P. Effects of silvicultural practices on genetic diversity and population structure of white spruce in Saskatchewan. Tree Genet. Genomes 2014, 10, 287–296. [Google Scholar] [CrossRef]
- Chhatre, V.E.; Byram, T.D.; Neale, D.B.; Wegrzyn, J.L.; Krutovsky, K.V. Genetic structure and association mapping of adaptive and selective traits in the east Texas loblolly pine (Pinus taeda L.) breeding populations. Tree Genet. Genomes 2013, 9, 1161–1178. [Google Scholar] [CrossRef]
- Yu, D.; Yuan, D.; Zhang, D.; Fan, Y.; Li, D.; Zhang, H.; Zhang, J. Genetic diversity of Larix principisrupprechtii Mayr. seed orchard among generations. J. Plant Genet. Resour. 2014, 15, 940–947. [Google Scholar]
- Jing, Y.; Bian, L.; Zhang, X.; Zhao, B.; Zheng, R.; Su, S.; Ye, D.; Zheng, X.; El-Kassaby, Y.A.; Shi, J. Genetic diversity and structure of the 4th cycle breeding population of Chinese fir (Cunninghamia lanceolata (lamb.) hook). Front. Plant Sci. 2023, 14, 1106615. [Google Scholar] [CrossRef] [PubMed]
- Chaisurisri, K.; El-Kassaby, Y.A. Genetic diversity in a seed production population vs. natural populations of Sitka spruce. Biodivers. Conserv. 1994, 3, 512–523. [Google Scholar] [CrossRef]
- Isik, F.; McKeand, S.E. Fourth cycle breeding and testing strategy for Pinus taeda in the NC State University Cooperative Tree Improvement Program. Tree Genet. Genomes 2019, 15, 70. [Google Scholar] [CrossRef]
- Pearse, D.E.; Crandall, K.A. Beyond F ST: Analysis of population genetic data for conservation. Conserv. Genet. 2004, 5, 585–602. [Google Scholar] [CrossRef]
- Stojnić, S.V.; Avramidou, E.; Fussi, B.; Westergren, M.; Orlović, S.; Matović, B.; Trudić, B.; Kraigher, H.A.; Aravanopoulos, F.; Konnert, M. Assessment of genetic diversity and population genetic structure of Norway spruce (Picea abies L.) Karsten at Its southern lineage in Europe. Implications for conservation of forest genetic resources. Forests 2019, 10, 258. [Google Scholar] [CrossRef]
- Lin, E.; Zhuang, H.; Yu, J.; Liu, X.; Huang, H.; Zhu, M.; Tong, Z. Genome survey of Chinese fir (Cunninghamia lanceolata): Identification of genomic SSRs and demonstration of their utility in genetic diversity analysis. Sci. Rep. 2020, 10, 4698. [Google Scholar] [CrossRef]
- Ithnin, M.; The, C.K.; Ratnam, W. Genetic diversity of Elaeis oleifera (HBK) Cortes populations using cross species SSRs: Implication’s for germplasm utilization and conservation. BMC Genet. 2017, 18, 37. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W. Allozyme diversity in plants. In Population Genetics, Breeding and Genetic Resources, 2nd ed.; Brown, A.D.H., Clegg, M.T., Kahler, A.L., Weir, B.S., Eds.; Sinauer & Associates: Sunderland, MA, USA, 1989; pp. 43–63. [Google Scholar]
- Petit, R.J.; Duminil, J.; Fineschi, S.; Hampe, A.; Salvini, D.; Vendramin, G.G. Invited review: Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol. Ecol. 2005, 14, 689–701. [Google Scholar] [CrossRef]
- Tong, Y.; Lewis, B.J.; Zhou, W.; Mao, C.; Wang, Y.; Zhou, L.; Yu, D.; Dai, L.; Qi, L. Genetic diversity and population structure of natural Pinus koraiensis populations. Forests 2019, 11, 39. [Google Scholar] [CrossRef]
- Wang, S. Genetic diversity and population structure of the endangered species Paeonia decomposita endemic to China and implications for its conservation. BMC Plant Biol. 2020, 20, 510. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liu, Q.; Zhou, Z.; Gao, K.; Luo, D. Genetic diversity analysis and core collection of pinewood nematodiasis-resistant Pinus massoniana germplasm resources. J. Zhejiang Agric. For. Univ. 2024, 41, 67–78. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).