Abstract
The mitochondrial genome (mitogenome) is essential for identifying species and tracing genetic variation, gene patterns, and evolutionary studies. Here, the mitogenome of Grateloupia turuturu was sequenced on the Illumina sequencing platform. This circular mitogenome (28,265 bp) contains 49 genes, including three rRNAs, twenty transfer RNAs (tRNAs), and twenty-six protein-coding genes (PCGs). Nucleotide composition indicates biased AT (68.8%) content. A Group II intronic sequence was identified between two exons of the cox1 gene, and this sequence comprises an open reading frame (ORF) that encodes a hypothetical protein. The gene content, annotation, and genetic makeup are identical to those of Halymeniaceae members. The complete mitogenome sequences of the Grateloupia and Polyopes species were used in a phylogenetic analysis, which revealed that these two genera are monophyletic and that G. turuturu and G. elliptica are closely related. This newly constructed mitogenome will help us better understand the general trends in the development of cox1 introns in Halymeniaceae, as well as the evolution of red algal mitogenomes within the Rhodophyta and among diverse algal species.
1. Introduction
Rhodophyta algae (red algae) are an evolutionarily significant eukaryotic lineage which inhabit marine and freshwater. Rhodophyta species are mostly multicellular, photoautotrophic, and abundant in marine habitats (around 98%) and rare in freshwater, with a few rare terrestrial or sub-aerial representatives [1]. The red alga have photosynthetic pigments, chlorophylls a and d, and characteristic red colors due to the phycoerythrin pigment. In the evolutionary sense, red algae are plant-like because they have a single shared parent with green algae (Chlorophyta) and higher plants (Embryophyta) [2,3]. Rhodophytes are divided into seven classes with around 7538 species, and among them, the Florideophyceae class possesses the maximum number of species (7141), which are mostly marine, multicellular algae including seaweeds [4].
A marine-habituated red macroalga, Grateloupia turuturu (Y. Yamada, 1941), classified under the phylum, Rhodophyta; class, Florideophyceae; subclass, Rhodymeniophycidae; order: Halymeniales; family, Halymeniaceae; and genus, Grateloupia [5]. There are 69 Grateloupia species that have been classified, and 36 species are still unclassified, and out of these, only 5 complete Grateloupia mitochondrial genomes are available on the National Center for Biotechnology Information (NCBI) website (https://www.ncbi.nlm.nih.gov, accessed on 15 June 2023). These species are found all around the world, including in the Atlantic islands, Caribbean islands, Europe, North and South America, Africa, Asia, Australia, and New Zealand [4]. Furthermore, there are 227 species listed under the family, Halymeniaceae, but to date, only 7 species (Grateloupia angusta, G. cornea, G. elliptica, G. filicina, G. taiwanensis, Polyopes affinis, and P. lancifolius) with complete mitogenome have been reported [6,7,8,9,10,11], as listed in Table 1.

Table 1.
An overview of the complete mitogenomes utilized in this study.
Relatively little is known about the mitogenome of Rhodophytes, and due to advancements in software and molecular technologies, more and more detailed studies are being reported. In fact, red algal mitogenomes are more complete than previously reported [12], and it has also been reported that red algae, Strylonematophyceae, contain multiple minicircular mitochondrial genomes that encode one or two genes [13]. These studies are made possible by applying bundles of software tools. The red algal mitogenomes have less molecular weight than other algae, and because of their maternal inheritance, they are a useful tool for evolutionary and phylogenetic studies. In addition, mitogenomes have a specific sequence that gives reliable data for studying the gene order, makeup, contents, and secondary structures of the encoded RNA [14,15], and it is also useful for making molecular kits (barcoding markers) for economically important species identification [16]. The Grateloupia species contain a characteristic intronic cox1 gene (Table 1), and such features are useful to understand evolutionary and phylogenetic studies [3,6,7,8,9,17]. Algae mitogenomes consist of introns in the genic region, tandem repeats, and large intergenic repeats, which create challenges for assembling complete circular mitogenomes [15] but due to revolutionary advances in sequencing technologies and bioinformatics tools, such issues can be overcome. So, utilizing modern, next-generation sequencing methods and bioinformatics tools, we provide here the full mitochondrial genome of red algae as well as a phylogenetic relationship based on the complete mitogenome sequence.
In this study, we used de novo assembly on the Illumina platform to sequence the complete circular mitogenome of G. turuturu. Gene annotation, genetic makeup, and gene order were confirmed using several bioinformatics tools and phylogenetic studies based on complete mitogenome sequencing. This study’s data were submitted to the NCBI GenBank and will be useful for future research on the evolution and phylogeny of red algae species.
2. Materials and Methods
2.1. Sample Collection and DNA Isolation
A deep-sea diver from the Marine Eco-Technology Institute in Busan, South Korea, collected Grateloupia turuturu from the coast of Gijang (35°28′ N, 129°25′ E) and then deposited it there under the voucher number PU-T01-S-MA-04 (contact person: Dr. Young-Ryun Kim, yykim@marine-eco.co.kr). Total DNA was extracted using the QIAGEN DNEasy Blood and Tissue Kit (QIAGEN, Hilden, Germany) as per the manufacturer’s protocol, and the purity and concentration of DNA were confirmed via a NanoDrop spectrophotometer (Thermo Fisher Scientific D1000, Waltham, MA, USA). Purified total genomic DNA samples were kept at −20 °C until required.
2.2. Whole Genome Sequencing
G. turuturu genome was sequenced using the Illumina Platform (Illumina Inc., San Diego, CA, USA). The library preparation and sequencing processes were carried out by the Macrogen Company in Daejeon, South Korea. Sequencing libraries were prepared using the TrueSeq Nano DNA Kit according to the manufacturer’s protocol, and sequencing was performed on the Illumina HiSeq 2500 Platform in paired-end 150 bp mode. Before downstream analysis, raw data initially underwent quality checks to obtain clean reads. The low-quality bases (phred quality score, Q < 20), empty reads, and Illumina adapters were removed to mitigate the analytical bias by Trimmomatic [18]. After filtering, 12,903,396 total reads (GC = 40.05%, Q20 = 99.26%) were produced from a total of 14,873,050 raw reads (GC = 40.23%, Q20 = 97.33%). The overall quality of the produced sequencing reads was verified using FastQC v0.11.5 (Babraham Institute, Bioinformatics) [19], and mitogenome de novo assembly was finished using various k-mers [20] and the SPAdes v3.13.0 program [21].
2.3. Mitogenome Assembly and Annotation
Mold/Protozoan Mitochondrial was selected for the genetic code; red algae belonging to the Florideophyceae and Bangiophyceae classes have demonstrated this method of codon translation [3,6,7,8,9,10,11]. The mitogenome annotation was performed using the MFannot tool (https://megasun.bch.umontreal.ca/apps/mfannot/, accessed on 10 May 2023) with genetic code 4 (Protozoan Mitochondrial Code) [22]. The final annotation was checked and verified using ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/, accessed on 10 May 2023), and predicted open reading frames (ORFs) were checked manually and annotated accordingly. Protein-coding genes (PCGs) were verified with previously sequenced red algal mitogenomes by BLAST homology searches against the NCBI database [23]. Transfer RNA was identified using tRNAscan-SE v2.0 (http://lowelab.ucsc.edu/tRNAscan-SE/, accessed on 10 May 2023) with the default setting (with Model: Mold & Protozoa Mito) [24]. The tRNA genes, rRNA genes, and introns were identified using RNAweasel (https://megasun.bch.umontreal.ca/apps/rnaweasel/, accessed on 10 May 2023) [25]. Tandem Repeat Finder (TRF) was used to identify and annotate the repeats in the mitogenome sequence [26]. The assembled contig was analyzed for identification by querying BlastN [23,27] for known red algae mitogenomes and comparing mitogenome sizes.
A physical map of the mitogenome was designed with OrganellarGenomeDRAW v. 1.3.1 (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html, accessed on 15 June 2023) [28]. The nucleotide composition of the mitogenome was estimated using MEGA11 v.11.2.8 [29]. Codon usage and relatively synonymous codon usage (RSCU) for collected ORFs of PCGs were analyzed by the Sequence Manipulation Suite (SMS) tool with genetic code 4 (http://www.bioinformatics.org/sms2/codon_usage.html, accessed on 10 May 2023) [30]. The following formula was used to calculate the asymmetric base composition of the mitochondrial genome: GC − skew = [G − C]/[G + C] and AT − skew = [A − T]/[A + T] [31].
2.4. Phylogenetic Analysis
The phylogenetic tree was made by using the complete circular mitogenome sequences of eight red algae from the family Halymeniaceae (Table 1) and one alga from the family Glaucocystaceae (Glaucocystis nostochinearum, GenBank accession number HQ908425) as an out-group member. All mitogenomes utilized in this investigation were obtained from the NCBI GenBank. The dataset was initially processed by ClustalW for multiple sequence alignment in MEGA11 [32]. Multiple sequenced aligned datasets were used to generate a maximum-likelihood phylogenetic tree using the Tamaru–Nei model and 1000 replicated bootstraps in MEGA11 with the default parameters [29,33].
2.5. Data Availability
The mitogenome sequence and related data were submitted to the NCBI GenBank (http://www.ncbi.nlm.nih.gov/, accessed on 12 May 2023 and 16 June 2023). The complete mitogenome sequence is available for the public under the accession number OQ972988, along with associated data including Sequence Read Archive (SRA), BioProject, and BioSample with the assigned numbers PRJNA984428, SAMN35767756, and SRR24947511, respectively.
3. Results and Discussion
3.1. Genome Size and Organization
The contig with a length of 28,265 bp was identified as the mitochondrial genome; based on BlastN analysis, it matches the reference species of Grateloupia, and the mitogenome size is comparable to that of other red algal mitogenomes (Table 1). The mitogenome sequence of Grateloupia turuturu is available in GenBank with accession number OQ972988. The complete circular mitogenome map with gene arrangement is shown in Figure 1. The contig is 28,265 bp long and is composed of A = 36.1%, T = 32.7%, G = 16.1%, and C = 15.5%, with a bias of 68.8% A + T contents. The G. turuturu mitogenome contains 3 rRNA, 20 tRNA, and 26 PCGs (including intronic and hypothetical protein genes), including 14 respiratory chain subunits (complexes 1–4), four ATP synthase subunits (complex 5), two each of LSU and SSU ribosomal proteins, one independent protein translocase (tatC), and two hypothetical protein genes (orf641 and orf173). Among these genes, 24 (12 PCGs, 10 tRNAs, and 2 rRNAs genes) are found on the heavy strand (H-strand), while the rest (14 PCGs, 10 tRNA, and 1 rRNA gene) are found on the light strand (L-strand). The positive AT skew (0.049) and GC skew (0.032) were observed in this study with the presence of more A and G than T and C, respectively (Table 1). In comparison to Grateloupia [6,7,8,9] and Polyopes [10,11] species with complete mitogenome features, the mitogenome of G. turuturu demonstrates no significant gene losses; however, G. elliptica (OP479979) [7] has closer mitogenome features in terms of nucleotide composition, bias AT content, and gene compositions. In Halymeniales, the typical complete mitogenome was circular and approximately 25 to 30 kb in length with correspondingly conserved gene content, which encoded 24 PCGs (excluding intronic and hypothetical genes), 2–3 rRNAs, and 18–23 tRNAs with A + T bias nucleotides (Table 1) [6,7,8,9,10,11].
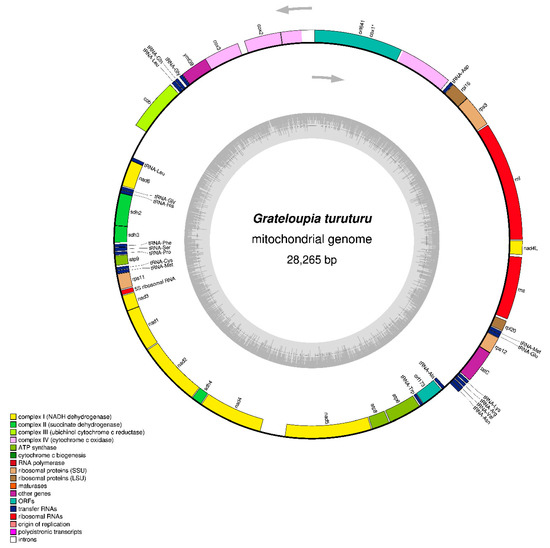
Figure 1.
Gene map of the Grateloupia turuturu (OQ972988) mitochondrial genome. Different categories of genes are represented by abbreviations and arrows outside and inside the circle, which indicates the direction of gene transcription. A gene (cox1) containing group II introns is denoted with an asterisks. The map was drawn using OrganellarGenomeDRAW (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html, accessed on 15 June 2023).
3.2. Protein-Coding Gene Features
The PCG area, which included intronic and hypothetical genes, made up 71.53% of the G. turuturu mitogenome and was 20,220 base pairs long. nad5 is the longest PCG with 1998 bp, while atp9 is the smallest with 231 bp. Each PCG was initiated by a canonical ATG codon, except for tatC, which was initiated by a TTG codon (Table 2). Similar results have been demonstrated in G. cornea (OQ910480), G. elliptica [7], and P. affinis [10]. Furthermore, out of 26 PCGs, 21 terminated with the TAA codon, except 5 PCGs (sdh2, cox2, atp8, atp6, and rps11) which terminated with the TAG codon, which was typical for Grateloupia [6,7,8,9] and P. lancifolius [11]. The G. turuturu mitogenome was analyzed for intergenic nucleotide, and it was noted that junctions of three gene pairs have an overlap; 1 bp each between trnL (number 2)–nad6 and trnH–sdh2, and 51 bp between cox3–ymf39. Furthermore, the intergenic gaps differ from 1 bp to 650 bp in length, with the longest gap of 650 bp between cob–trnL (number 2) (Table 2).
Table 2.
Sequence characteristics of G. turuturu (OQ972988) mitochondrial genome.
Analysis of the complete mitogenome sequence of G. turuturu revealed the presence of a group II intron segment (position: 5042–7298) between two exons of cox1, which encodes an ORF (orf641; position: 5084–7009). Hypothetical genes orf641 and orf173 (Table 2) with an unknown function were identified and encoded hypothetical proteins. Similar outcomes have been documented for all G. angusta, G. cornea, G. elliptica, G. filicina, and G. taiwanensis, but not for P. affinis and P. lancifolius (Table 1) [6,7,8,9,10,11]. Group II intronic cox1 and trnI are the unique features of red algal mitogenomes [12,13,15]. The ymf39 gene is transcribed between cox3 and trnG genes in the mitogenome of G. turuturu and encodes an ATP synthase B chain precursor. Similar annotations were reported for Grateloupia species [6,7,8,9] and P. lancifolius [11], but the annotation for P. affinis is atp4 [10]. In recent studies, a reanalysis of red mitogenome sequences revealed that the atp4 gene was annotated with the name, ymf39, instead of its original name [12,34]. It is suggested that the ymf39 ORF encodes for ATP synthase chain b; therefore, Florideophyceae mitogenome annotation should change ymf39 to atp4 to avoid further confusion [35]. Likewise, the conserved sequence of PCG encodes a sec-independent protein translocase protein annotated (identified) with tatC [7,10] and secY [6,8,9,11] names within the species of Florideophyceae. In the review of red algae, scientists noted that secY is not found in the algal mitogenome and recommended that the secY annotation be changed to tatC [12,34,35,36]. The rpl20 gene is located between rrs and trnM in the G. turuturu mitogenome (Figure 1). The gene content of the mitogenomes of G. turuturu and other species of Grateloupia [6,7,8,9] and Polyopes [10,11] is identical, with the exception of the absence of rpl20 in G. cornea (OQ910480).
Codon usage analysis of the 26 PCGs of the G. turuturu mitogenome (intronic and hypothetical ORFs included) showed that 6714 amino acid triplets were expressed (Table 3), not including stop codons. Leucine (N = 990, 14.74%) and cysteine (N = 85, 1.27%) are the most and least abundant amino acids, respectively. Furthermore, the most frequently used codons in PCGs include TTA (N = 544, 8.10%, Leu), TTT (N = 507, 7.55%, Phe), ATT (N = 390, 5.80%, Ile), AAA (N = 291, 4.33%, Lys), and GTT (N = 199, 2.96%, Val). The present study results are in line with the mitogenome of G. cornea (OQ910480).
Table 3.
Codon usage of PCGs in the mitogenome of G. turuturu (OQ972988).
3.3. Ribosomal RNA and Transfer RNA
The mitogenome of G. turuturu consists of three rRNAs (Table 4): two small subunits (rns = 1367 bp and rrn5 = 108 bp) and one large subunit (rnl = 2596 bp). Two rRNAs (rnl and rns) are transcribed on the H-strand and separated by the nad4L gene. However, the rrn5 gene is located between nad3 and rps11 and is transcribed on the L-strand. Similar annotations have been reported for the mitogenomes of Grateloupia [6,7,8,9] and Polyopes [10,11] species, except for the absence of rrn5 in the mitogenomes of G. angusta [6], G. filicina [8], G. taiwanensis [9], and P. lancifolius [11].
Table 4.
Mitochondrial rRNA and tRNA in Halymeniaceae.
Twenty tRNAs were identified in the mitogenome of G. turuturu (Table 2), accounting for 5.23% (1495 bp) of the total length of the mitogenome; the length of individual tRNAs ranges from 70 (trnQ-TTG) to 85 bp (trnL-TAA and trnS-TGA). In addition, an equal number of tRNAs were transcribed on both strands (H- and L-strands). The number of tRNAs ranged from 18 to 24, with small variations in the tRNA gene content among the Halymeniaceae family members shown in Table 4. The mitogenome of G. turuturu contains double copies of three tRNA (trnG, trnL, and trnM), of which two tRNA (trnG, trnL) use different anticodons. Additionally, the mitogenome lacks trnI and trnY, and there is no intronic tRNAs. The trnI is the intronic tRNA gene, present in the G. angusta [6], G. filicina [8], G. taiwanensis [9], and P. lancifolius [11]. At least two copies of trnM-CAT (except three copies in G. angusta [6]) were present in all examined species, suggesting a major role for this tRNAs in Halymeniaceae mitogenomes. It should be noted that trnR-TCT (Arg) and trnS-GCT (Ser) were absent in G. turuturu although they could be found in other known Rhodophyte mitogenomes.
3.4. Phylogenetic Analysis
The mitogenome maximum-likelihood (ML) phylogenetic tree was constructed using a complete mitogenome sequence of Halymeniaceae members obtained from GenBank and G. nostochinearum as an out-group member (Figure 2). Results indicate that G. turuturu is positioned next to G. elliptica, suggesting a close relationship. All members of the Halymeniaceae family are monophyletic, and the clade is strongly supported (99–100 percent bootstrap values). The ML phylogenetic relationships based on complete mitogenome sequences [7,10] and PCGs [8,11] indicate that the Grateloupia (intronic cox1 gene-containing) and Polyopes species are monophyletic. Our phylogenetic analysis results are consistent with previous studies. The findings of this study will be helpful for taxonomic and phylogenetic research on red algae.
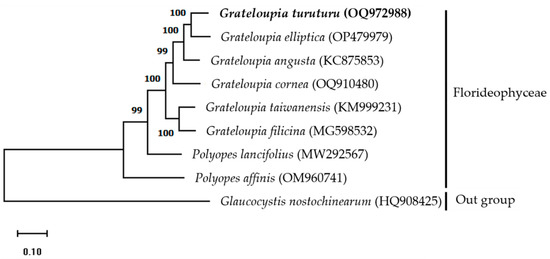
Figure 2.
Maximum-likelihood (ML) phylogenetic tree based on complete mitogenome sequences indicating the relationship between red algae (family Halymeniaceae). A alga from the Glaucocystaceae family was used as an outgroup member. Bootstrap support values are indicated at nodes. NCBI GenBank accession numbers are listed next to the corresponding species names.
4. Conclusions
In this study, we reported the complete mitogenome of G. turuturu (OQ972988), which is circular, 28,265 bp in length, with AT bias (68.8%) composition, encoding 49 genes including 26 PCGs, 20 tRNA, and 3 rRNA genes. This mitogenome contains the intronic cox1 gene with functional ORF similar to those in G. angusta, G. cornea, G. elliptica, G. filicina, and G. taiwanensis. The G. turuturu mitogenome lacks the intronic tRNA-Ile (trnI) that is present in the mitogenomes of G. angusta, G. filicina, G. taiwanensis, and P. lancifolius. We may learn more about the evolution of red algal mitogenomes within the Rhodophyta species and across different algal species with the help of this newly constructed mitogenome, as well as about the general patterns in the development of cox1 introns in Halymeniaceae.
Author Contributions
M.P.P. and J.-O.K. performed the experiments, analyzed the data, were involved in the data analysis and the drafting of the paper, and approved the final draft. Y.-R.K., S.Y. and K.K. were involved in data analysis, organizing the results, and preparing figures. J.-O.K. and K.K. were involved in the conception and design of the work, funding acquisition, revising it critically for intellectual content, and the final approval of the version to be published. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a grant from the National Institute of Fisheries Science, Korea (R2023005) and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (Grant 2021R1I1A306037411).
Institutional Review Board Statement
Not applicable. This study did not involve humans or animals.
Informed Consent Statement
Not applicable. This study did not involve humans.
Data Availability Statement
The mitogenome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/ under accession number OQ972988.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gurgel:, C.F.D.; Lopez-Bautista, J. Red algae. Encyclopedia of Life Sciences; John Willey & Sons, Ltd.: Hoboken, NJ, USA, 2007. [Google Scholar] [CrossRef]
- Adl, S.M.; Simpson, A.G.; Farmer, M.A.; Andersen, R.A.; Anderson, O.R.; Barta, J.R.; Bowser, S.S.; Brugerolle, G.U.Y.; Fensome, R.A.; Fredericq, S.; et al. The new higher level classification of eukaryotes with emphasis on the taxonomy of protists. J. Eukaryot. Microbiol. 2005, 52, 399–451. [Google Scholar] [CrossRef] [PubMed]
- Burger, G.; Saint-Louis, D.; Gray, M.W.; Lang, B.F. Complete sequence of the mitochondrial DNA of the red alga Porphyra purpurea: Cyanobacterial introns and shared ancestry of red and green algae. Plant Cell 1999, 11, 1675–1694. [Google Scholar] [CrossRef] [PubMed]
- Guiry, M.D.; AlgaeBase. World-Wide Electronic Publication, National University of Ireland, Galway. 2017. Available online: https://www.algaebase.org (accessed on 15 June 2023).
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Yang, E.C.; Boo, S.M.; Yoon, H.S. Complete mitochondrial genome of the marine red alga Grateloupia angusta (Halymeniales). Mitochondrial DNA 2014, 25, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.P.; Kim, J.O.; Kim, K.; Kim, Y.R. The complete sequence of the mitochondrial DNA and phylogenetic analysis of the marine red alga Grateloupia elliptica (Rhodophyta: Halymeniales). Mitochondrial DNA Part B Resour. 2023, 8, 222–223. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Meinita, M.D.N.; Liu, T.; Chi, S.; Yin, H. Complete sequences of the mitochondrial DNA of the Grateloupia filicina (Rhodophyta). Mitochondrial DNA B Resour. 2018, 3, 76–77. [Google Scholar] [CrossRef] [PubMed]
- DePriest, M.S.; Bhattacharya, D.; Lopez-Bautista, J.M. The mitochondrial genome of Grateloupia taiwanensis (Halymeniaceae, Rhodophyta) and comparative mitochondrial genomics of red algae. Biol. Bull. 2014, 227, 191–200. [Google Scholar] [CrossRef]
- Patil, M.P.; Kim, J.O.; Kim, K.; Kim, Y.R.; Yoon, S. Complete mitochondrial genome and phylogenetic analysis of the marine red alga Polyopes affinis (Rhodophyta: Halymeniales). Mitochondrial DNA Part B Resour. 2022, 7, 1387–1388. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Cho, C.H.; Yang, E.C.; Yoon, H.S.; Kim, M.S. Complete mitochondrial genome of Polyopes lancifolius and comparison with related species in Halymeniales (Rhodophyta). Mitochondrial DNA Part B Resour. 2021, 6, 1365–1366. [Google Scholar] [CrossRef] [PubMed]
- Salomaki, E.D.; Lane, C.E. Red algal mitochondrial genomes are more complete than previously reported. Genome Biol. Evol. 2017, 9, 48–63. [Google Scholar] [CrossRef][Green Version]
- Lee, Y.; Cho, C.H.; Noh, C.; Yang, J.H.; Park, S.I.; Lee, Y.M.; West, J.A.; Bhattacharya, D.; Jo, K.; Yoon, H.S. Origin of minicircular mitochondrial genomes in red algae. Nat. Commun. 2023, 14, 3363. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Yoon, H.S.; Yi, G.; Shin, W.; Archibald, J.M. Comparative mitochondrial genomics of cryptophyte algae: Gene shuffling and dynamic mobile genetic elements. BMC Genom. 2018, 19, 275. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.; Cho, C.H.; Kim, E.J.; Bhattacharya, D.; Yoon, H.S. Group II intron and repeat-rich red algal mitochondrial genomes demonstrate the dynamic recent history of autocatalytic RNAs. BMC Biol. 2022, 20, 2. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Yuan, C.; Tao, L.; Cai, Y.; Zhang, W. Life barcoded by DNA barcodes. Conserv. Genet. Resour. 2022, 14, 351–365. [Google Scholar] [CrossRef]
- Fang, J.; Xu, X.; Chen, Q.; Lin, A.; Lin, S.; Lei, W.; Zhong, C.; Huang, Y.; He, Y. The complete mitochondrial genome of Isochrysis galbana harbors a unique repeat structure and a specific trans-spliced cox1 gene. Front. Microbiol. 2022, 13, 966219. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Beck, N.; Lang, B. MFannot, Organelle Genome Annotation Webserver; Université de Montréal: Montréal, QC, USA, 2010; Available online: https://megasun.bch.umontreal.ca/apps/mfannot/ (accessed on 10 May 2023).
- Gish, W.; States, D.J. Identification of protein coding regions by database similarity search. Nat. Genet. 1993, 3, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Search and Contextual Analysis of Transfer RNA Genes. Nucl. Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Lang, B.F.; Laforest, M.J.; Burger, G. Mitochondrial introns: A critical view. Trends Genet. 2007, 23, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P. The Sequence Manipulation Suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinf. 2003, 1, 2–3. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.C.; Kim, K.M.; Kim, S.Y.; Lee, J.; Boo, G.H.; Lee, J.H.; Nelson, W.A.; Yi, G.; Schmidt, W.E.; Fredericq, S.; et al. Highly conserved mitochondrial genomes among multicellular red algae of the Florideophyceae. Genome Biol. Evol. 2015, 7, 2394–2406. [Google Scholar] [CrossRef] [PubMed]
- Burger, G.; Lang, B.F.; Braun, H.P.; Marx, S. The enigmatic mitochondrial ORF ymf39 codes for ATP synthase chain b. Nucleic Acids Res. 2003, 31, 2353–2360. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Burger, G.; Nedelcu, A.M. Mitochondrial Genomes of Algae. In Genomics of Chloroplasts and Mitochondria (Advances in Photosynthesis and Respiration); Bock, R., Knoop, V., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 127–157. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).