Abstract
The central nervous system is metabolically very demanding and consequently vulnerable to defects of the mitochondrial respiratory chain. While the clinical manifestations and the corresponding radiological findings of the brain involvement in mitochondrial diseases (e.g., stroke-like episodes, signal changes of the basal ganglia, cerebral and cerebellar atrophy) are well known, at present there are few data on the spinal-cord abnormalities in these pathologies, in particular in adult subjects. In this study, we present a cross-sectional cohort study on the prevalence and characterization of spinal-cord involvement in adult patients with genetically defined mitochondrial diseases.
1. Introduction
Mitochondrial diseases (MDs) are a common group of genetic disorders characterized by a primary defect in mitochondrial oxidative phosphorylation, which is the main source of cellular ATP, and are under the dual genetic control of mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) working in concert [1]. Tissues with a high dependence on the oxidative metabolism are the most frequently affected. In particular, the central nervous system (CNS), including the brain and spinal cord, is metabolically very demanding and consequently vulnerable to defects of the mitochondrial respiratory chain [2]. While the clinical manifestations and the corresponding radiological findings of the brain involvement of mitochondrial diseases (e.g., stroke-like episodes, signal changes of the basal ganglia, cerebral and cerebellar atrophy) are well known [3,4], at present there are few data on the spinal-cord abnormalities in these pathologies, especially in adult subjects [5,6]. A detailed evaluation of this clinical aspect has only been performed for pediatric or juvenile patients in small case studies with a single phenotype or in retrospective studies [7,8,9].
Here, we present a cross-sectional cohort study on the prevalence and characterization of spinal-cord involvement in adult patients with genetically defined MDs.
2. Materials and Methods
We included all of the consecutive adult mitochondrial patients (18 years and above) who came to our hospital, and performed a brain and spinal-cord MRI. In particular, the protocol for the spinal cord included sagittal and axial T1 and T2 weighted images with contrast administration. Two neurologists with experience in neuroradiology independently reviewed the MRI images. All of the patients, evaluated by a neurologist specialized in mitochondrial medicine, had a defined mitochondrial disease based on molecular, morphological and biochemical analysis. The study was approved by the Ethics Committee of the Università Cattolica del Sacro Cuore (Rome, Italy; ID: 3754) and all of the participants signed an informed consent prior to inclusion. All procedures were conducted in compliance to the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
3. Results
3.1. Patients
Fifty one patients (mean age: 45, range 18–78; 33 females) were enrolled during this period (Table 1): 20 patients had progressive external ophthalmoplegia (PEO) associated with single mtDNA deletion (n = 8), recessive or dominant mutations in the nuclear genes POLG (n = 6), TWNK (n = 2) and SLC25A4 (n = 1) or multiple mtDNA deletions without an identified nuclear gene defect (n = 3); 3 had POLG mutations and ataxia neuropathy spectrum (ANS); 11 patients carried the m.3243A > G mutation affected by mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS; n = 3), maternally inherited diabetes and deafness (MIDD; n = 6), cardiomyopathy (n = 1) and PEO (n = 1); 5 patients had the m.8344A > G mutation and myoclonic epilepsy with ragged-red fibers (MERRF); 3 patients had Leber hereditary optic neuropathy (LHON) (2 associated with m.14484T > C mutation e and 1 with m.11778G > A) and 1 was affected by Leigh syndrome (LS) with m.13513G > A mutation; 2 patients presented with autosomal dominant optic atrophy (ADOA) and mutations in the OPA1 gene; 1 subject was diagnosed with leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL) and mutations in DARS2 gene; 2 mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) harbored recessive TYMP mutations; 3 patients had other mtDNA point mutations including m.8356T > G, m.8356T > C (both associated with the MERRF phenotype) and m.13042G > A.

Table 1.
Clinical, demographic and molecular features in patients with mitochondrial diseases undergoing spinal-cord MRI.
3.2. Spinal-Cord Imaging Findings
We found spinal cord abnormalities in five patients (9.8%; Table 2 and Figure 1). A 45-year-old man with the m.3243A > G mutation in the MT-TL1 gene and the MIDD phenotype presented a significant thinning of the spinal cord at the level of the soma D3, where a focal area of signal hyperintensity was present in the T2 images with a cystic-like appearance located in the white matter of the dorsal and lateral columns (patient 1). The MRI showed symmetrical T2 hyperintensities in the spinal grey matter in the cervical and the upper-thoracic spinal-cord segments and a medullary atrophy in an 18-year-old girl with Leigh syndrome and m.13513G > A mutation (patient 2). A medullary atrophy without signal alteration was detected in one patient who was affected by ANS associated with a mutation in the POLG gene (c.3293A > G) (patient 3). The neuroimaging study in a 27-year-old female with the m.11778G > A mutation affected by LHON and clinically definite multiple sclerosis (MS) showed focal abnormalities in the dorsal and lateral columns of the spinal cord (patient 4). Finally, a 41-year-old female with mutations in the DARS2 gene (IVS2-20_-21delTTinsC, c.374G > A) and a diagnosis of LBSL had diffuse abnormalities of the T2 signal of the cervical and thoracic spinal cord involving the dorsal columns and the lateral corticospinal tracts (patient 5).
Table 2.
Clinical diagnoses, genetic and magnetic resonance imaging findings in patients with mitochondrial disorders and spinal-cord involvement.
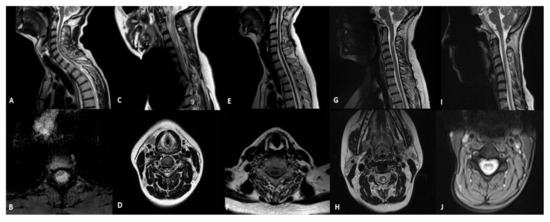
Figure 1.
Sagittal and axial T2-weithing images of MRI in patients with mitochondrial diseases and spinal cord involvement. (A,B) MIDD with m.3243A > G mutation (patient 1): selective thinning of the spinal cord with a cystic-like lesion located in the white matter of dorsal and lateral columns; (C,D) Leigh syndrome with m.13513G > A (patient 2): hyperintensity spinal grey matter in the cervical and the upper-thoracic spinal cord segments and medullary atrophy; (E,F) ANS with POLG mutation (patient 3): medullary atrophy without signal alterations; (G,H) LHON with m.11778G > A (patient 4): focal hyperintensity in the cervical posterior columns; (I,J) LBSL with DARS2 mutation (patient 5): abnormalities of the T2 signal of the cervical and thoracic spinal cord involving dorsal columns and lateral corticospinal tracts.
3.3. Brain Imaging Findings
Brain MRI imaging that was performed on the five patients with spinal cord involvement revealed the following abnormalities: a generalized cortical atrophy in the ANS patient; multiple T2-hyperintense white-matter periventricular lesions and an enhancing lesion with an ovoid/round shape in the subject affected by LHON; brain radiological findings of the LBSL patient were characterized by bilateral periventricular T2 white-matter hyperintensities, sparing the U-fibers, associated with signal abnormalities in the superior and inferior cerebellum peduncles and in cerebellar white matter with subcortical preponderance; diffuse cerebral and cerebellar white matter involvement in combination with left putamen and right globus pallidus lesions, cerebellar cortical atrophy and brainstem abnormalities were found in LS. In the m.3243A > G carrier, no pathological findings were detected.
3.4. Clinical and Laboratory Findings
Patient 1 (m.3243A > G mutation) had asymptomatic spinal cord lesions. On the contrary, a mild spasticity was present in the lower limbs of patient 4 (LHON), and a severe spasticity was present in the upper and lower limbs of patient 2 (LS) and 5 (LBSL). In the last two cases we also observed the Babinski sign, which is an increased deep tendon reflex and, in patient 5, absent vibratory sensation in both legs below the knees. Finally, with regard to the ANS patient with a POLG mutation, we can hypothesize that the spinal-cord atrophy contributes to the ataxic phenotype. A cerebrospinal fluid analysis was performed on three patients: patient 4 revealed the presence of oligoclonal bands, while it was normal in patient 1 and 2. There were no significant common features (e.g., diet, previous treatments) among the patients with spinal-cord involvement.
4. Discussion
In patients with proven mitochondrial diseases, the abnormalities of the CNS are well known for cerebral and cerebellar involvement with frequently reported MRI findings [3]. Spinal cord abnormalities are rarely described in these neurogenetic disorders, with the exception of LBSL, which is a rare autosomal recessive mitochondrial disease that is associated with mutations in the DARS2 gene and characterized by a slowly progressive pyramidal and cerebellar dysfunction, presenting a distinct and uniform MRI pattern with signal abnormalities in the cerebral white matter and the selective involvement of specific brainstem and spinal-cord tracts (dorsal columns and lateral corticospinal tracts) [10]. Furthermore, case studies have described patients affected by LHON and multiple sclerosis (MS). This association is defined as “LHON-MS”, which is a distinct clinical phenotype with specific differences from LHON and MS, but also in this case the spinal cord abnormalities are rarely reported [5,11]. Similarly, spinal cord involvement rarely characterizes subjects with MELAS or Leigh syndrome [12,13,14]. The few publications in which this clinical aspect has been examined in detail were small case studies or retrospective studies of pediatric or juvenile patients. In particular, three different research articles recently reported the high prevalence of spinal cord involvement in a case series of Kearns-Sayre syndrome (54.5%, 6/11) and cohorts of pediatric patients (74%, 14/19 and 58%, 19/33), respectively [7,8,9]. Our cross-sectional study analyzed the prevalence and the radiological characterization of spinal cord involvement in adult patients with genetically defined mitochondrial diseases. The prevalence of neuroradiological abnormalities was 9.8% and the five patients were characterized by different clinical phenotypes and mutations (MIDD, LS, ANS, LHON and LBSL were associated with m.3243A > G, m.13513G > A, POLG, m.11778G > A and DARS2 mutations, respectively; Table 2). Although the prevalence in our cohort was lower than documented in pediatric or juvenile populations, it confirmed that spinal cord lesions are common in primary mitochondrial diseases. The different prevalence of spinal cord involvement between the pediatric and adult population reflects the well-known data of frequent CNS involvement in the first group that is often characterized by clinically more severe phenotypes and with a worse prognosis. Furthermore, our data confirmed the coexistence of different pathogenic mechanisms that underlie this clinical aspect, reporting both a case in which the autoimmunity played a key role (LHON-MS patient), as also suggested by the presence of oligoclonal bands in the cerebrospinal fluid, and a case in which the degenerative mechanisms play a predominant role (ANS). Interestingly, spinal-cord atrophy without signal alterations was described in a subject affected by mitochondrial diseases (patient 3). This spinal MRI feature has been previously reported in other genetic disorders such as hereditary spastic paraplegia, Friedreich’s ataxia or adult polyglucosan body disease, but never in patients with a primary defect in mitochondrial oxidative phosphorylation [6]. This observation is supported by postmortem findings in a POLG patient with gait ataxia characterized by posterior column degeneration [15]. Regarding the latter aspect, compared to the spinal cord more data are available on the biogenetics of the brain. In particular, the neuron-rich grey matter has a very high ATP turnover (30 μmol ATP/g tissue per min) [16] and the white matter consumes approximately one third of the energy of the gray matter [17]. Therefore, it is clear that the CNS is metabolically very demanding and consequently vulnerable to defects of the mitochondrial respiratory chain [2].
5. Conclusions
Our cross-sectional study analyzed the prevalence of the spinal cord involvement in a cohort of adult patients with primary mitochondrial disorders. The analysis of the data suggests that this clinical aspect is not infrequent and that the combination of brain and spinal MRI imaging associated with other symptoms suggestive of these neurogenic disorders is useful for a correct molecular diagnosis, which remains a considerable diagnostic challenge even in the omics era.
Author Contributions
Conceptualization, G.P. and S.S.; methodology, G.P. and S.S.; investigation, G.P., P.M., I.T., C.S., A.S., A.T., R.C. and S.S.; data curation, G.P., P.M., I.T., C.S., A.S., A.T., R.C. and S.S.; writing—original draft preparation, G.P.; writing—review and editing, G.P., P.M., I.T., C.S., A.S., A.T., R.C. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was approved by the Ethics Committee of the Università Cattolica del Sacro Cuore (Rome, Italy; ID: 3754) and all participants signed an informed consent prior to inclusion. All procedures were conducted in compliance to the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
G.P. and S.S. are members of the European Reference Network for Neuromuscular Diseases-Project ID N° 870177.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ng, Y.S.; Bindoff, L.A.; Gorman, G.S.; Klopstock, T.; Kornblum, C.; Mancuso, M.; McFarland, R.; Sue, C.M.; Suomalainen, A.; Taylor, R.W.; et al. Mitochondrial disease in adults: Recent advances and future promise. Lancet Neurol. 2021, 20, 573–584. [Google Scholar] [CrossRef]
- Lax, N.Z.; Gorman, G.S.; Turnbull, D.M. Review: Central nervous system involvement in mitochondrial disease. Neuropathol. Appl. Neurobiol. 2017, 43, 102–118. [Google Scholar] [CrossRef] [PubMed]
- Mascalchi, M.; Montomoli, M.; Guerrini, R. Neuroimaging in mitochondrial disorders. Essays Biochem. 2018, 62, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Cerebral imaging in adult mitochondrial disorders. J. Neurol. Sci. 2019, 404, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Bindu, P.S.; Arvinda, H.; Taly, A.B.; Govindaraju, C.; Sonam, K.; Chiplunkar, S.; Kumar, R.; Gayathri, N.; Bharath, S.; Nagappa, M.; et al. Magnetic resonance imaging correlates of genetically characterized patients with mitochondrial disorders: A study from south India. Mitochondrion 2015, 25, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Marelli, C.; Salsano, E.; Politi, L.S.; Labauge, P. Spinal cord involvement in adult-onset metabolic and genetic diseases. J. Neurol. Neurosurg. Psychiatry 2019, 90, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Luca, P.; Alessia, G.; Camilla, R.M.; Antonio, N.; Diego, M.; Federica, D.; Daria, D.; Rosalba, C.; Carlo, D.V.; Daniela, L. Spinal cord involvement in Kearns-Sayre syndrome: A neuroimaging study. Neuroradiology 2020, 62, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.A.P.F.; Teixeira, S.R.; Martin-Saavedra, J.S.; Guimarães Gonçalves, F.; Lo Russo, F.; Muraresku, C.; McCormick, E.M.; Falk, M.J.; Zolkipli-Cunningham, Z.; Ganetzky, R.; et al. Pediatric Leigh Syndrome: Neuroimaging Features and Genetic Correlations. Ann. Neurol. 2020, 88, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.A.P.F.; Goldstein, A.; Teixeira, S.R.; Martin-Saavedra, J.S.; de Barcelos, I.P.; Fadda, G.; Caschera, L.; Kidd, M.; Gonçalves, F.G.; McCormick, E.M.; et al. Involvement of the Spinal Cord in Primary Mitochondrial Disorders: A Neuroimaging Mimicker of Inflammation and Ischemia in Children. AJNR Am. J. Neuroradiol. 2021, 42, 389–396. [Google Scholar] [CrossRef] [PubMed]
- van Berge, L.; Hamilton, E.M.; Linnankivi, T.; Uziel, G.; Steenweg, M.E.; Isohanni, P.; Wolf, N.I.; Krägeloh-Mann, I.; Brautaset, N.J.; Andrews, P.I.; et al. Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation: Clinical and genetic characterization and target for therapy. Brain 2014, 137, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, G.; Burke, A.; Yu-Wai-Man, P.; Compston, D.A.; Chinnery, P.F. Clinical features of MS associated with Leber hereditary optic neuropathy mtDNA mutations. Neurology 2013, 81, 2073–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascalchi, M.; Bartolini, E.; Bianchi, A.; Gulino, P.; Procopio, E. Teaching NeuroImages: Spinal cord gray matter involvement in complex I deficiency mitochondriopathy. Neurology 2016, 87, 106–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, P.V.; Pinto, W.B.; Oliveira, A.S. Teaching NeuroImages: Longitudinally extensive transverse myelitis in MELAS. Neurology 2016, 86, e37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, P.V.S.; Bortholin, T.; Teixeira, C.A.C.; Seneor, D.D.; Marin, V.D.G.B.; Dias, R.B.; Farias, I.B.; Badia, B.M.L.; Silva, L.H.L.; Pinto, W.B.V.R.; et al. Leigh syndrome caused by mitochondrial DNA-maintenance defects revealed by whole exome sequencing. Mitochondrion 2019, 49, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.E.; Somoza, A.; Gilbert, D.L. Rare autosomal dominant POLG1 mutation in a family with metabolic strokes, posterior column spinal degeneration, and multi-endocrine disease. J. Child. Neurol. 2010, 2, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Kunz, W.S. Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. 2015, 14, 956–966. [Google Scholar] [CrossRef]
- Harris, J.J.; Attwell, D. The energetics of CNS white matter. J. Neurosci. 2012, 32, 356–371. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).