Characterization of Sensorineural Hearing Loss in Children with Alport Syndrome

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Patient Characteristics
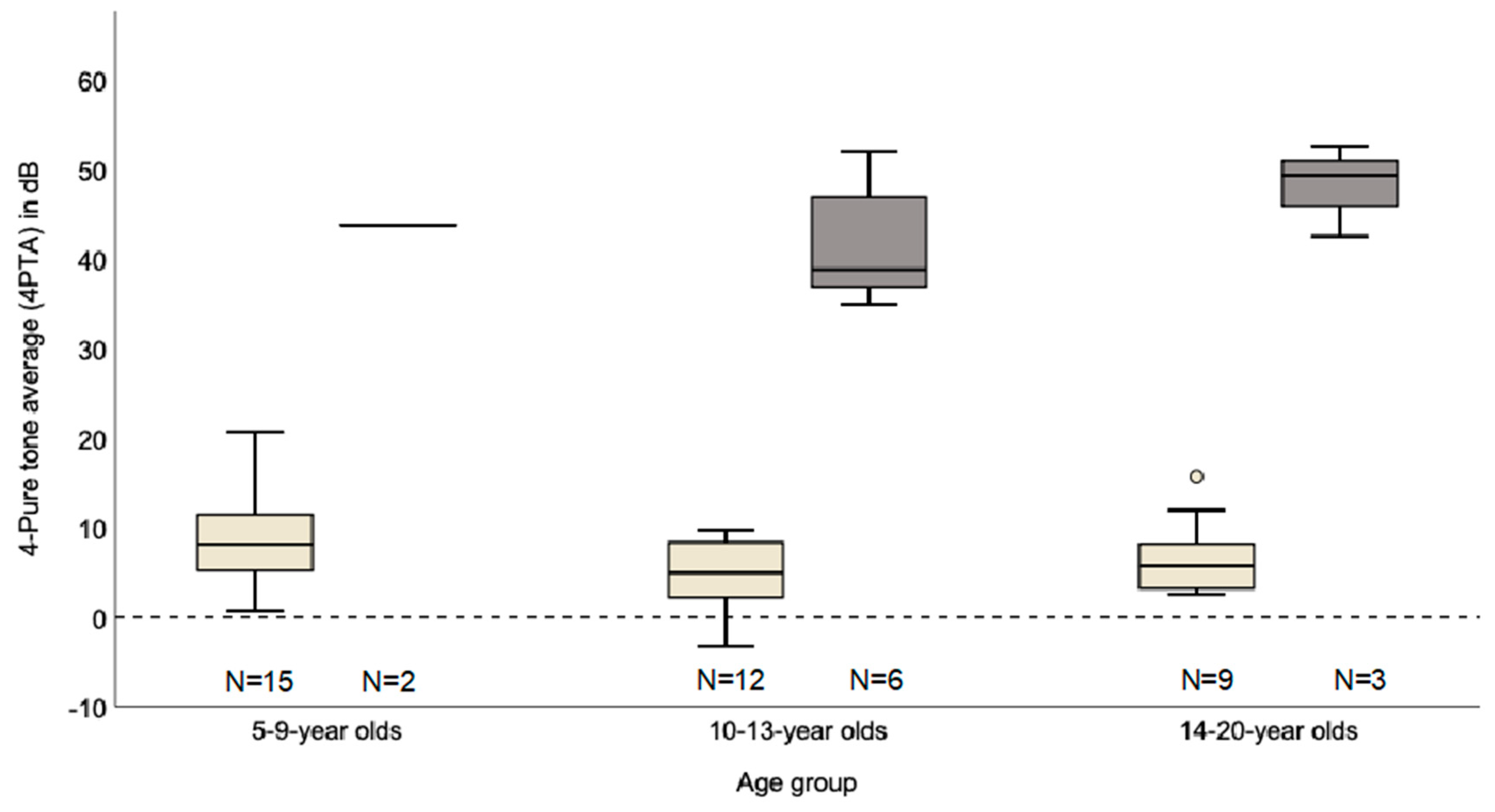
2.2. Clinical Audiological Characteristics
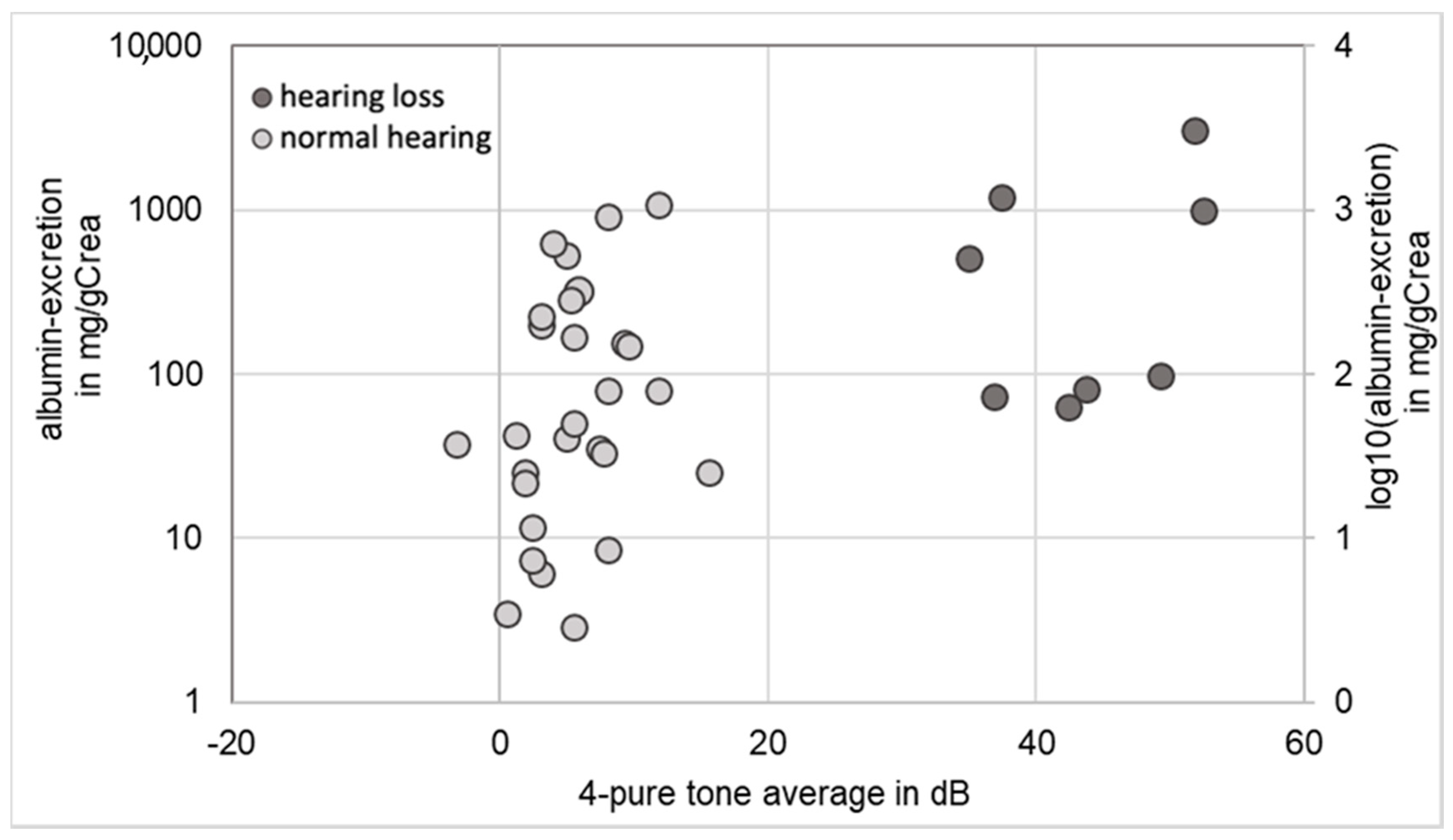
2.3. Correlation between Renal Function and Hearing
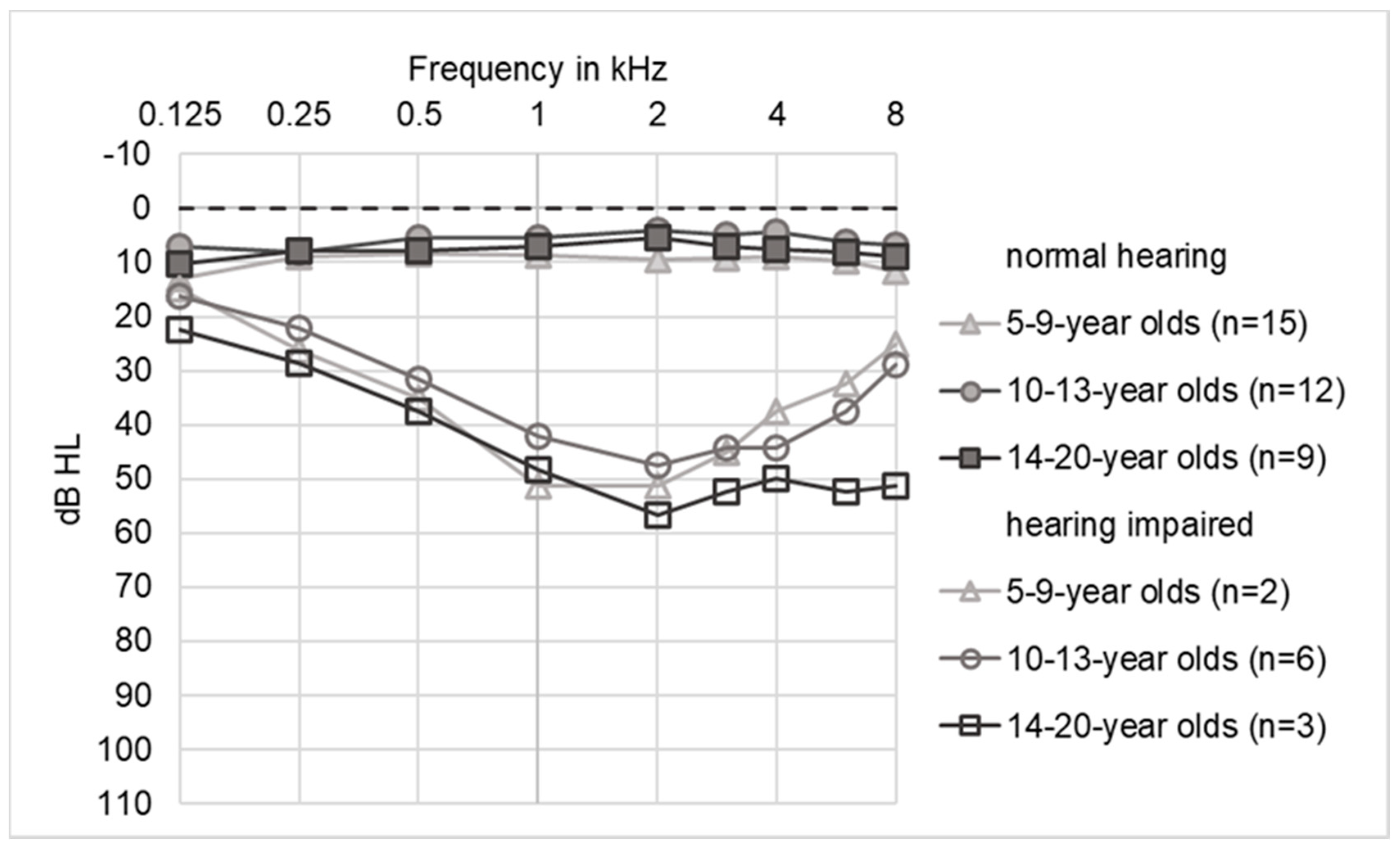
2.4. Audiograms
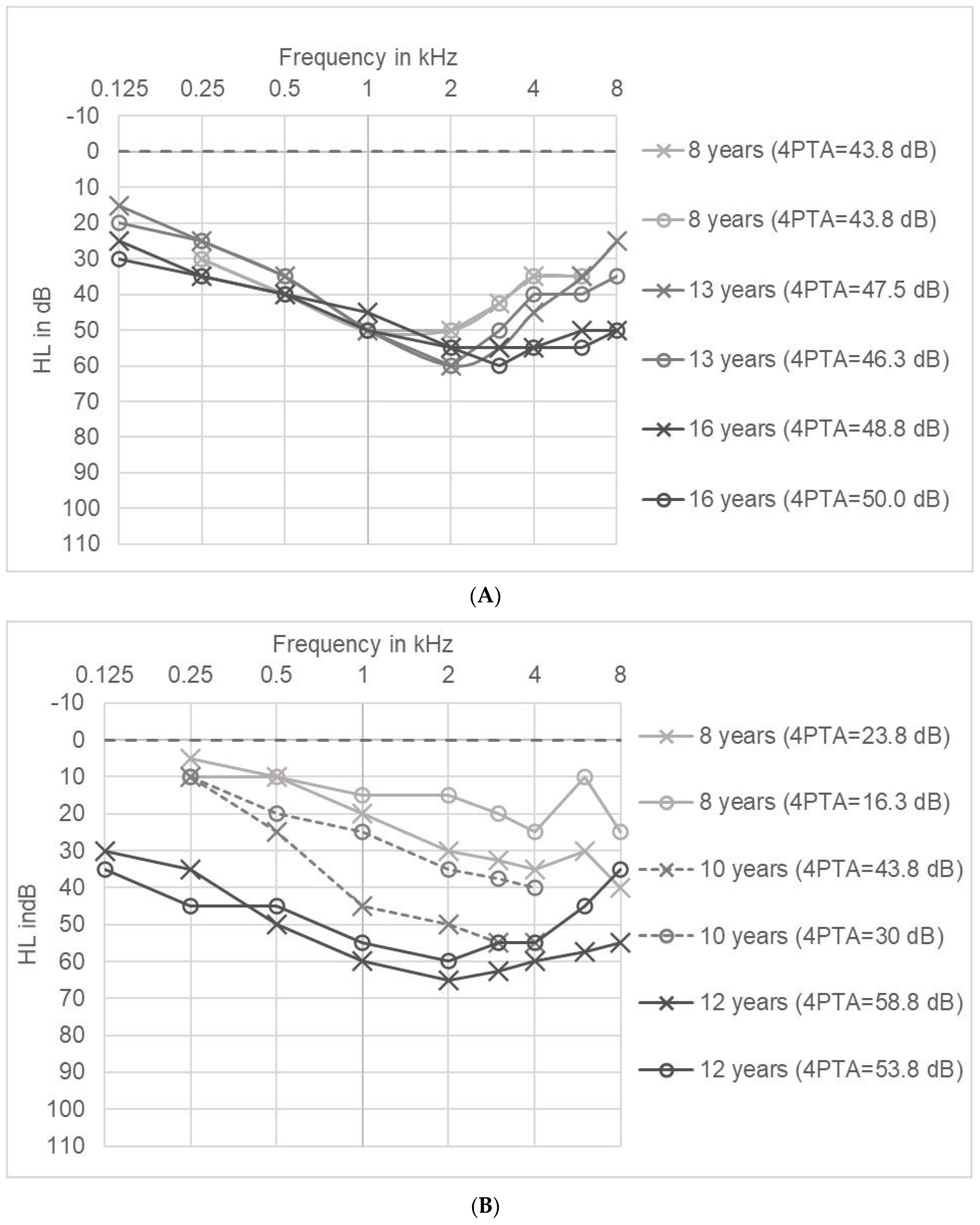
2.5. Long Term Tracking of Hearing Impairment in Individual Patients
3. Discussion
4. Materials and Methods
4.1. Patients
- Stage 0: Microhematuria without microalbuminuria
- Stage I: Microalbuminuria: 30–300 mg albumin/g creatinine (gCrea)
- Stage II: Proteinuria: >300 mg albumin/gCrea
4.2. Audiograms
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4PTA | 4-pure tone average |
| AS | Alport syndrome |
| ACEi | Angiotensin-converting enzyme inhibitors |
| ADAS | Autosomal dominant Alport syndrome |
| ARAS | Autosomal recessive Alport syndrome |
| AS | Alport syndrome |
| ESRF | End stage renal failure |
| GBM | Glomerular basement membrane |
| HL | Hearing loss |
| IQR | Interquartile ratio |
| RAAS | Renin angiotensin aldosterone system |
| TEOAE | Transient Evoked Otoacoustic Emissions |
| XLAS | X-linked Alport syndrome |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinic of Nephrology and Rheumatology, University Medical Center Goettingen, Goettingen, Germany | |
| Michael Koziolek | deputy of coordinating principal investigator |
| Carsten Paul Bramlage | deputy of coordinating principal investigator |
| Frauke Weber | trial assistant to coordinating principal investigator |
| Tanja Albrecht-Nock | trial assistant to coordinating principal investigator |
| Joseph Sonntag | trial assistant to coordinating principal investigator |
| Jenny Frese | trial assistant to coordinating principal investigator |
| Department of Pediatrics and Pediatric Neurology, University Medical Center Goettingen, Goettingen, Germany | |
| Hildegard Zappel | physician investigator |
| Matthias Kettwig | physician investigator |
| Department of Medical Statistics, University Medical Center Goettingen, Goettingen, Germany | |
| Tim Friede | medical statistics and trial design |
| Markus Harden | medical statistics and trial design |
| Reinhard Hilgers | medical statistics and trial design |
| Clementine Children’s Hospital, Frankfurt, Germany | |
| Kay Latta | physician investigator |
| Matthias Hansen | physician investigator |
| Department of Pediatric Nephrology, Hannover Medical School, Hannover, Germany | |
| Lars Pape | physician investigator |
| Christian Lerch | physician investigator |
| Mirja Wedekin | physician investigator |
| Nicole Meyer | trial assistant |
| Department of Pediatrics I, University Children’s Hospital Heidelberg, Heidelberg, Germany | |
| Burkhard Tönshoff | physician investigator |
| Britta Hoecker | physician investigator |
| Susanne Klaiber | trial assistant |
| Pediatric Nephrology, Children’s and Adolescents’ Hospital, University Hospital Cologne, Cologne, Germany | |
| Lutz T. Weber | physician investigator |
| Rasmus Ehren | physician investigator |
| Michaela Gessner | physician investigator |
| Max Liebau | physician investigator |
| Anne-Kristin Vogt-Weigeldt | trial assistant |
| Division of Pediatric Nephrology, Children’s Hospital Memmingen, Memmingen, Germany | |
| Henry Fehrenbach | physician investigator |
| Therese Jungraithmayr | physician investigator |
| Dr v. Haunersches Children’s Hospital, Ludwig Maximilians University Munich, Munich, Germany | |
| Baerbel Lange-Sperandio | physician investigator |
| Sabine Ponsel | physician investigator |
| Pediatric Nephrology, University Children’s Hospital Rostock, Rostock, Germany | |
| Hagen Staude | physician investigator |
| Ulrike Jacoby | physician investigator |
| Pediatric Nephrology, University Children’s Hospital Muenster, Muenster, Germany | |
| Sabine König | physician investigator |
| Martin Konrad | physician investigator |
| Brigitta Kranz | physician investigator |
| Jens Koenig | physician investigator |
| Lisa Loechtermann | trial assistant |
| Division of Pediatric Nephrology, University Children’s Hospital Jena, Jena, Germany | |
| Ulrike John | physician investigator |
| Michael Pohl | physician investigator |
| Ralf Husain | physician investigator |
| Katrin Mueller | trial assistant |
| Department of Pediatrics, University Children’s Hospital Berlin, University Hospital Berlin Charité, Berlin, Germany | |
| Jutta Gellermann | physician investigator |
| Julia Thumfart | physician investigator |
| Department of Pediatrics, Division of Pediatric Nephrology, University of Bonn, Bonn, Germany | |
| Bernd Hoppe | physician investigator |
| Gesa Schalk | physician investigator |
| Markus Feldkoetter | physician investigator |
| Sabine Schmidt | trial assistant |
| Department of Pediatrics, University Hospital Erlangen, Erlangen, Germany | |
| Matthias Galiano | physician investigator |
| Katja Sauerstein | physician investigator |
| Evelin Muschiol | trial assistant |
| Paediatrics I, University Children’s Hospital Tuebingen, Tuebingen, Germany | |
| Heiko Billing | referring physician investigator |
| Pediatric Nephrology, Helios Kliniken Schwerin, Schwerin, Germany | |
| Frauke Wilkening | referring physician investigator |
References
- Hudson, B.G.; Tryggvason, K.; Sundaramoorthy, M.; Neilson, E.G. Alport’s Syndrome, Goodpasture’s Syndrome, and Type IV Collagen. N. Engl. J. Med. 2003, 348, 2543–2556. [Google Scholar] [CrossRef] [PubMed]
- Kruegel, J.; Rubel, D.; Gross, O. Alport syndrome—Insights from basic and clinical research. Nat. Rev. Nephrol. 2013, 9, 170–178. [Google Scholar] [CrossRef]
- Hertz, J.M.; Thomassen, M.; Storey, H.; Flinter, F. Clinical utility gene card for: Alport syndrome—Update 2014. Eur. J. Hum. Genet. 2015, 23, 1269. [Google Scholar] [CrossRef]
- Kashtan, C.E. Alport syndromes: Phenotypic heterogeneity of progressive hereditary nephritis. Pediatr. Nephrol. 2000, 14, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.F.; Pruchno, C.J.; Jiang, X.; Atkin, C.L.; Stone, E.M.; Denison, J.C.; Fain, P.R.; Gregory, M.C. A mutation causing Alport syndrome with tardive hearing loss is common in the western United States. Am. J. Hum. Genet. 1996, 58, 1157–1165. [Google Scholar]
- Govan, J.A. Ocular manifestations of Alport’s syndrome: A hereditary disorder of basement membranes? Br. J. Ophthalmol. 1983, 67, 493–503. [Google Scholar] [CrossRef]
- Nielsen, C.E. Lenticonus Anterior and Alport’s Syndrome. Acta Ophthalmol. (Copenh) 1978, 56, 518–530. [Google Scholar] [CrossRef]
- Pöschl, E.; Schlötzer-Schrehardt, U.; Brachvogel, B.; Saito, K.; Ninomiya, Y.; Mayer, U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 2004, 131, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Bekheirnia, M.R.; Reed, B.; Gregory, M.C.; McFann, K.; Shamshirsaz, A.A.; Masoumi, A.; Schrier, R.W. Genotype–Phenotype Correlation in X-Linked Alport Syndrome. J. Am. Soc. Nephrol. 2010, 21, 876–883. [Google Scholar] [CrossRef]
- Morinière, V.; Dahan, K.; Hilbert, P.; Lison, M.; Lebbah, S.; Topa, A.; Bole-Feysot, C.; Pruvost, S.; Nitschke, P.; Plaisier, E.; et al. Improving Mutation Screening in Familial Hematuric Nephropathies through Next Generation Sequencing. J. Am. Soc. Nephrol. 2014, 25, 2740–2751. [Google Scholar] [CrossRef]
- Jais, J.-P.; Knebelmann, B.; Giatras, I.; De Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, K.O.; Flinter, F.; et al. X-linked Alport Syndrome Natural History in 195 Families and Genotype- Phenotype Correlations in Males. J. Am. Soc. Nephrol. 2000, 11, 649–657. [Google Scholar]
- Gross, O.; Kashtan, C.E.; Rheault, M.N.; Flinter, F.; Savige, J.; Miner, J.H.; Torra, R.; Ars, E.; Deltas, C.; Savva, I.; et al. Advances and unmet needs in genetic, basic and clinical science in Alport syndrome: Report from the 2015 International Workshop on Alport Syndrome. Nephrol. Dial. Transplant. 2017, 32, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Haas, M. Alport Syndrome and Thin Glomerular Basement Membrane Nephropathy: A Practical Approach to Diagnosis. Arch. Pathol. Lab. Med. 2009, 133, 224–232. [Google Scholar] [PubMed]
- Khoshnoodi, J.; Pedchenko, V.; Hudson, B.G. Mammalian collagen IV. Microsc. Res. Tech. 2008, 71, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, Y.; Kagawa, M.; Iyama, K.; Naito, I.; Kishiro, Y.; Seyer, J.M.; Sugimoto, M.; Oohashi, T.; Sado, Y. Differential expression of two basement membrane collagen genes, COL4A6 and COL4A5, demonstrated by immunofluorescence staining using peptide- specific monoclonal antibodies. J. Cell Biol. 1995, 130, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Shield, C.F.; Todd, P.; Hudson, B.G.; Neilson, E.G. Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J. Clin. Investig. 1997, 99, 2470–2478. [Google Scholar] [CrossRef] [PubMed]
- Zehnder, A.F.; Adams, J.C.; Santi, P.A.; Kristiansen, A.G.; Wacharasindhu, C.; Mann, S.; Kalluri, R.; Gregory, M.C.; Kashtan, C.E.; Merchant, S.N. Distribution of Type IV Collagen in the Cochlea in Alport Syndrome. Arch. Otolaryngol. Neck Surg. 2005, 131, 1007–1013. [Google Scholar] [CrossRef]
- Gleeson, M.J. Alport’s syndrome: Audiological manifestations and implications. J. Laryngol. Otol. 1984, 98, 449–465. [Google Scholar] [CrossRef]
- Harvey, S.J.; Mount, R.; Sado, Y.; Naito, I.; Ninomiya, Y.; Harrison, R.; Jefferson, B.; Jacobs, R.; Thorner, P.S. The Inner Ear of Dogs with X-Linked Nephritis Provides Clues to the Pathogenesis of Hearing Loss in X-Linked Alport Syndrome. Am. J. Pathol. 2001, 159, 1097–1104. [Google Scholar] [CrossRef]
- Alves, F.R.A.; de A Quintanilha Ribeiro, F. Revision about hearing loss in the Alport’s syndrome, analyzing the clinical, genetic and bio-molecular aspects. Braz. J. Otorhinolaryngol. 2005, 71, 813–819. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Zhang, Y.; Gu, H.; Chen, Z.; Ren, L.; Lu, X.; Chen, L.; Wang, F.; Liu, Y.; et al. X-linked Alport syndrome: Pathogenic variant features and further auditory genotype-phenotype correlations in males. Orphanet, J. Rare Dis. 2018, 13, 229. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Licht, C.; Anders, H.J.; Hoppe, B.; Beck, B.B.; Tönshoff, B.; Höcker, B.; Wygoda, S.; Ehrich, J.; Pape, L.; et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012, 81, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Friede, T.; Hilgers, R.; Görlitz, A.; Gavénis, K.; Ahmed, R.; Dürr, U. Safety and Efficacy of the ACE-Inhibitor Ramipril in Alport Syndrome: The Double-Blind, Randomized, Placebo-Controlled, Multicenter Phase III EARLY PRO-TECT Alport Trial in Pediatric Patients. ISRN Pediatr. 2012, 2012, 436046. [Google Scholar] [CrossRef]
- Gross, O.; Tönshoff, B.; Weber, L.T.; Pape, L.; Latta, K.; Fehrenbach, H.; Lange-Sperandio, B.; Zappel, H.; Hoyer, P.; Staude, H.; et al. A multicenter, randomized, placebo-controlled, double-blind phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport’s syndrome. Kidney Int. 2020, 97, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Alves, F.R.A.; de Andrade Quintanilha Ribeiro, F. Clinical data and hearing of individuals with Alport syndrome. Braz. J. Otorhinolaryngol. 2008, 74, 807–814. [Google Scholar] [CrossRef]
- Meehan, D.T.; Delimont, D.; Dufek, B.; Zallocchi, M.; Phillips, G.; Gratton, M.A.; Cosgrove, D. Endothelin-1 mediated induction of extracellular matrix genes in strial marginal cells underlies strial pathology in Alport mice. Hear. Res. 2016, 341, 100–108. [Google Scholar] [CrossRef]
- Johnsson, L.G.; Arenberg, I.K. Cochlear abnormalities in Alport’s syndrome. Arch. Otolaryngol. 1981, 107, 340–349. [Google Scholar] [CrossRef]
- Cosgrove, D.; Kornak, J.M.; Samuelson, G. Expression of basement membrane type IV collagen chains during postnatal development in the murine cochlea. Hear. Res. 1996, 100, 21–32. [Google Scholar] [CrossRef]
- Cosgrove, D.; Samuelson, G.; Meehanet, D.T.; Miller, C.; McGee, J.; Walsh, E.J.; Siegel, M. Ultrastructural, physiological, and molecular defects in the inner ear of a gene-knockout mouse model for autosomal Alport syndrome. Hear. Res. 1998, 121, 84–98. [Google Scholar] [CrossRef]
- Merchant, S.N.; Burgess, B.J.; Adams, J.C.; Kashtan, C.E.; Gregory, M.C.; Santi, P.A.; Colvin, R.; Collins, B.; Nadol, J.B. Temporal Bone Histopathology in Alport Syndrome. Laryngoscope 2004, 114, 1609–1618. [Google Scholar] [CrossRef]




| Total (n = 51) | without Hearing Loss (n = 39) | Unknown Hearing Loss (n = 3) | with Hearing Loss (n = 9) | |
|---|---|---|---|---|
| n (%) | n (%) | n (%) | n (%) | |
| Study group | ||||
| Randomised | 16 (31%) | 14 (36%) | 1 (33%) | 1 (11%) |
| Open | 35 (67%) | 25 (64%) | 2 (67%) | 8 (89%) |
| Treatment | ||||
| Placebo | 7 (14%) | 6 (15%) | 0 (0%) | 1 (11%) |
| Ramipril | 44 (86%) | 33 (85%) | 3 (100%) | 8 (89%) |
| Mean age in years ± SD | ||||
| Baseline | 9 ± 4.2 | 8.9 ± 4.1 | 3.7 ± 1.2 | 10.8 ± 4 |
| At last ear examination | 11.5 ± 4.3 | 11.5 ± 4.1 | 5 ± 2.6 | 13.7 ± 3.7 |
| Sex | ||||
| Male | 49 (96%) | 38 (97%) | 3 (100%) | 8 (89%) |
| Female | 2 (4%) | 1 (3%) | 0 (0%) | 1 (11%) |
| Mode of heritage | ||||
| X-linked Alport Syndrome (XLAS) | 42 (82%) | 34 (87%) | 2 (67%) | 6 (67%) |
| Autosomal recessive Alport Syndrome (ARAS) | 8 (16%) | 5 (13%) | 0 (0%) | 3 (33%) |
| n/a | 1 (2%) | 0 (0%) | 1 (33%) | 0 (0%) |
| Positive family history | 18 (35%) | 15 (38%) | 2 (67%) | 1 (11%) |
| Median albuminuria at baseline in mg albumin/gCrea (IQR) | 61 (227.4) | 34.7 (161.1) | 82.9 (54,5) | 272.5 (505) |
| Maximum Hearing Loss Per Ear | 5–9 Year Old (n = 2) | 10–13 Year Old (n = 6) | 14–20 Year Old (n = 3) |
|---|---|---|---|
| low frequencies (0.125, 0.25 and 0.5 kHz) | 0 | 0 | 0 |
| mid-frequencies (1, 2 and 3 kHz) | 4 | 5 | 5 |
| middle-high frequencies (2, 3 and 4 kHz) | 0 | 3 | 1 |
| high frequencies (4, 6 and 8 kHz) | 0 | 4 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boeckhaus, J.; Strenzke, N.; Storz, C.; Gross, O.; on behalf of the GPN Study Group; EARLY PRO-TECT Alport Investigators. Characterization of Sensorineural Hearing Loss in Children with Alport Syndrome. Life 2020, 10, 360. https://doi.org/10.3390/life10120360
Boeckhaus J, Strenzke N, Storz C, Gross O, on behalf of the GPN Study Group, EARLY PRO-TECT Alport Investigators. Characterization of Sensorineural Hearing Loss in Children with Alport Syndrome. Life. 2020; 10(12):360. https://doi.org/10.3390/life10120360
Chicago/Turabian StyleBoeckhaus, Jan, Nicola Strenzke, Celine Storz, Oliver Gross, on behalf of the GPN Study Group, and EARLY PRO-TECT Alport Investigators. 2020. "Characterization of Sensorineural Hearing Loss in Children with Alport Syndrome" Life 10, no. 12: 360. https://doi.org/10.3390/life10120360
APA StyleBoeckhaus, J., Strenzke, N., Storz, C., Gross, O., on behalf of the GPN Study Group, & EARLY PRO-TECT Alport Investigators. (2020). Characterization of Sensorineural Hearing Loss in Children with Alport Syndrome. Life, 10(12), 360. https://doi.org/10.3390/life10120360