Spectroscopic and Crystal-Chemical Features of Sodalite-Group Minerals from Gem Lazurite Deposits

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Chemical Composition
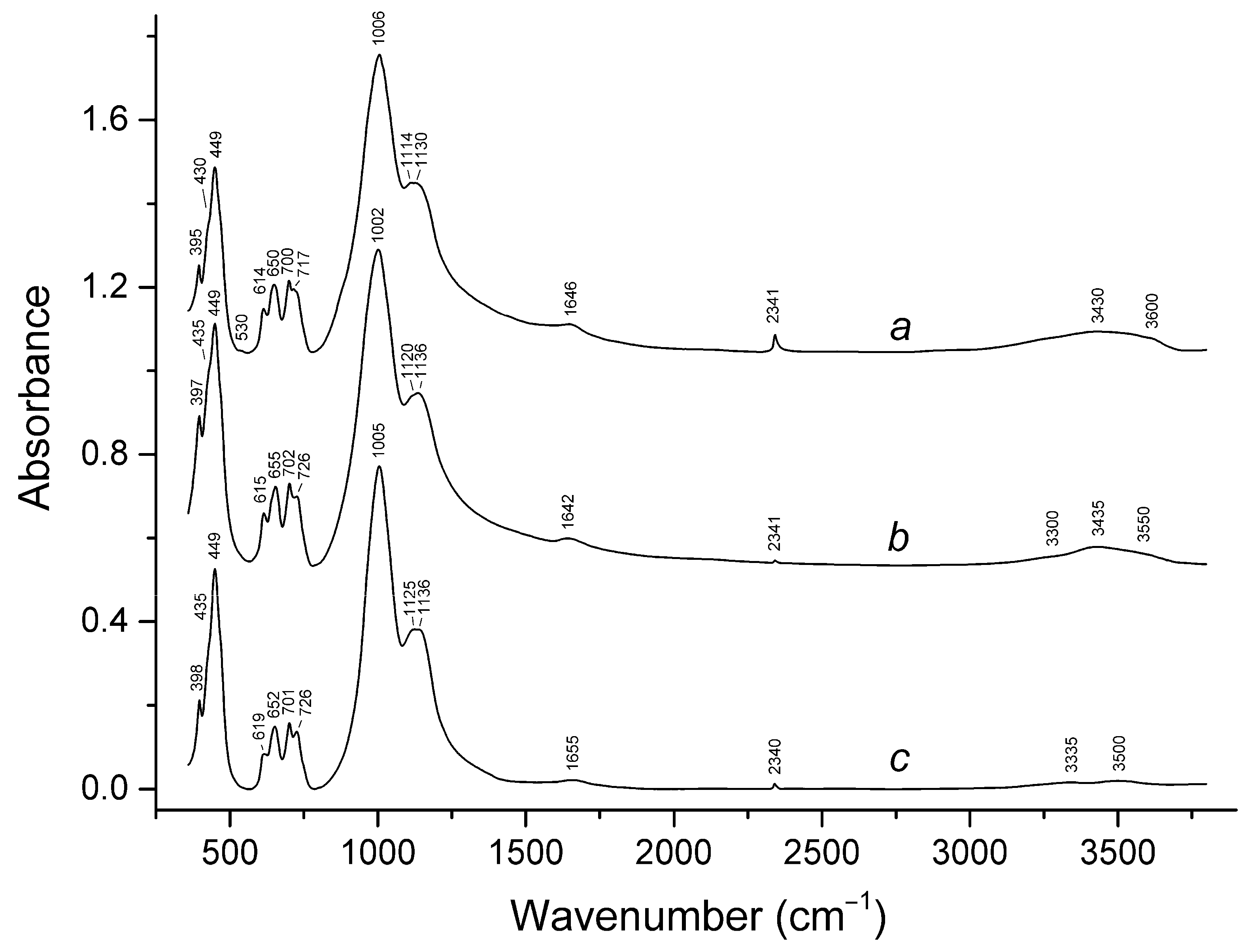
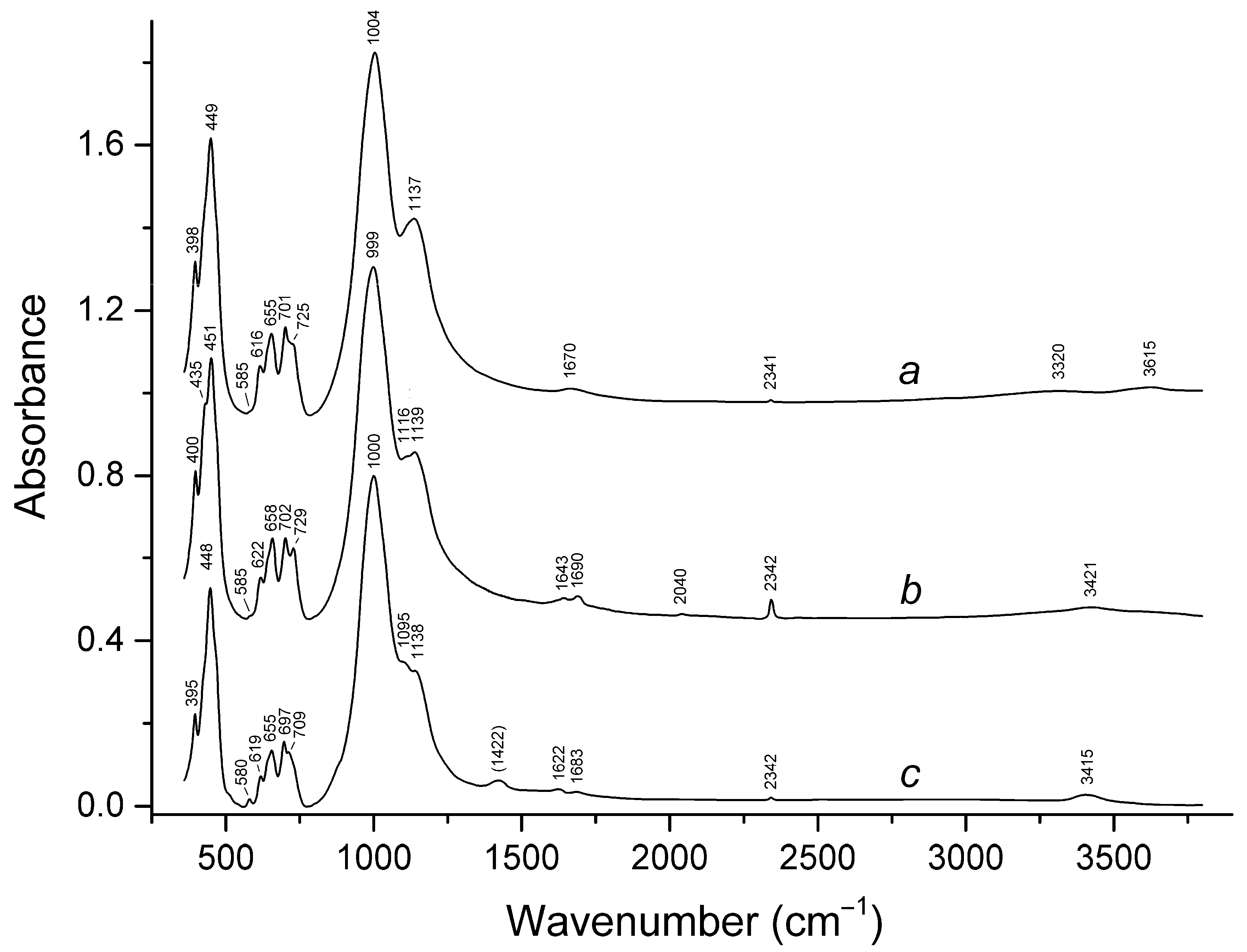
3.2. Infrared Spectroscopy
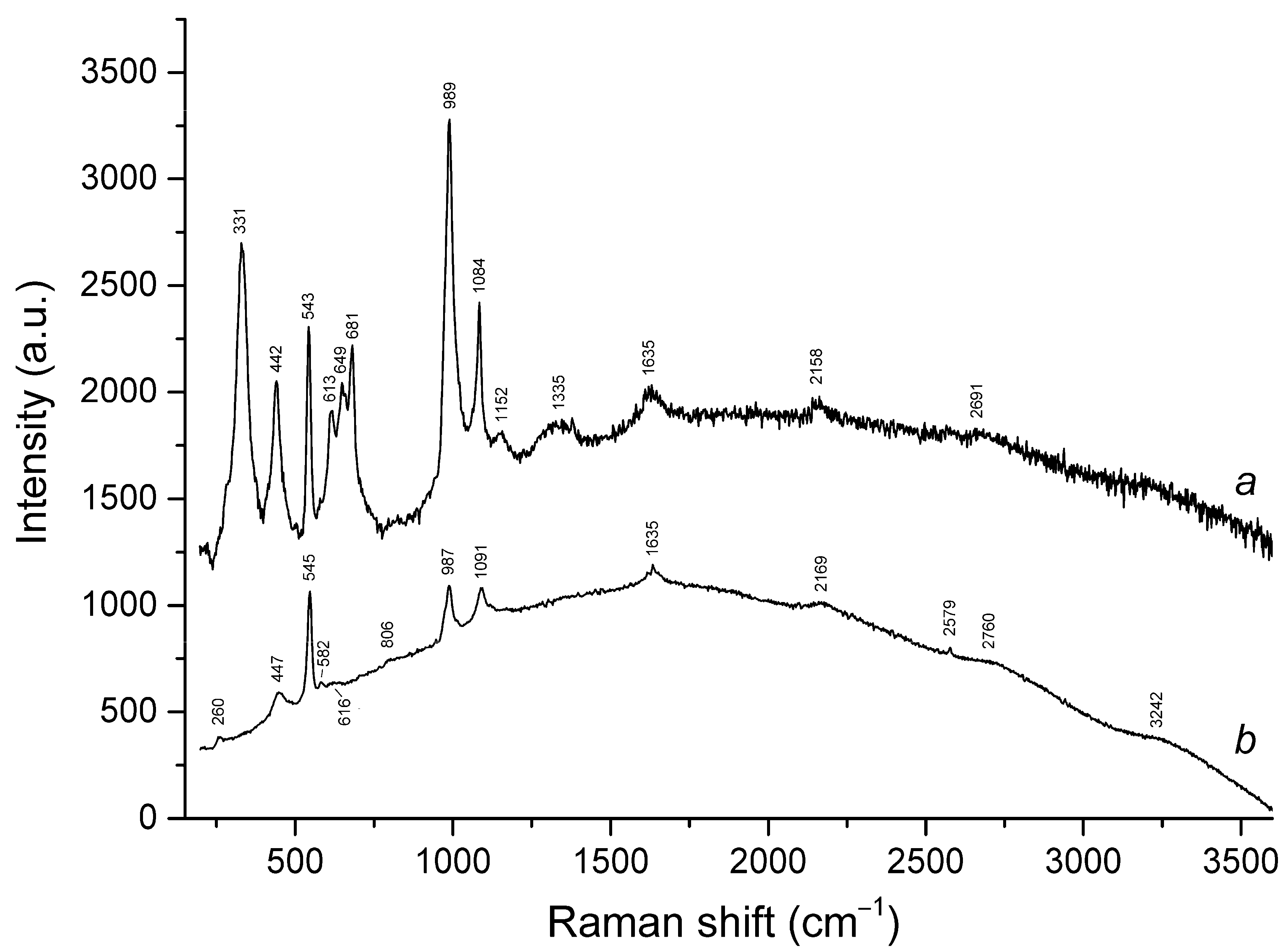
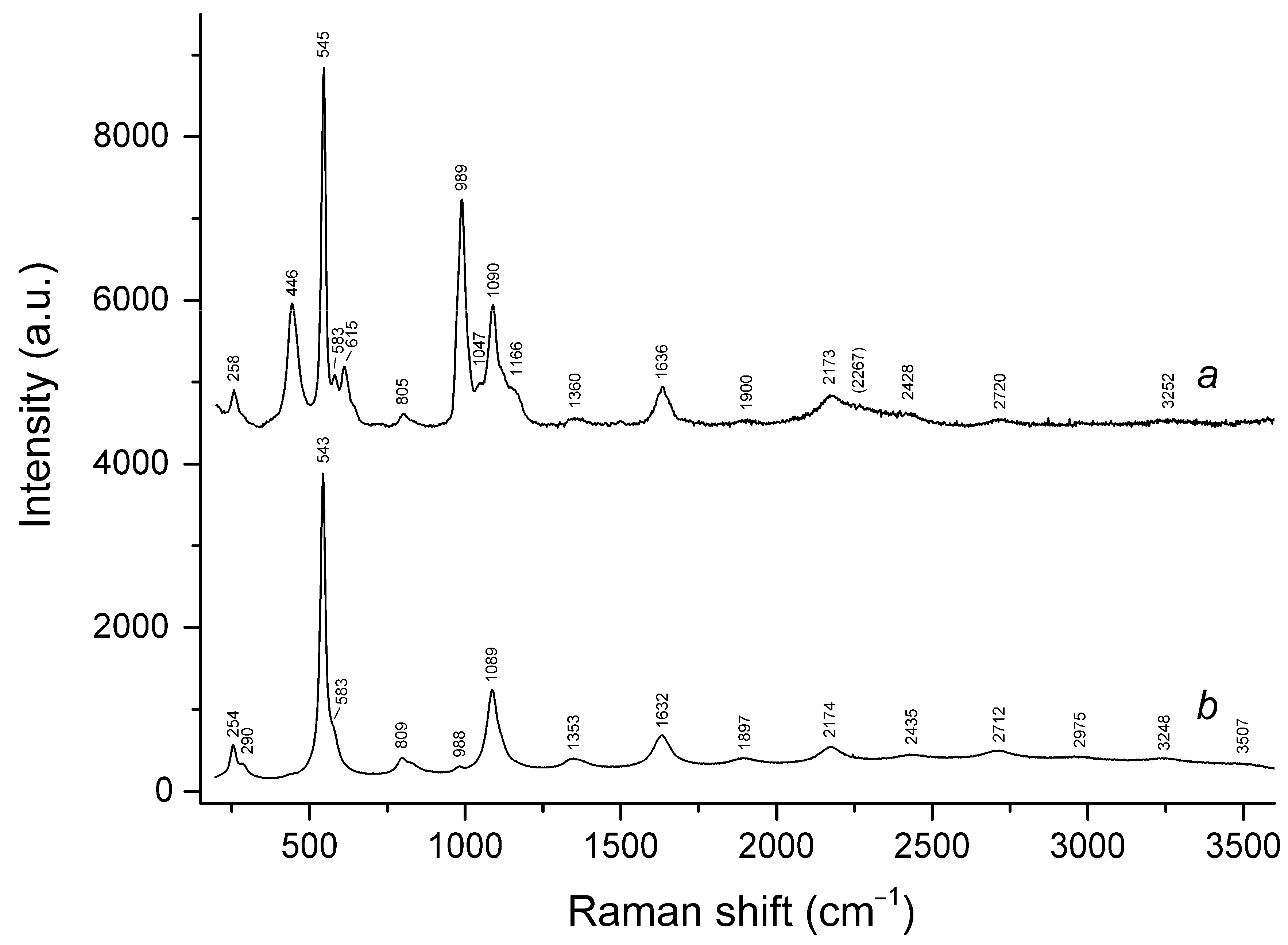
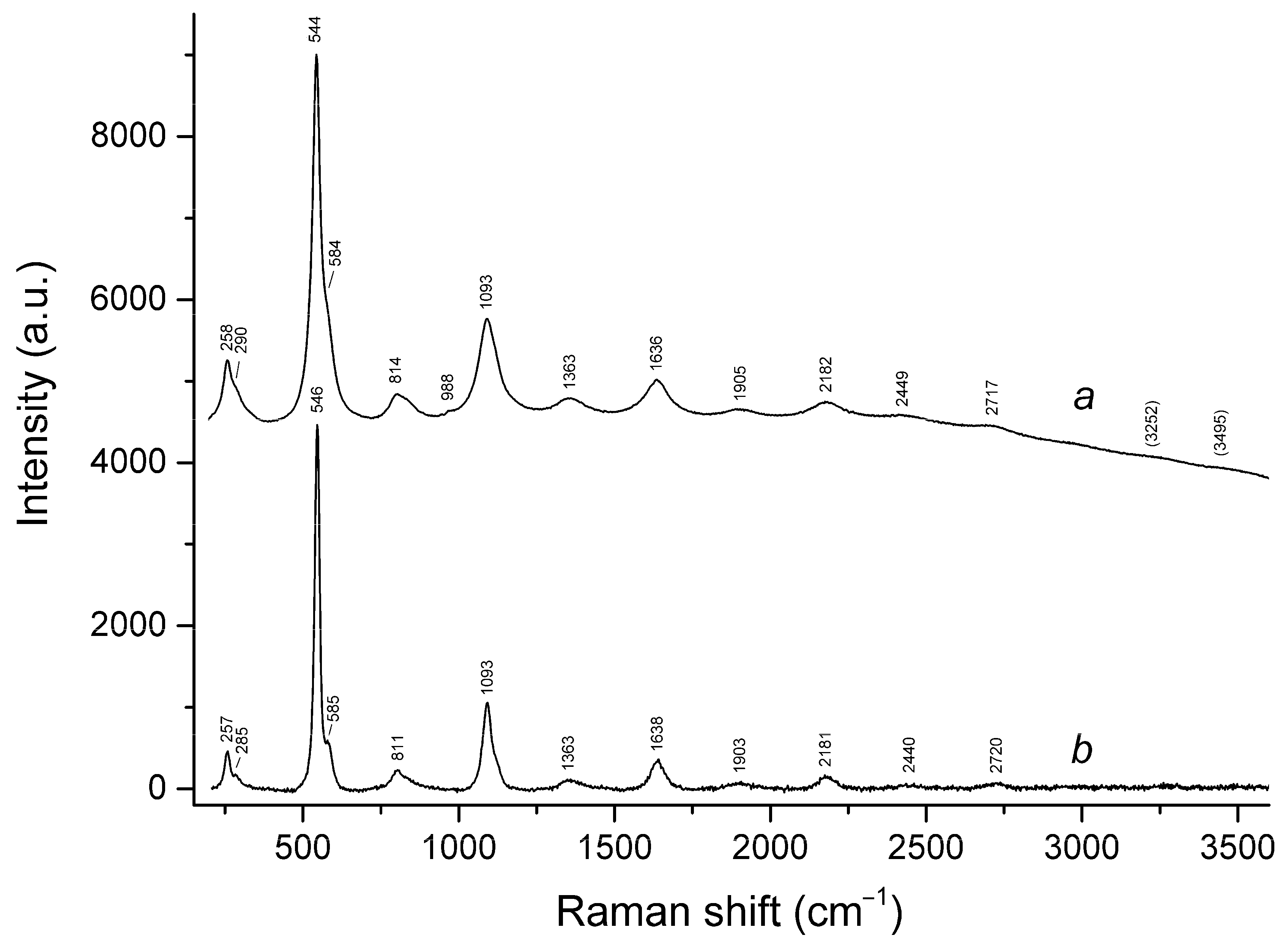
3.3. Raman Spectroscopy
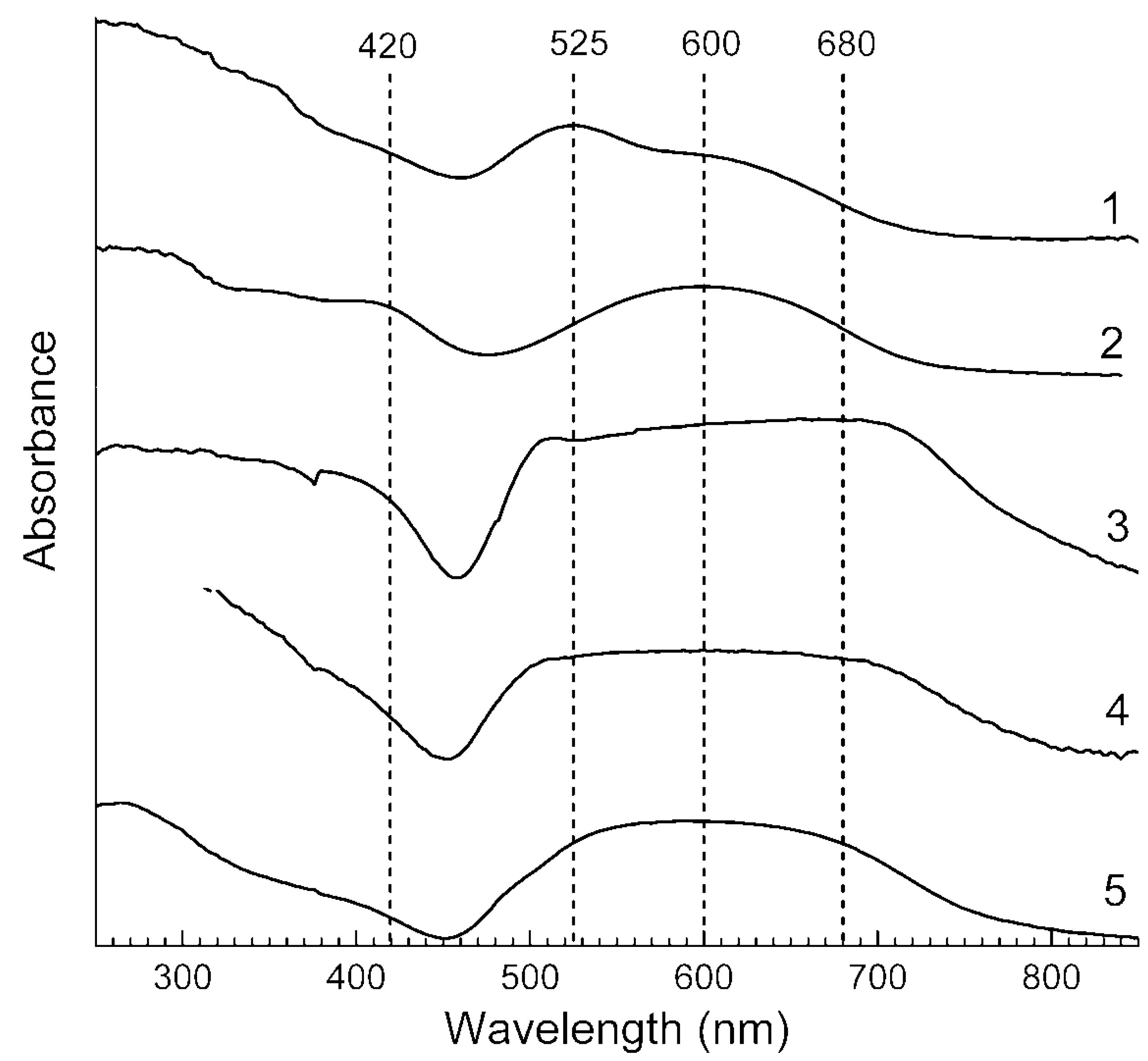
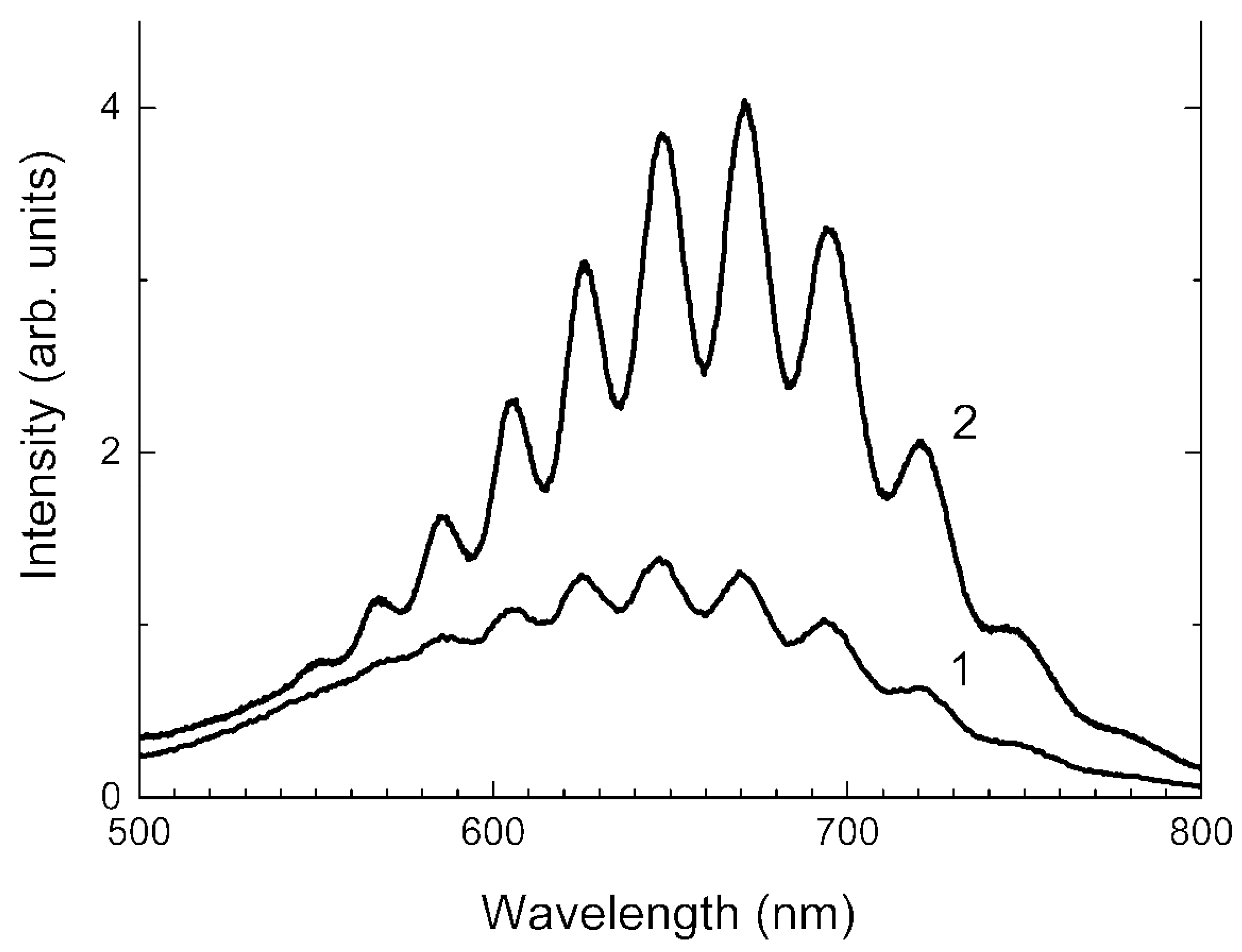
3.4. Absorption Spectra in the Range of 250–1000 nm
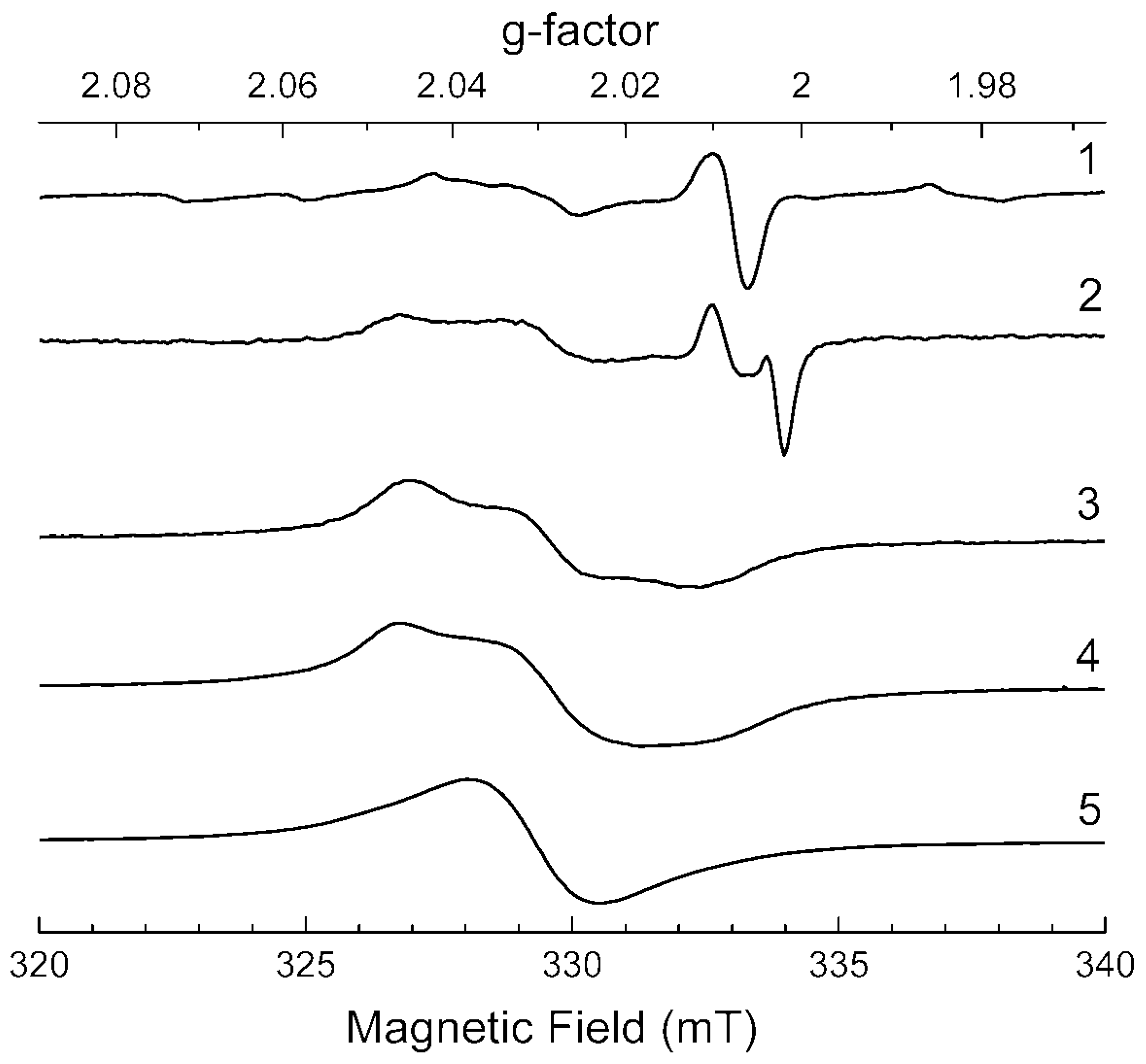
3.5. ESR Spectroscopy
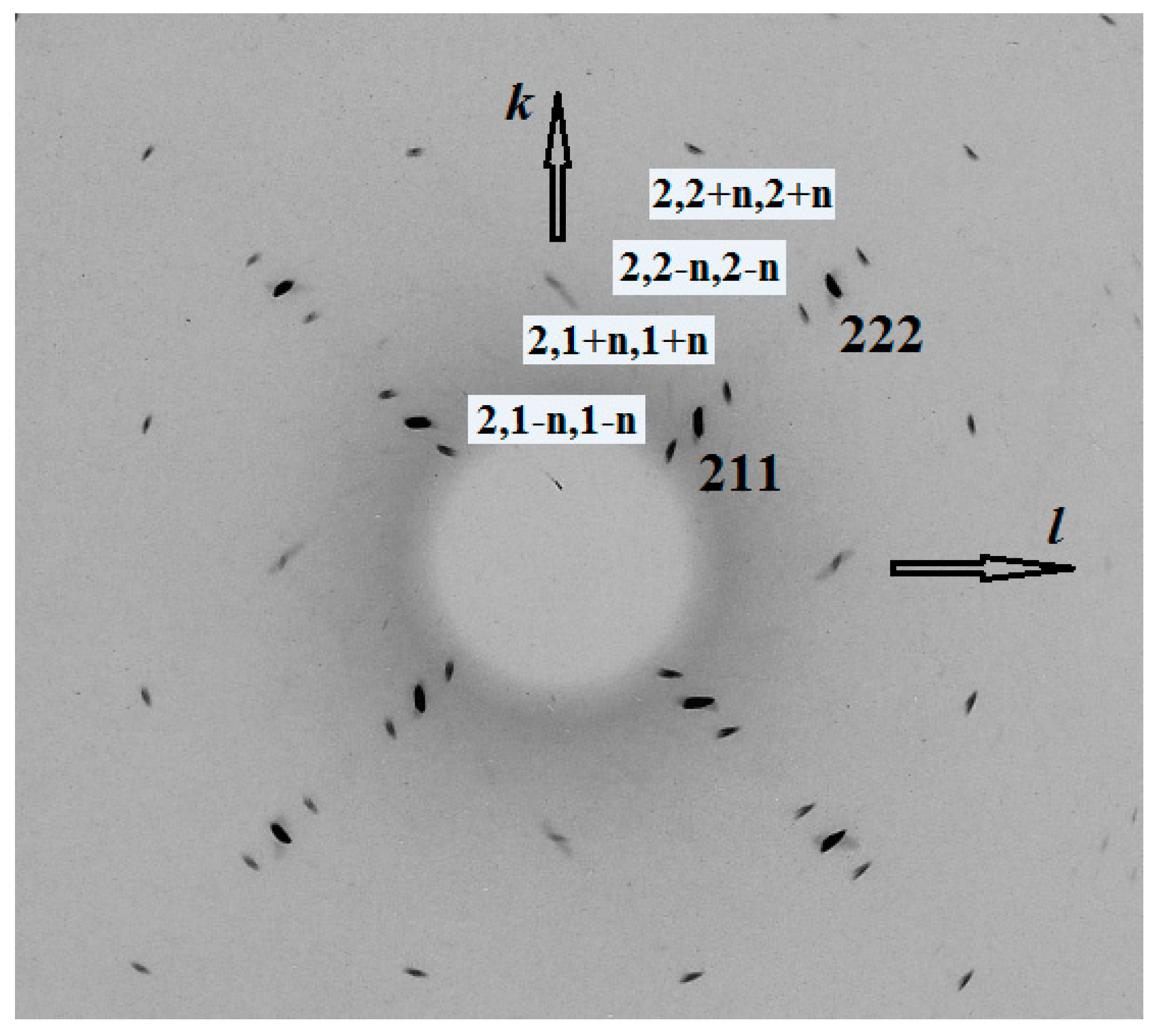
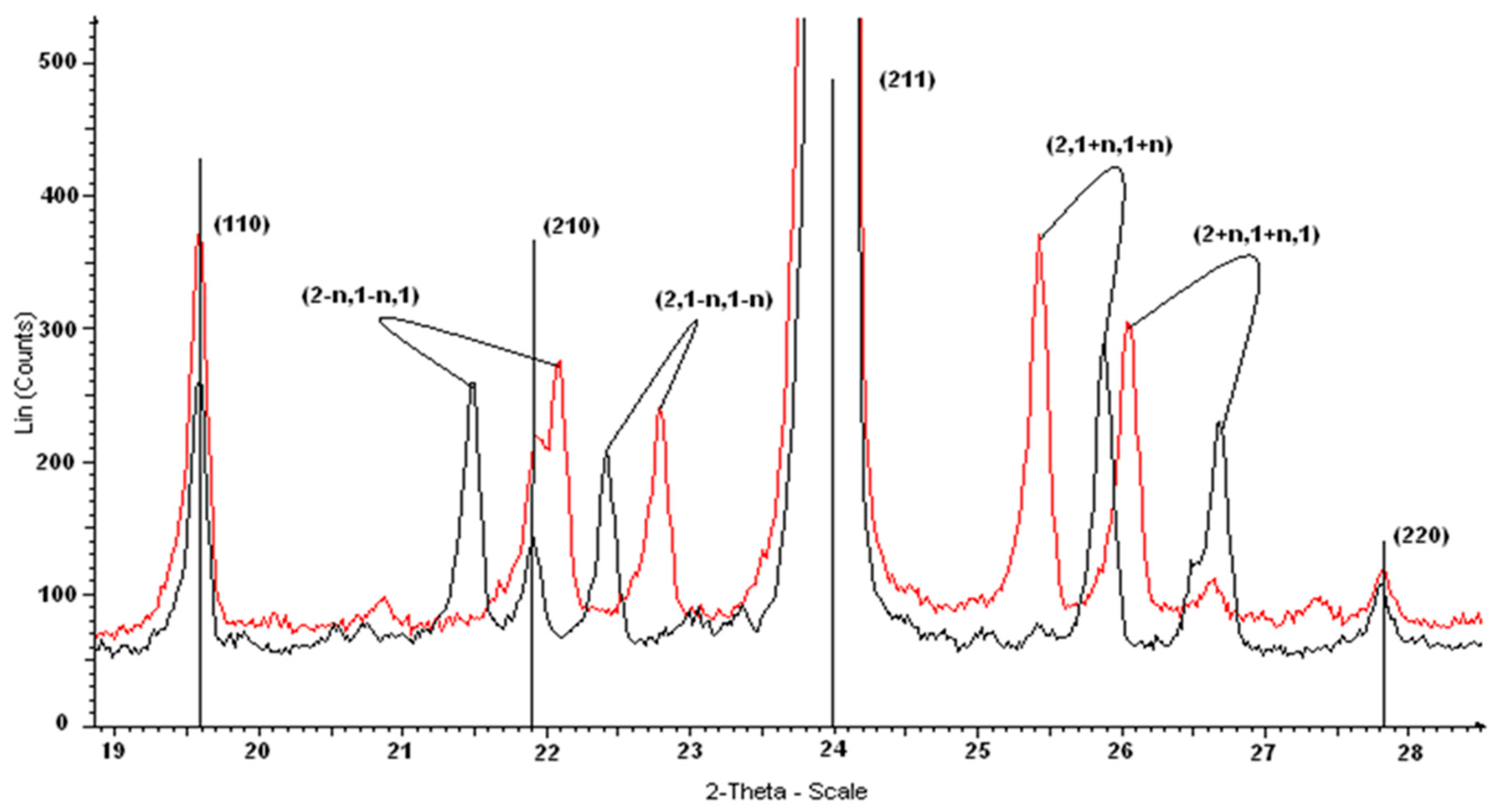
3.6. X-Ray Diffraction
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taylor, D. The sodalite group of minerals. Contr. Mineral. Petrol. 1967, 16, 172–188. [Google Scholar] [CrossRef]
- Sahl, K.; Chatterjee, N.D. The crystal structure of bicchulite, Ca2[Al2SiO6](OH)2. Z. Krist. 1977, 146, 35–41. [Google Scholar] [CrossRef]
- Sahl, K. Refinement of the crystal structure of bicchulite, Ca2[Al2SiO6](OH)2. Z. Krist. 1980, 152, 13–21. [Google Scholar] [CrossRef]
- Uchida, E.; Iiyama, J.T. On kamaishilite, Ca2Al2SiO6(OH)2; A new mineral (tetragonal), dimorphous with bicchulite, from the Kamaishi mine, Japan. Proc. Jpn. Acad. 1981, 57B, 239–243. (In English) [Google Scholar] [CrossRef][Green Version]
- Peterson, R.C. The structure of hackmanite, a variety of sodalite, from Mont St-Hilaire, Quebec. Can. Mineral. 1983, 21, 549–552. [Google Scholar]
- Hassan, I.; Grundy, H.D. The crystal structures of sodalite-group minerals. Acta Cryst. 1984, 40, 6–13. [Google Scholar] [CrossRef]
- Hassan, I.; Buseck, P. Cluster ordering and antiphase domain boundaries in hauyne. Can. Mineral. 1989, 27, 173–180. [Google Scholar]
- Sapozhnikov, A.N.; Tauson, V.L.; Lipko, S.V.; Shendrik, R.Y.; Levitskii, V.I.; Suvorova, L.F.; Chukanov, N.V.; Vigasina, M.F. On the crystal chemistry of sulfur-rich lazurite, ideally Na7Ca(Al6Si6O24)(SO4)(S3)−nH2O. Am. Mineral. 2020. [Google Scholar] [CrossRef]
- Hassan, I.; Peterson, R.C.; Grundy, H.D. The structure of lazurite, ideally Na6Ca2(Al6Si6O24)S2, a member of the sodalite group. Acta Cryst. C 1985, 41, 827–832. [Google Scholar] [CrossRef]
- Gobeltz-Hautecoeur, N.; Demortier, A.; Lede, B.; Lelieur, J.P.; Duhayon, C. Occupancy of the sodalite cages in the blue ultramarine pigments. Inorg. Chem. 2002, 41, 2848–2854. [Google Scholar] [CrossRef]
- Bellatreccia, F.; Della Ventura, G.; Piccinini, M.; Cavallo, A.; Brilli, M. H2O and CO2 in minerals of the hauyne-sodalite group: A FTIR spectroscopy study. Mineral. Mag. 2009, 73, 399–413. [Google Scholar] [CrossRef]
- Fechtelkord, M. Structural study of Na8[AlSiO4]6(CO3)x(HCOO)2-2x(H2O)4x, 0.2 ≤ x ≤ 1, synthesized in organic solvents: Order and disorder of carbonate and formate anions in sodalite. Microporous Mesoporous Mater. 1999, 28, 335–351. [Google Scholar] [CrossRef]
- Gesing, T.M.; Buhl, J.C. Crystal structure of a carbonate-nosean Na8[AlSiO4]6CO3. Eur. J. Mineral. 1998, 10, 71–77. [Google Scholar] [CrossRef]
- Yakubovich, O.V.; Kotel’nikov, A.R.; Shchekina, T.I.; Gramenitskiy, E.N.; Zubkov, E.S. New representative in the sodalite structure type with extraframework anions [AlF6]3–. Crystallogr. Rep. 2011, 56, 190–197. [Google Scholar] [CrossRef]
- Gramenitskii, E.N.; Kotel’nikov, A.R.; Shchekina, T.I.; Yakubovich, O.V.; Devyatova, V.N.; Zubkov, E.S.; Suk, N.I.; Vigasina, M.F.; Kotel’nikova, Z.A. Composition, structure and conditions of formation of fluorine-containing sodalite (experimental data). Geochem. Int. 2018, 56, 521–534. [Google Scholar] [CrossRef]
- Hassan, I.; Grundy, H.D. The crystal structure of basic cancrinite, ideally Na8[Al6Si6O24](OH)2·3H2O. Can. Mineral. 1991, 29, 123–130. [Google Scholar]
- Ivanov, V.G.; Sapozhnikov, A.N. Lazurites of the USSR; Nauka: Novosibirsk, Russia, 1985. (In Russian) [Google Scholar]
- IMA List of Minerals. Available online: http://cnmnc.main.jp/ (accessed on 22 November 2020).
- Deer, W.A.; Howie, R.A.; Wise, W.S.; Zussman, J. Rock-Forming Minerals. Volume 4B. Framework Silicates: Silica Minerals. Feldspathoids and the Zeolites; The Geological Society: London, UK, 2004. [Google Scholar]
- Tauson, V.L.; Goettlicher, J.; Sapozhnikov, A.N.; Mangold, S.; Lustenberg, E.E. Sulfur speciation in lazurite-type minerals (Na,Ca)8[Al6Si6O24](SO4,S)2 and their annealing products: A comparative XPS and XAS study. Eur. J. Miner. 2012, 24, 133–152. [Google Scholar] [CrossRef]
- Chukanov, N.V.; Vigasina, M.F.; Zubkova, N.V.; Pekov, I.V.; Schäfer, C.; Kasatkin, A.V.; Yapaskurt, V.O. New aspects of the application of infrared and Raman spectroscopy to the characterization of extra-framework components in sodalite-group minerals. Minerals 2020, 10, 363. [Google Scholar] [CrossRef]
- Chivers, T.; Elder, P.J.W. Ubiquitous trisulfur radical anion: Fundamentals and applications in materials science, electrochemistry, analytical chemistry and geochemistry. Chem. Soc. Rev. 2013, 42, 5996–6005. [Google Scholar] [CrossRef]
- Tauson, V.L.; Sapozhnikov, A.N.; Shinkareva, S.N.; Lustenberg, E.E. Indicative properties of lazurite as a member of clathrasil mineral family. Doklady Earth Sci. 2011, 441, 1732–1737. [Google Scholar] [CrossRef]
- Platonov, A.N.; Tarashchan, A.N.; Belichenko, V.P.; Povarennikh, A.S. Spectroscopic study of sulfide sulfur in some framework aluminosilicates. Const. Prop. Miner. 1971, 5, 61–72. (In Russian) [Google Scholar]
- Samoilovich, M.I. An ESR study of sulfur-bearing radical ions in minerals. Geokhimiya 1971, 4, 477–483. (In Russian) [Google Scholar]
- Steudel, R. Inorganic polysulfides Sn2− and radical anions Sn•−. In Elemental Sulfur und Sulfur-Rich Compounds II. Topics in Current Chemistry; Steudel, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2003; Volume 231. [Google Scholar]
- Climent-Pascual, E.; de Paz, J.R.; Rodríguez-Carvajal, E.; Suard, E.; Sáez-Puche, R. Synthesis and characterization of the ultramarine-type analog Na8-x[Si6Al6O24]·(S−2,S−3,CO3)(1–2). Inorg. Chem. 2009, 48, 6526–6533. [Google Scholar] [CrossRef] [PubMed]
- Steudel, R.; Chivers, T. The role of polysulfide dianions and radical anions in the chemical, physical and biological sciences, including sulfur-based batteries. Chem. Soc. Rev. 2019, 48, 3279–3319, 4338. [Google Scholar] [CrossRef]
- Evsyunin, V.G.; Sapozhnikov, A.N.; Kashaev, A.A.; Rastsvetaeva, R.K. Crystal Structure of Triclinic Lazurite. Crystallogr. Rep. 1997, 42, 938–945. [Google Scholar] [CrossRef]
- Ostroumov, E.; Fritsch, E.; Faulques, E.; Chauvet, O. Etude spectrometrique de la lazurite du Pamir, Tajikistan. Can. Mineral. 2002, 40, 885–893. (In French) [Google Scholar] [CrossRef][Green Version]
- Eckert, B.; Steudel, F. Molecular spectra of sulfur molecules and solid sulfur allotropes. Top. Curr. Chem. 2003, 231, 31–97. [Google Scholar] [CrossRef]
- Rejmak, P. Computational refinement of the puzzling red tetrasulfur chromophore in ultramarine pigments. Phys. Chem. Chem. Phys. 2020. [Google Scholar] [CrossRef]
- Sapozhnikov, A.N. Modulated structure of lazurite from deposits in southwestern Pamir. Sov. Phys. Crystallogr. 1992, 37, 470–472. [Google Scholar]
- Sofich, D.; Tushinova, Y.L.; Shendrik, R.; Bazarov, B.G.; Dorzhieva, S.G.; Chimitova, O.D.; Bazarova, J.G. Optical spectroscopy of molybdates with composition Ln2Zr3(MoO4)9 (Ln: Eu, Tb). Opt. Mater. 2018, 81, 71–77. [Google Scholar] [CrossRef]
- Kaneva, E.; Shendrik, R.; Mesto, E.; Bogdanov, A.; Vladykin, N. Spectroscopy and crystal chemical properties of NaCa2[Si4O10]F natural agrellite with tubular structure. Chem. Phys. Lett. 2020, 738, 136868. [Google Scholar] [CrossRef]
- Kaneva, E.V.; Shendrik, R.; Radomskaya, T.A.; Suvorova, L.F. Fedorite from Murun Alkaline Complex (Russia): Spectroscopy and crystal chemical features. Minerals 2020, 10, 702. [Google Scholar] [CrossRef]
- Chukanov, N.V.; Chervonnyi, A.D. Infrared Spectroscopy of Minerals and Related Compounds; Springer: Cham, Switzerland, 2016; 1109p. [Google Scholar] [CrossRef]
- Clark, R.J.H.; Cobbold, D.G. Characterization of sulfur radical-ions in solutions of alkalipolysulfides in dimethylformamide and hexamethylphosphoramide and in solid-state in ultramarine blue, green, and red. Inorg. Chem. 1978, 17, 3169–3174. [Google Scholar] [CrossRef]
- Li, S.; Liu, M.; Sun, L. Preparation of acid-resisting ultramarine blue by novel two-step silica coating process. Ind. Eng. Chem. Res. 2011, 50, 7326–7331. [Google Scholar] [CrossRef]
- Chukanov, N.V. Infrared Spectra of Mineral Species: Extended Library; Springer-Verlag GmbH: Dordrecht, The Netherlands, 2014; 1716p, ISBN 9400771274. [Google Scholar]
- Wong, M.W.; Steudel, R. Structure and spectra of tetrasulfur S4—An ab initio MO Study. Chem. Phys. Lett. 2003, 379, 162–169. [Google Scholar] [CrossRef]
- Tubergen, M.J.; Lavrich, R.J.; McCargar, J.W. Infrared Spectrum and Group Theoretical Analysis of the Vibrational Modes of Carbonyl Sulfide. J. Chem. Ed. 2000, 77, 1637–1639. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 4th ed.; A Wiley-Interscience Publication: New York, NY, USA, 1986. [Google Scholar]
- Ledé, B.; Demortier, A.; Gobeltz-Hautecœur, H.; Lelieur, J.-P.; Picquenard, E.; Duhayo, C. Observation of the ν3 Raman band of S3− inserted into sodalite cages. J. Raman Spectrosc. 2007, 38, 1461–1468. [Google Scholar] [CrossRef]
- Rolfe, J. Emission spectra of S2−, Se2−, and SeS− ions in KI Crystals. J. Chem. Phys. 1968, 49, 4193–4197. [Google Scholar] [CrossRef]
- Goslar, J.; Lijewski, S.; Hoffmann, S.K.; Jankowska, A.; Kowalak, S. Structure and dynamics of S3− radicals in ultramarine-type pigment based on zeolite A: Electron spin resonance and electron spin echo studies. J. Chem. Phys. 2009, 130, 204504. [Google Scholar] [CrossRef]
- Pinon, V.; Levillain, E.; Lelieur, J. The S3– radical as a standard for ESR experiments. J. Magn. Reson. 1992, 96, 31–39. [Google Scholar] [CrossRef]
- Kowalak, S.; Jankowska, A.; Zeidler, S.; Wiećkowski, A.B. Sulfur radicals embedded in various cages of ultramarine analogs prepared from zeolites. J. Solid State Chem. 2007, 180, 1119–1124. [Google Scholar] [CrossRef]
- Vassilikou-Dova, A.B.; Lehmann, G. Paramagnetic defects in the mineral hauyne. Cryst. Res. Technol. 1990, 25, 525–529. [Google Scholar] [CrossRef]
- Barabas, M. The nature of the paramagnetic centres at g = 2.0057 and g = 2.0031 in marine carbonates. Intern. J. Radiat. Appl. Instrum. Part D Nucl. Tracks Radiat. Meas. 1992, 20, 453–464. [Google Scholar] [CrossRef]
- Sapozhnikov, A.N. Indication of additional reflections on lazurite debyegrams in connection with the study of modulation of its structure. Zapiski Vsesoyuznogo Mineral. Obs. 1990, 119, 110–116. (In Russian) [Google Scholar]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | LSh | MD | Hn7981 | PH | KL | ZK |
|---|---|---|---|---|---|---|
| Color | Lilac | Light blue | Light blue | Bright blue | Deep blue | Dark blue |
| Contents of components (wt%) | ||||||
| Na2O | 17.61 | 18.09 | 15.06 | 18.05 | 20.01 | 19.94 |
| K2O | 0.25 | 0.13 | 1.42 | 0.02 | 0.20 | 0.43 |
| CaO | 7.81 | 6.84 | 9.84 | 6.91 | 5.18 | 4.56 |
| Al2O3 | 26.86 | 27.35 | 26.07 | 27.35 | 28.25 | 28.02 |
| SiO2 | 32.49 | 33.01 | 32.25 | 32.91 | 34.20 | 33.48 |
| CO2 a | 0.62 | 0.10 | 0.21 | 0.09 | 0.62 | 0.13 |
| SO3 | 13.78 | 12.43 | 13.89 b | 13.89 | 8.67 | 8.08 |
| S | 0.06 | 0.11 | No data | 1.31 | 2.34 | 5.07 |
| Cl | 0.17 | 0.50 | 0.34 | 0.28 | 0.71 | 0.13 |
| −O=(Cl,S) | −0.04 | −0.12 | −0.08 | −0.07 | −0.07 | −0.33 |
| Total | 99.61 | 98.44 | 100.08 c | 100.74 | 100.11 | 100.71 d |
| Formula coefficients calculated on Al + Si + Fe = 12 atoms per formula unit and unit-cell parameter a (Å) | ||||||
| Na | 6.39 | 6.45 | 5.52 | 6.45 | 6.90 | 6.97 |
| K | 0.06 | 0.03 | 0.35 | 0.01 | 0.05 | 0.10 |
| Ca | 1.57 | 1.35 | 1.99 | 1.36 | 0.99 | 0.88 |
| Al | 5.92 | 5.93 | 5.81 | 5.94 | 5.92 | 5.96 |
| Si | 6.08 | 6.07 | 6.10 | 6.06 | 6.08 | 6.04 |
| C | 0.15 | 0.02 | 0.05 | 0.02 | 0.15 | 0.03 |
| SO4 | 1.93 | 1.72 | 1.97 | 1.56 | 1.16 | 1.09 |
| S | 0.02 | 0.04 | No data | 0.45 | 0.78 | 1.70 |
| Cl | 0.07 | 0.16 | 0.11 | 0.09 | 0.21 | 0.04 |
| a | 9.076 | 9.067 | No data | 9.071 | 9.066 | 9.087 |
| Excess of negative charge in the empirical formula calculated on [(SO4)2−, S2−] | ||||||
| −0.32 | −0.43 | −0.10 | −0.88 | −1.07 | −2.77 | |
| Kinds and amounts (pfu) of S-bearing extra-framework components determined based on spectroscopic data (see below) and the charge-balance condition | ||||||
| SO42− | 1.78 | 1.35 | 1.91 | 1.56 | 1.16 | 1.09 |
| SO3•− | 0.37 | |||||
| S2− | 0.02 | 0.12 | 0.05 | |||
| S2•− | 0.02 | 0.03 | ||||
| S3•− | 0.03 | 0.22 | 0.55 | |||
| S4 | 0.03 | 0.09 | ||||
| Sample | Assignment | |||||
|---|---|---|---|---|---|---|
| LSh | MD | Hn7981 | PH | KL | ZK | |
| Raman Shift (cm−1) | ||||||
| 260sh | 260w | 258 | 254w | 258 | 257 | S3•− bending (ν2) |
| 290sh | 290sh | 285w | Combination of low-frequency lattice modes | |||
| 331s | cis-S4 symmetric stretching (ν1 − A1) | |||||
| 442s | 447 | 446s | 441s | SO4 bending and/or δ[O–Si(Al)–O] | ||
| 543s | 545s | 545s | 543s | 544s | 546s | S3•− symmetric stretching (ν1) (possibly, overlapping with the stretching band of gauche- or trans-S4) |
| 582w | 583w | 584sh | 584sh | 585sh | S3•− antisymmetric stretching (ν3) (possibly, overlapping with the stretching band of S2•−) | |
| 613 | 616w | 615 | SO4 bending (ν4 − F2) | |||
| 649 | trans-S4 or gauche-S4 symmetric stretching (ν1 − A1) | |||||
| 681s | cis-S4 antisymmetric stretching (ν3 − E) | |||||
| 806w | 805 | 809 | 814 | 811 | S3•− combination mode (ν1 + ν2) | |
| 989s | 987s | 989s | 988w | 988w | SO4 symmetric stretching (ν1 − A1) | |
| 1047w | Framework stretching vibrations ? | |||||
| 1084s | 1091s | 1090s | 1089s | 1093s | 1093s | S3•− overtone (2 × ν1) [possibly, overlapping with SO4•− stretching band (ν3 − F2)] |
| 1152w | SO4 asymmetric (ν3 − F2) mode [possibly, overlapping with S2•− overtone (2 × ν1)] | |||||
| 1160sh | 1166sh | S2•− overtone (2 × ν1) | ||||
| 1335br | cis-S4 antisymmetric stretching (2 × ν3) | |||||
| 1360 | 1353 | 1362 | 1363 | S3•− combination mode (2ν1 + ν2) | ||
| 1635 | 1635 | 1636 | 1632 | 1636 | 1638 | S3•− overtone (3 × ν1) |
| 1900w | 1897 | 1905w | 1903 | S3•− combination mode (3 × ν1 + ν2) | ||
| 2158w | 2169 | 2173 | 2174 | 2182 | 2181 | S3•− overtone (4 × ν1) |
| 2267sh | Assignment is ambiguous | |||||
| 2428 | 2434 | 2449w | 2440w | S3•− combination mode (4 × ν2 + ν1) | ||
| 2579w | S–H stretching ? | |||||
| 2691w | cis-S4 antisymmetric stretching (4 × ν3) | |||||
| 2760br | 2720w | 2712w | 2717w | 2720 | S3•− overtone (5 × ν1) | |
| 2975w | S3•− combination mode (5 × ν1 + ν2) | |||||
| 3242br | 3252br | 3249w | 3251w | S3•− overtone (6 × ν1) | ||
| 3647br | 3507 | 3495w | H2O stretching | |||
| h, k, l * | dmeas (Å) | dcalc (Å) | I (%) | h, k, l * | dmeas (Å) | dcalc (Å) | I (%) |
|---|---|---|---|---|---|---|---|
| 1 1 0 | 6.42 | 6.41 | 13.5 | 3 + n, 3, n | 2.060 | 2.060 | 3.8 |
| 1 + n, 1, n | 5.72 | 5.71 | 4.4 | 4 + n, 1 + n, 1 | 2.015 | 2.016 | 2.5 |
| 2–0.5, 1–0.5, 1 | 4.859 | 4.847 | 3.4 | 3 3 2 | 1.9328 | 1.9331 | 3.3 |
| 2 0 0 | 4.538 | 4.534 | 5.8 | 4 2 2 | 1.8487 | 1.8508 | 2.2 |
| 2 − n, 1 − n, 1 | 4.139 | 4.134 | 4.5 | 4 + n, 2 − n, 2 | 1.8144 | 1.8154 | 1.9 |
| 2 1 0 | 4.058 | 4.055 | 3.6 | 5 1 0, 4, 3, 1 | 1.7781 | 1.7782 | 7.6 |
| 2, 1 − n, 1 − n | 3.961 | 3.962 | 4.2 | 4, 3 + n, 1 − n | 1.7482 | 1.7466 | 1.8 |
| 2–0.5, 1 + 0.5, 1 | 3.862 | 3.866 | 2.9 | 5 + n, 1 − n, 0 | 1.7188 | 1.7197 | 1.6 |
| 2 1 1 | 3.703 | 3.702 | 100 | 5 2 1 | 1.6553 | 1.6554 | 2.8 |
| 2 + n, 1 − n, 1 | 3.551 | 3.551 | 3 | 4 − n, 4, n | 1.6446 | 1.6449 | 2 |
| 2, 1 + n, 1 + n | 3.440 | 3.441 | 4.5 | 4 − n, 3 − n, 3 | 1.6253 | 1.6258 | 1.6 |
| 2 + n,1 + n,1 | 3.339 | 3.339 | 4.3 | 4 4 0 | 1.6028 | 1.6029 | 7.2 |
| 2 2 0 | 3.203 | 3.206 | 3.2 | 4 − n, 3 + n, 3 | 1.5551 | 1.5596 | 3.8 |
| 2–0.5, 2 + 0.5, 0 | 3.111 | 3.110 | 2.9 | 4 4 2 | 1.5109 | 1.5112 | 3.5 |
| 2 + 0.5, 1 + 0.5, 1 | 2.943 | 2.942 | 2.9 | 6 1 1 | 1.4710 | 1.4709 | 4.3 |
| 3 1 0 | 2.868 | 2.867 | 13.9 | 4 + n, 4, 2 + n | 1.4586 | 1.4584 | 1.7 |
| 2, 2 − n, 2 − n | 2.816 | 2.814 | 4.2 | 5, 4 − n, 1 + n | 1.4199 | 1.4193 | 1.7 |
| 3 + n, 1 − n, 0 | 2.732 | 2.740 | 2.7 | 5 4 1 | 1.3988 | 1.3991 | 1.6 |
| 2 2 2 | 2.618 | 2.617 | 25.9 | 6 − n, 2, 2 + n | 1.3926 | 1.3927 | 1.6 |
| 2 + n, 2 + n, 2 | 2.441 | 2.441 | 3.8 | 6 2 2 | 1.3669 | 1.3669 | 5.4 |
| 3 2 1 | 2.423 | 2.423 | 7.9 | 6 3 1 | 1.3371 | 1.3369 | 2.2 |
| 4 0 0 | 2.267 | 2.267 | 6.9 | 4 4 4 | 1.3085 | 1.3087 | 3.5 |
| 4 − n, 1 + n, 1 | 2.211 | 2.211 | 3.3 | 7 1 0 | 1.2825 | 1.2823 | 2.2 |
| 4, 1 − n, 1 − n | 2.183 | 2.184 | 2.6 | 7, 2 − n, 1 − n | 1.2479 | 1.2477 | 1.4 |
| 4 1 1 | 2.137 | 2.137 | 16.1 | 7 2 1 | 1.2339 | 1.2339 | 3.7 |
| 3 + 2n, 2 + 2n, 1 | 2.092 | 2.100 | 2.7 | 7, 2 + n, 1 + n | 1.2184 | 1.2185 | 1.4 |
| h, k, l * | dmeas (Å) | dcalc (Å) | I (%) | h, k, l * | dmeas (Å) | dcalc (Å) | I (%) |
|---|---|---|---|---|---|---|---|
| 1 1 0 | 6.43 | 6.41 | 12,6 | 5 − n, 2 + n, 1 | 1.6817 | 1.6813 | 0.9 |
| 1 + n, 1, n | 5.89 | 5.88 | 1,2 | 5, 2 − n, 1 + n | 1.6636 | 1.6627 | 0.8 |
| 2 0 0 | 4.538 | 4.533 | 4,4 | 5 2 1 | 1.6554 | 1.6551 | 2.8 |
| 2 − n, 1 − n, 1 | 4.030 | 4.025 | 3.3 | 4 − n, 4, n | 1.6346 | 1.6349 | 1.9 |
| 2, 1 − n, 1 − n | 3.905 | 3.901 | 2.9 | 5 + n, 2 − n, 1 | 1.6274 | 1.6272 | 0.8 |
| 2 1 1 | 3.704 | 3.701 | 100 | 4 4 0 | 1.6024 | 1.6025 | 8.9 |
| 2, 1 + n, 1 + n | 3.503 | 3.500 | 4.6 | 5, 3 − n, n | 1.5765 | 1.5763 | 0.8 |
| 2 + n, 1 + n, 1 | 3.421 | 3.418 | 3.7 | 4 + n, 4, n | 1.5691 | 1.5695 | 2.1 |
| 2 2 0 | 3.207 | 3.205 | 1.4 | 4 3 3, 5 3 0 | 1.5544 | 1.5547 | 3.8 |
| 3, 1 − n, n | 2.907 | 2.907 | 1.3 | 5, 3 + n, n | 1.5318 | 1.5315 | 0.8 |
| 3 1 0 | 2.869 | 2.867 | 12.7 | 6 0 0, 4 4 2 | 1.5108 | 1.5109 | 4 |
| 3, 1 + n, n | 2.817 | 2.814 | 1.3 | 4 + n, 3 + n, 3 | 1.5033 | 1.5037 | 1 |
| 2, 2 − n, 2 − n | 2.767 | 2.766 | 3.1 | 5 + n, 3 + n, 0 | 1.4970 | 1.4970 | 0.8 |
| 2 2 2 | 2.618 | 2.617 | 27.6 | 5 − n, 3, 2 + n | 1.4887 | 1.4889 | 0.8 |
| 3, 2 − n, 1 − n | 2.508 | 2.507 | 1.2 | 5 3 2 | 1.4704 | 1.4706 | 6 |
| 2 + n, 2 + n, 2 | 2.482 | 2.480 | 3.9 | 5, 3 + n, 2 − n | 1.4638 | 1.4633 | 1 |
| 3 2 1 | 2.424 | 2.423 | 7.3 | 4 + n, 4 + n, 2 | 1.4577 | 1.4577 | 1 |
| 3 + n, 2 − n, 1 | 2.391 | 2.390 | 0.9 | 5 + n, 3, 2 − n | 1.4515 | 1.4509 | 0.9 |
| 3, 2 + n, 1 + n | 2.339 | 2.338 | 1.1 | 5, 3 + n, 2 + n | 1.4388 | 1.4389 | 1 |
| 3 + n, 2, 1 + n | 2.314 | 2.313 | 1 | 5 + n, 3 + n, 2 | 1.4208 | 1.4215 | 0.8 |
| 4 0 0 | 2.267 | 2.266 | 6.2 | 5, 4 − n, 1 + n | 1.4141 | 1.4145 | 0.8 |
| 4 − n, 1 − n, 1 | 2.238 | 2.237 | 1.1 | 5 4 1 | 1.3988 | 1.3988 | 1 |
| 4 − n, 1 + n, 1 | 2.194 | 2.194 | 2.6 | 6 − n, 2 + n, 2 | 1.3862 | 1.3866 | 1.3 |
| 4, 1 − n, 1 − n | 2.174 | 2.173 | 1.2 | 6 2 2 | 1.3665 | 1.3666 | 6 |
| 4 1 1 | 2.137 | 2.137 | 16.8 | 6 + n, 2 − n, 2 | 1.3455 | 1.3460 | 1.6 |
| 4 − n, 2, n | 2.094 | 2.096 | 1.3 | 6 3 1 | 1.3366 | 1.3366 | 1.9 |
| 4 + n, 1 − n, 1 | 2.079 | 2.078 | 4 | 6 + n, 2 + n, 2 | 1.3264 | 1.3269 | 0.9 |
| 4 + n, 1 + n, 1 | 2.044 | 2.043 | 2.2 | 4 4 4 | 1.3081 | 1.3085 | 4 |
| 3 − n, 3 − n, 2 | 2.023 | 2.026 | 1 | 5 − n, 5, n | 1.3027 | 1.3028 | 0.7 |
| 3, 3 − n, 2 − n | 2.006 | 2.006 | 1 | 5 − n, 4, 3 + n | 1.2898 | 1.2898 | 0.8 |
| 3 3 2 | 1.9333 | 1.9327 | 2.7 | 7 1 0, 5 5 0 | 1.2819 | 1.2820 | 1.8 |
| 4 − n, 2 + n, 2 | 1.8733 | 1.8741 | 1 | 7, 1 + n, n | 1.2770 | 1.2772 | 0.8 |
| 3, 3 + n, 2 + n | 1.8622 | 1.8625 | 1.2 | 6, 4 − n, n | 1.2725 | 1.2726 | 0.7 |
| 3 + n, 3 + n, 2 | 1.8492 | 1.8498 | 1.3 | 7 − n, 2 − n, 1 | 1.2676 | 1.2680 | 0.6 |
| 4 − n, 3, 1 − n | 1.8344 | 1.8346 | 1.1 | 6 − n, 4 + n, 0 | 1.2645 | 1.2645 | 0.6 |
| 4, 3 − n, 1 − n | 1.8228 | 1.8224 | 1.4 | 6 4 0 | 1.2567 | 1.2571 | 0.8 |
| 4, 3 − n, 1 + n | 1.7991 | 1.7988 | 0.9 | 7, 2 − n, 1 − n | 1.2441 | 1.2444 | 1.2 |
| 4 3 1 | 1.7776 | 1.7778 | 9.1 | 7 2 1 | 1.2334 | 1.2336 | 4.4 |
| 4, 3 + n, 1 − n | 1.7543 | 1.7541 | 0.8 | 7, 2 + n, 1 + n | 1.2218 | 1.2220 | 1.2 |
| 4, 3 + n, 1 + n | 1.7336 | 1.7330 | 1.2 | 6 4 2 | 1.2112 | 1.2114 | 0.9 |
| 4 + n, 3, 1 + n | 1.7229 | 1.7227 | 1.1 | 7 + n, 2, 1 + n | 1.2041 | 1.2042 | 0.7 |
| 5 − n, 2, 1 + n | 1.6922 | 1.6908 | 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chukanov, N.V.; Sapozhnikov, A.N.; Shendrik, R.Y.; Vigasina, M.F.; Steudel, R. Spectroscopic and Crystal-Chemical Features of Sodalite-Group Minerals from Gem Lazurite Deposits. Minerals 2020, 10, 1042. https://doi.org/10.3390/min10111042
Chukanov NV, Sapozhnikov AN, Shendrik RY, Vigasina MF, Steudel R. Spectroscopic and Crystal-Chemical Features of Sodalite-Group Minerals from Gem Lazurite Deposits. Minerals. 2020; 10(11):1042. https://doi.org/10.3390/min10111042
Chicago/Turabian StyleChukanov, Nikita V., Anatoly N. Sapozhnikov, Roman Yu. Shendrik, Marina F. Vigasina, and Ralf Steudel. 2020. "Spectroscopic and Crystal-Chemical Features of Sodalite-Group Minerals from Gem Lazurite Deposits" Minerals 10, no. 11: 1042. https://doi.org/10.3390/min10111042
APA StyleChukanov, N. V., Sapozhnikov, A. N., Shendrik, R. Y., Vigasina, M. F., & Steudel, R. (2020). Spectroscopic and Crystal-Chemical Features of Sodalite-Group Minerals from Gem Lazurite Deposits. Minerals, 10(11), 1042. https://doi.org/10.3390/min10111042