Mirror Symmetry Breaking in Liquids and Their Impact on the Development of Homochirality in Abiogenesis: Emerging Proto-RNA as Source of Biochirality?


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
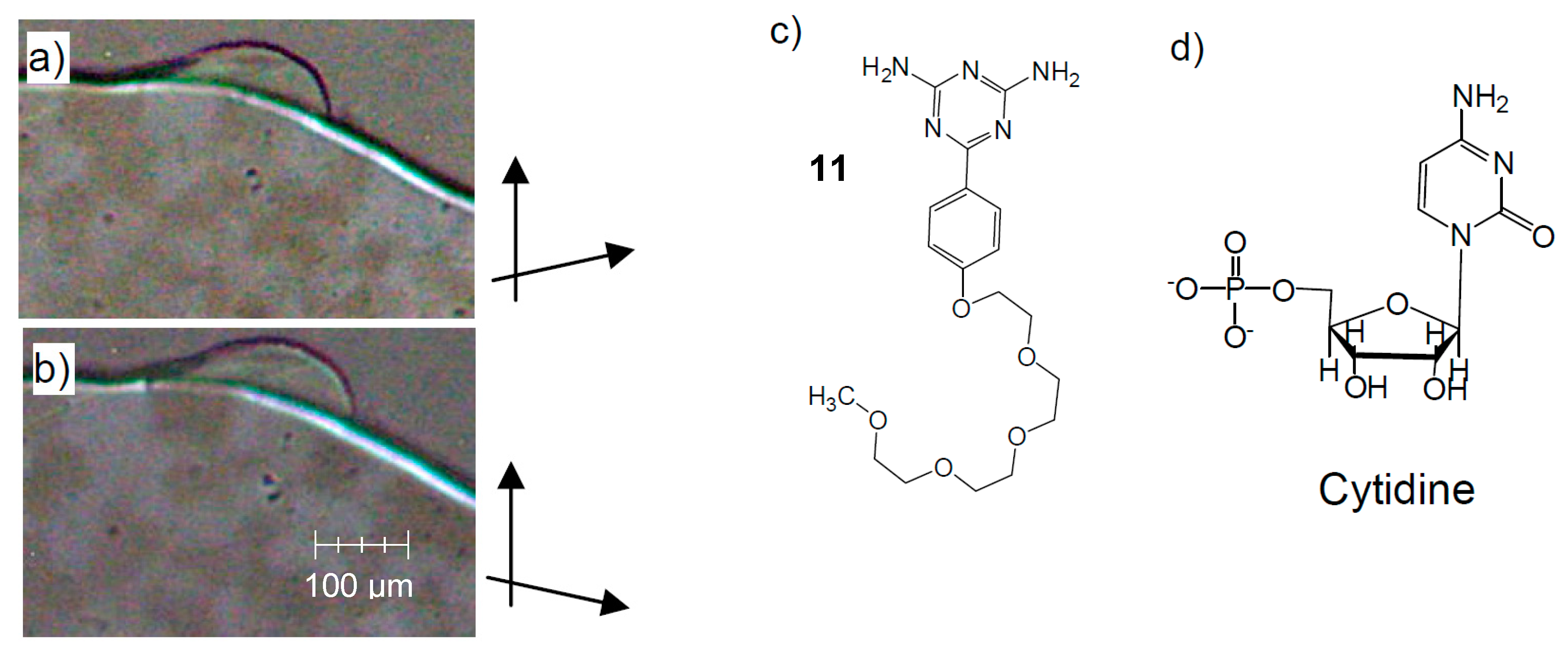{kind=link}
{kind=link}
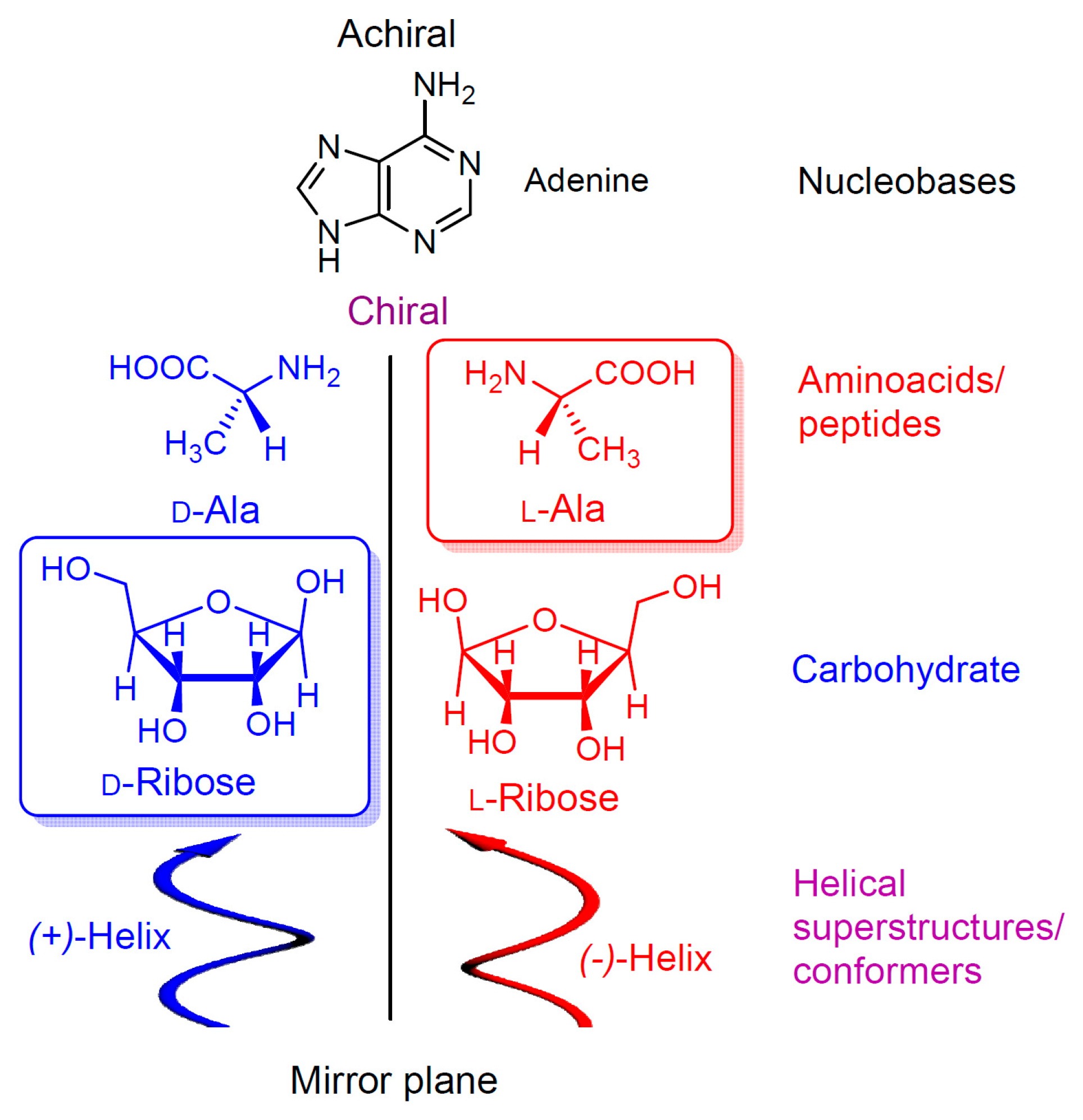
1. Introduction-Homochirality and Life
2. Emergence of Homochirality in Non-Biological Systems-Artificial Chirogenesis
2.1. Racemates vs. Conglomerates
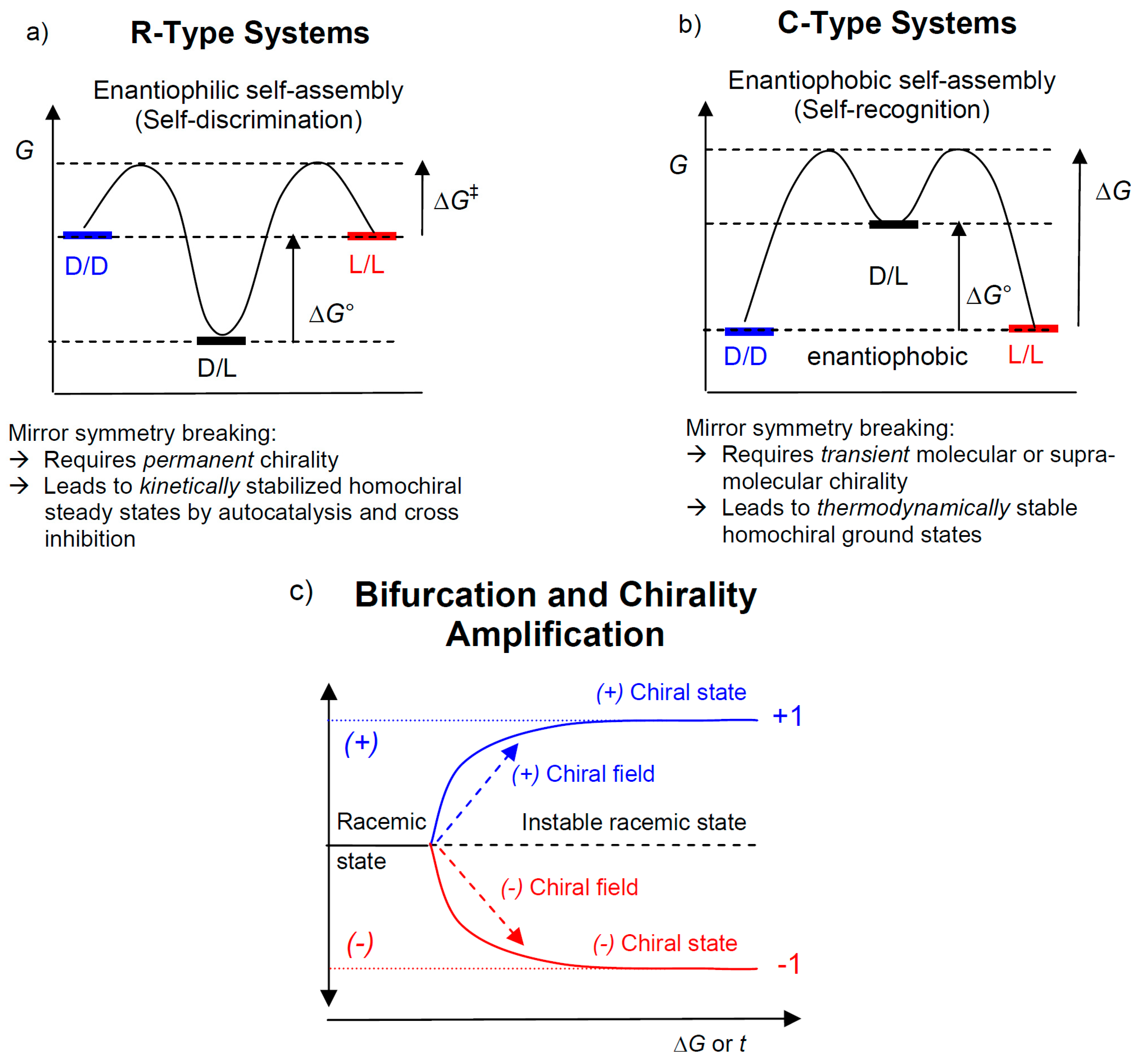
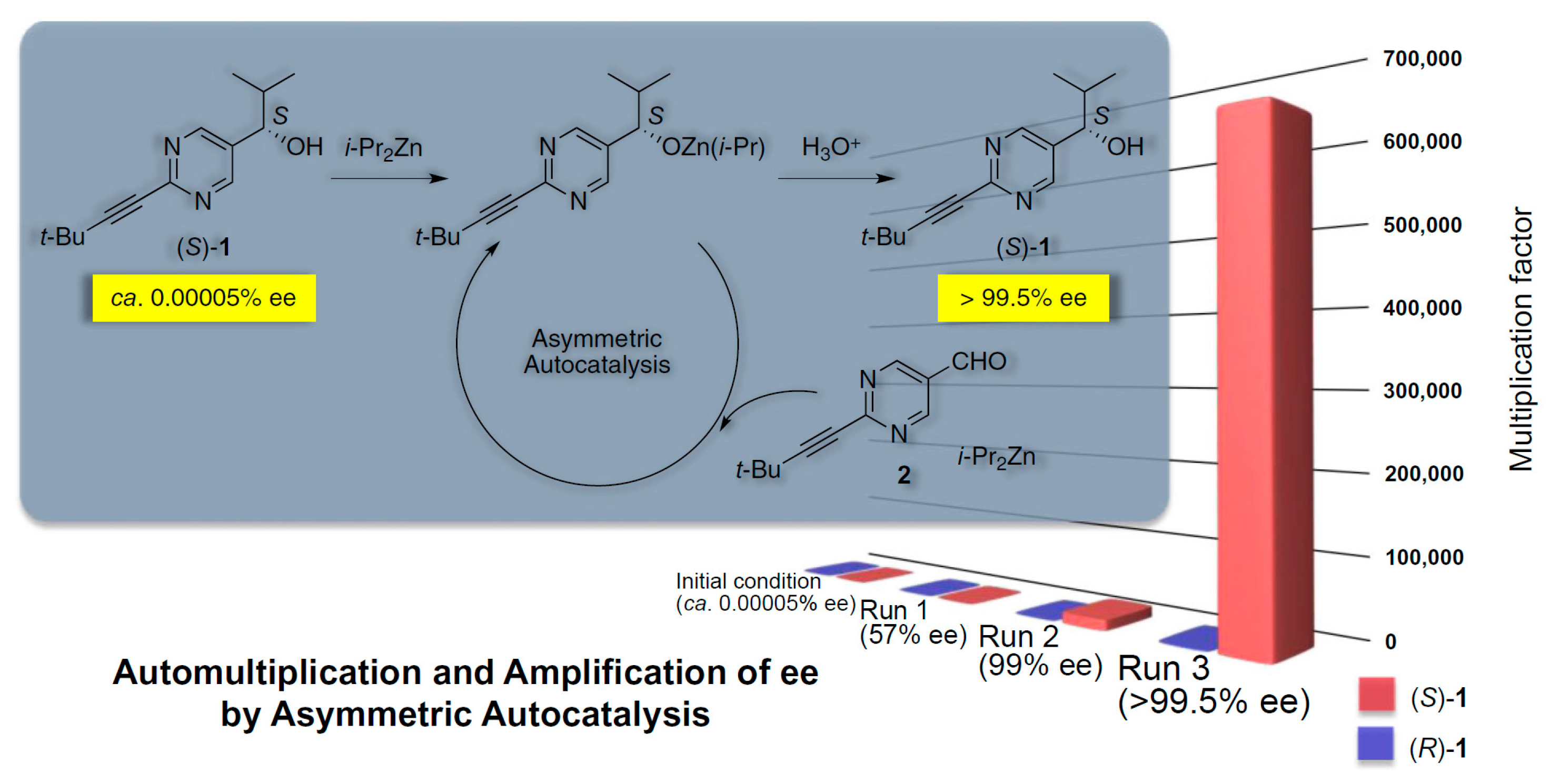
2.2. R-Type Systems: Mirror Symmetry Breaking by Autocatalysis And Enantiomeric Cross-Inhibition in Chemical Reaction Cycles-Soai Reaction
2.3. C-Type Systems: Spontaneous Homochirality
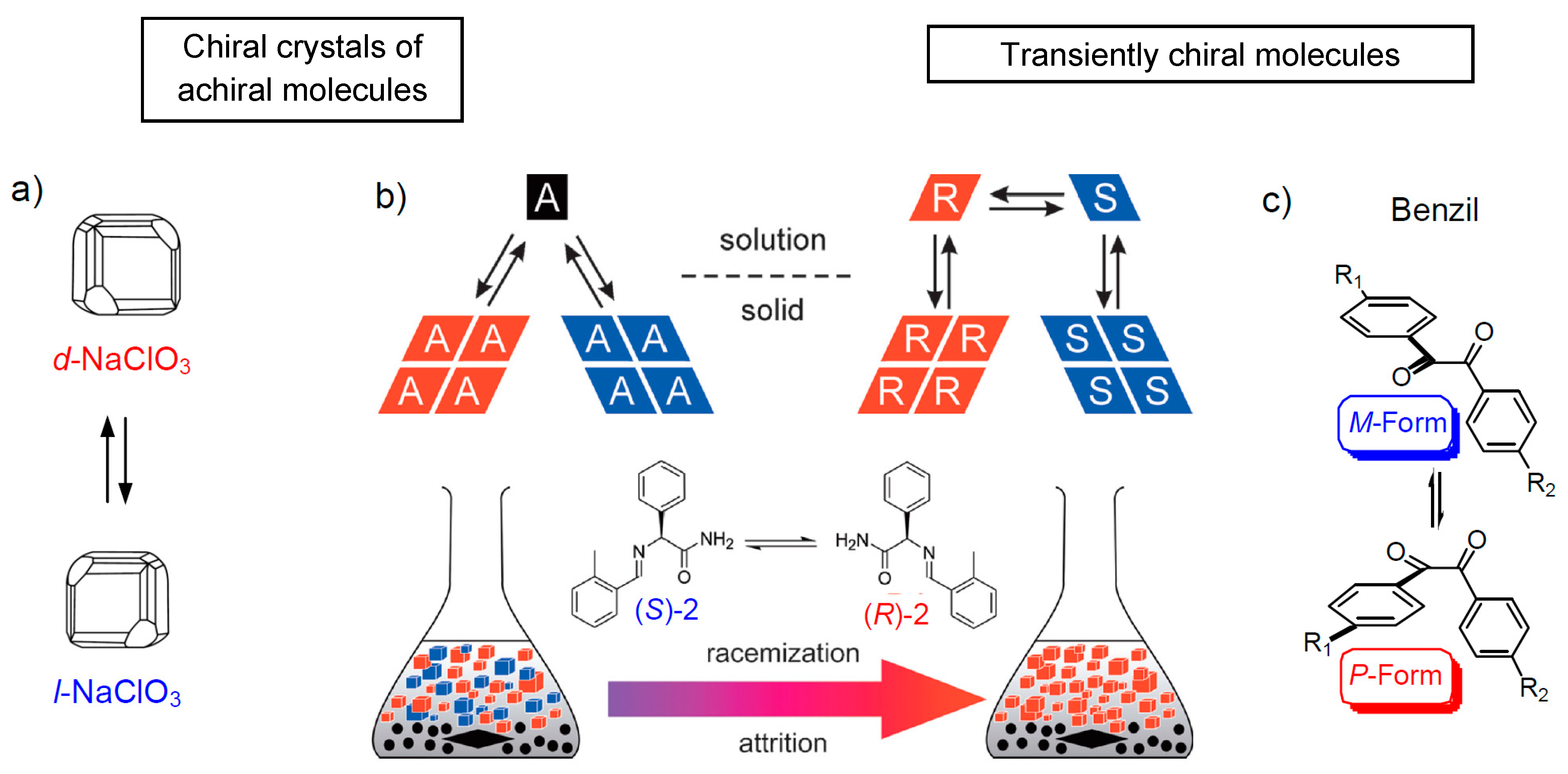
2.3.1. Spontaneous Homochirality by Phase Transitions-Viedma Ripening
2.3.2. Spontaneous Mirror Symmetry Breaking in Liquids
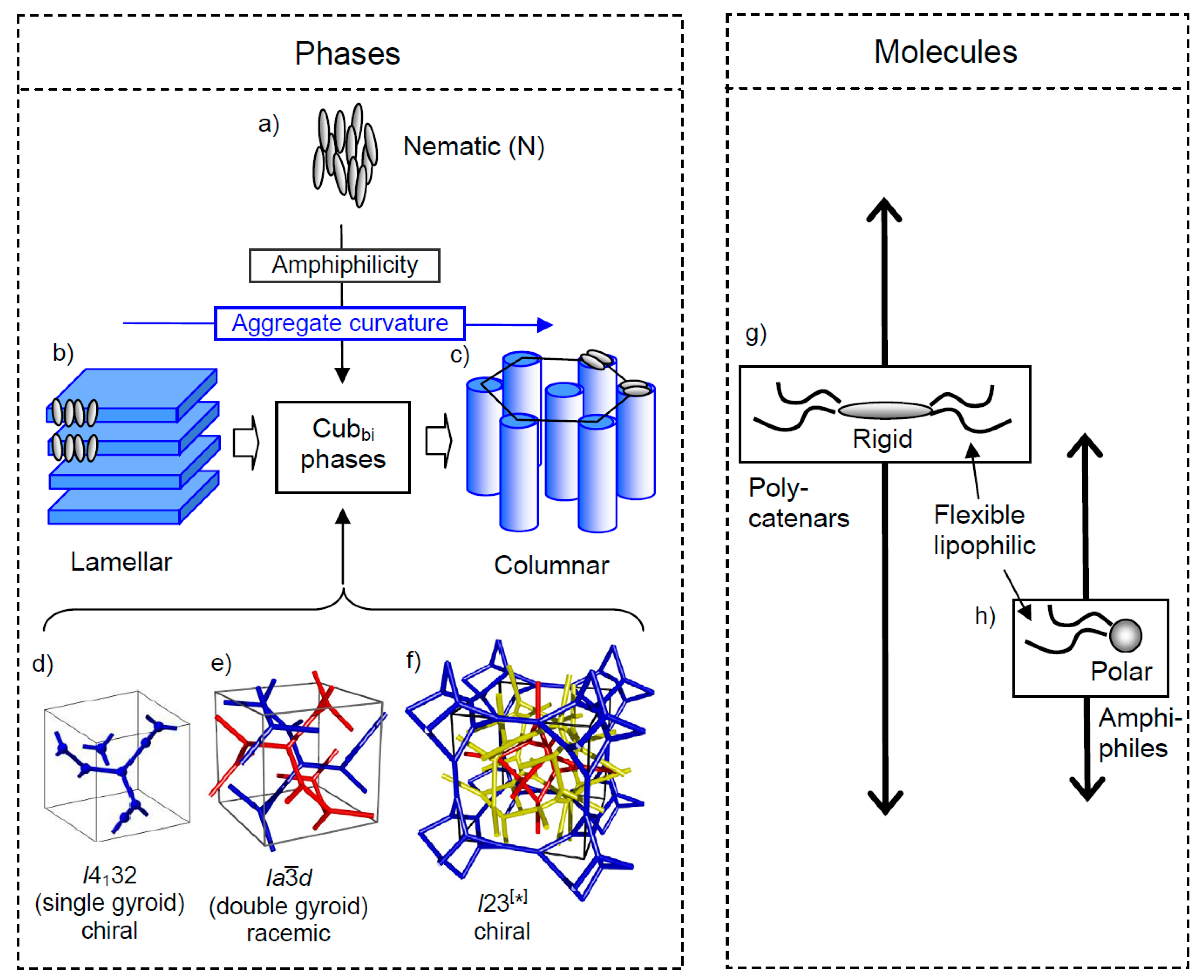
3. From Crystals via Liquid Crystals to Liquids
4. Mirror Symmetry Breaking in Cubic Network Phases
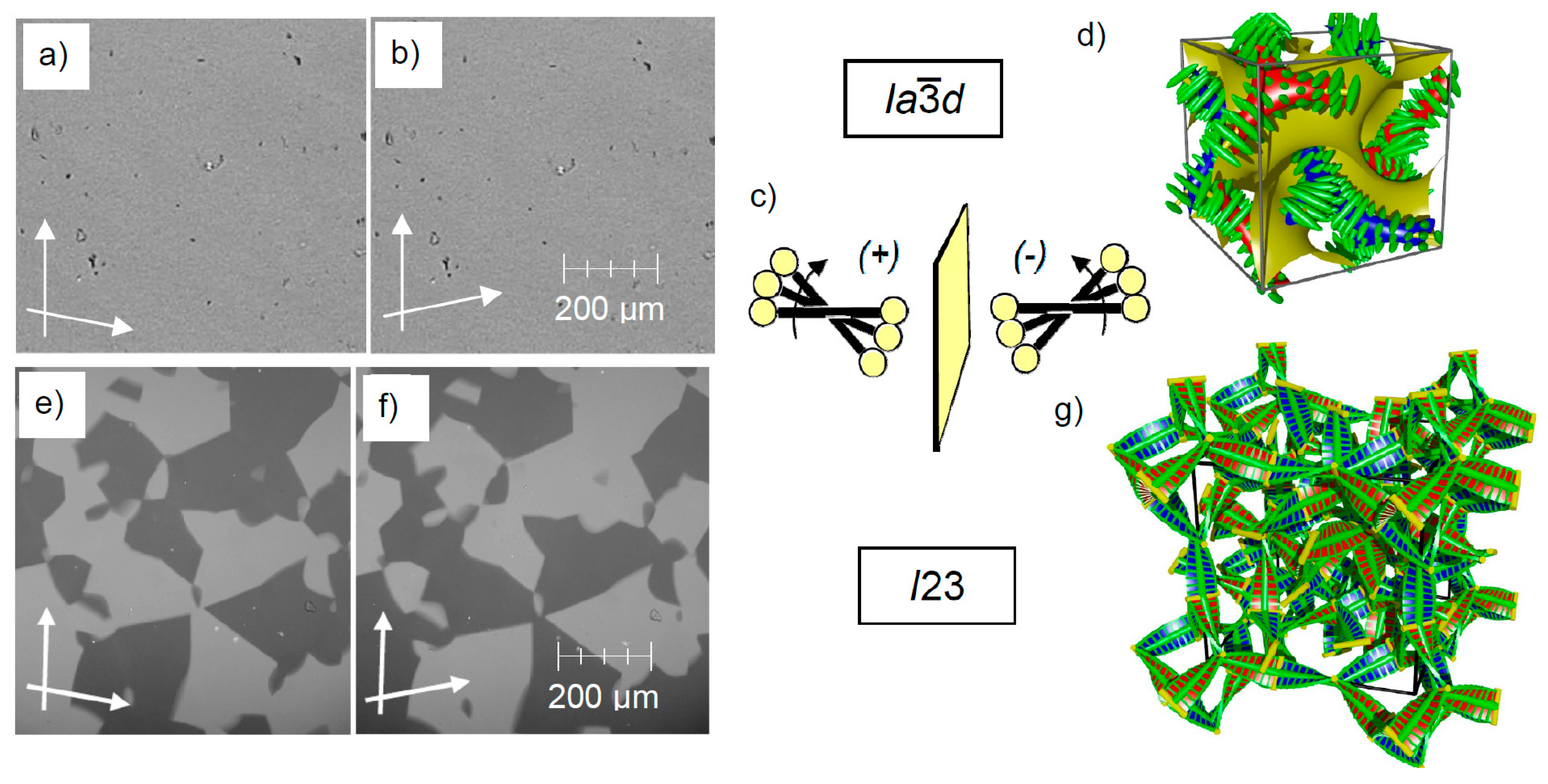
4.1. Racemic d Phases and Chiral Conglomerate Type I23 Phases
4.2. Conglomerate Formation vs. Complete Mirror Symmetry Breaking in Cubic Pases
5. Experimental Demonstration of Mirror Symmetry Breaking in Isotropic Liquids
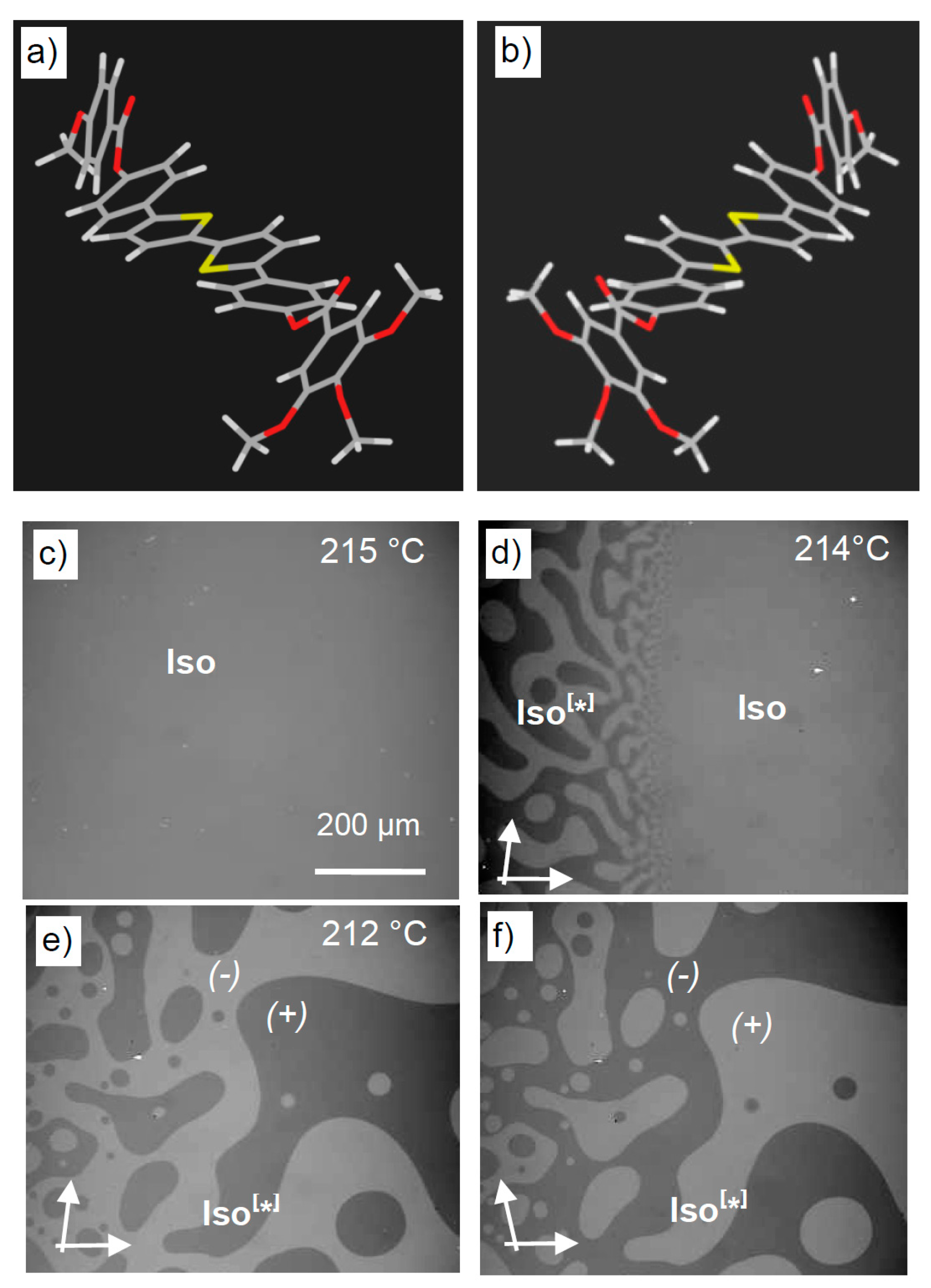
5.1. Local Mirror Symmetry Breaking by Conglomerate Formation
5.2. Total Mirror Symmetry Breaking in Isotropic Liquids
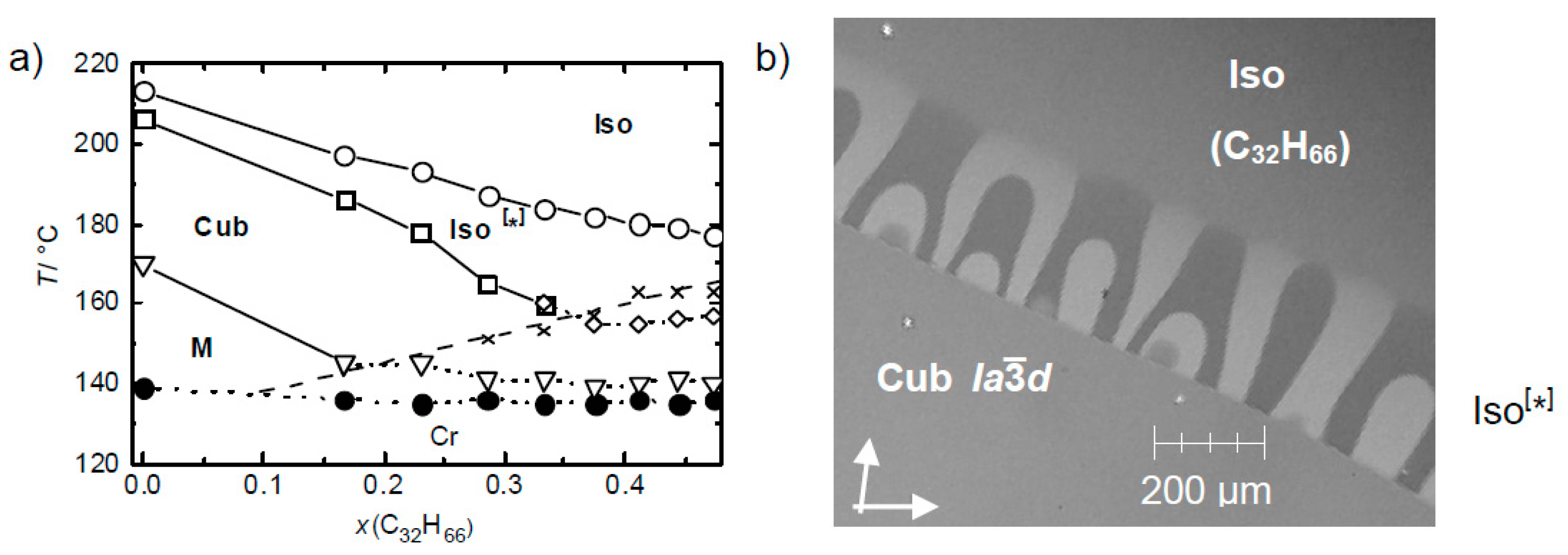
5.3. Effects of Dilution on Mirror Symmetry Breaking in Isotropic Liquids
5.4. Chirality Amplification in Isotropic Liquids
5.5. Total Mirror Symmetry Breaking in the Cubbi/I23 Phase
5.6. Cubbi/I23 Phase-Assisted Total Mirror Symmetry Breaking in Isotropic Liquids
5.7. Complete Mirror Symmetry Breaking in Isotropic Liquds Due to Compartmentalization
5.8. Impact of Network Formation on Mirror Symmetry Breaking in Isotropic Liquids
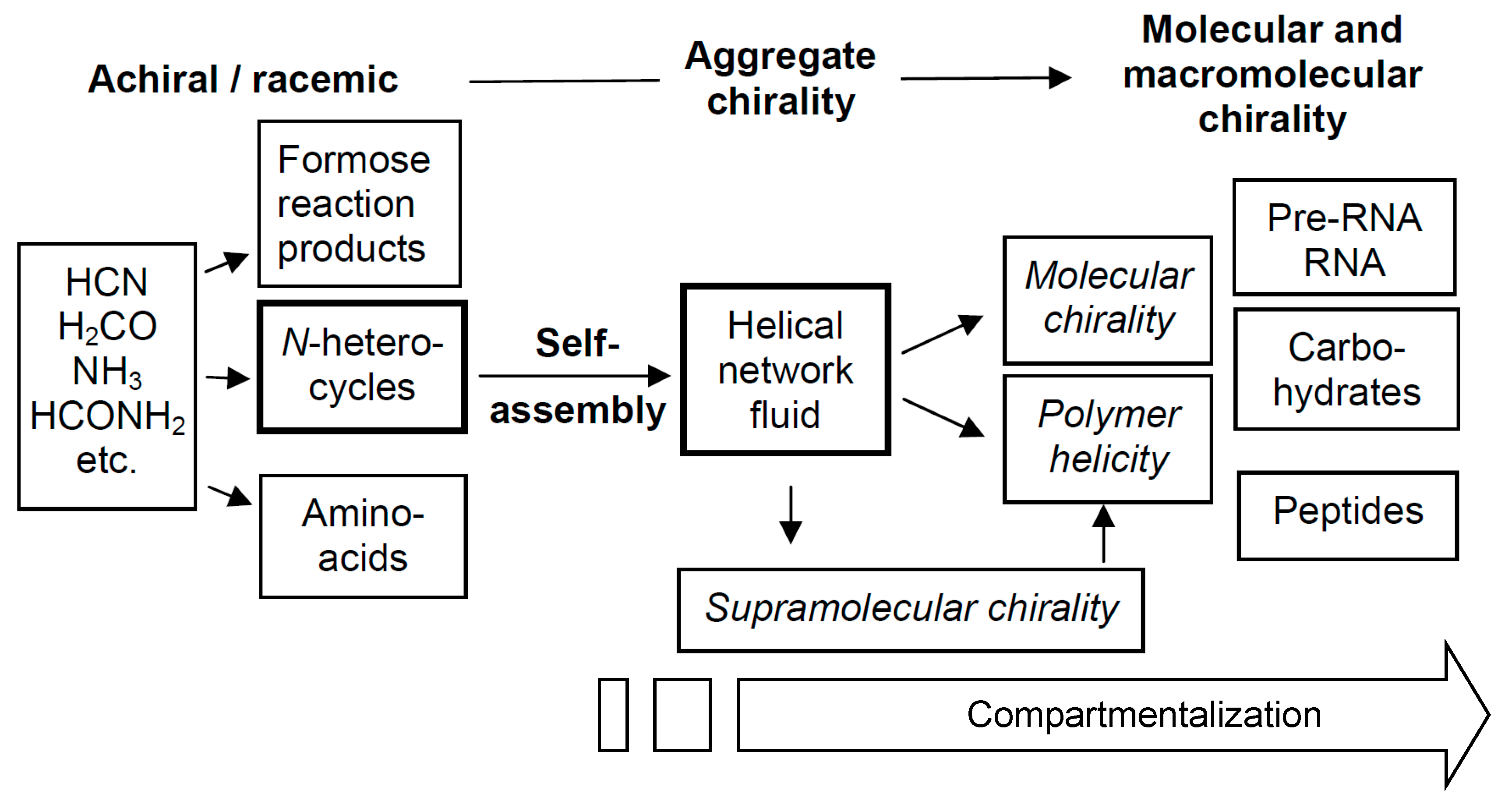
6. Possible Scenario for Chirogenesis Based on Mirror Symmetry Breaking in Network Liquids
7. Conclusions
Funding
Conflicts of Interest
References
- Pasteur, L. Recherches sur les relations qui peuvent exister entre la forme crystalline, la composition chimique et le sens de la polarisation rotatoire. Ann. Chim. Phys. 1848, 24, 442–459. [Google Scholar]
- Palyi, G. Biological Chirality; Academic Press, Elsevier: London, UK, 2020. [Google Scholar]
- Weiss, M.C.; Preiner, M.; Xavier, J.C.; Zimorski, V.; Martin, W.F. The last universal common ancestor between ancient Earth chemistry and the onset of Genetics. PLoS Genet. 2018, 14, e1007518. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Powner, M.W. Prebiotic Systems Chemistry: Complexity Overcoming Clutter. Chem 2017, 2, 470–501. [Google Scholar] [CrossRef]
- Green, M.M.; Jain, V. Homochirality in Life: Two Equal Runners, One Tripped. Orig. Life Evol. Biosph. 2010, 40, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.I. Origins of life: A problem for physics, a key issues review. Rep. Prog. Phys. 2017, 80, 092601. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Mirazo, K.; Briones, C.; de la Escosura, A. Prebiotic Systems Chemistry: New Perspectives for the Origins of Life. Chem. Rev. 2014, 114, 285–366. [Google Scholar] [CrossRef] [PubMed]
- Rauchfuss, H. Chemical Evolution and the Origin of Life; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Guijarro, A.; Yus, M. The Origin of Chirality in the Molecules of Life; RSC publishing: Cambridge, UK, 2009. [Google Scholar]
- Luisi, P.L. The Emergence of Life; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Meierhenrich, U. Amino Acids and the Asymmetry of Life; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Cintas, P. Biochirality, Origins, Evolution and Molecular Recognition; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Cronin, J.; Reisse, J. Chirality and the Origin of Homochirality. In Lectures in Astrobiology; Gargaud, M., Barbier, B., Martin, H., Reise, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 1, pp. 473–515. [Google Scholar]
- Blackmond, D.G. The Origin of Biological Homochirality. Cold Spring Harb. Perspect. Biol. 2019, 11, a032540. [Google Scholar] [CrossRef] [PubMed]
- Coveney, P.V.; Swadling, J.B.; Wattis, J.A.D.; Greenwell, H.C. Theory, modelling and simulation in origins of life studies. Chem. Soc. Rev. 2012, 41, 5430–5446. [Google Scholar] [CrossRef] [PubMed]
- Ribo, J.M.; Hochberg, D.; Crusats, J.; El-Hachemi, Z.; Moyano, A. Spontaneous mirror symmetry breaking and origin of biological homochirality. J. R. Soc. Interface 2017, 14, 20170699. [Google Scholar] [CrossRef] [PubMed]
- Ribó, J.M.; Hochberg, D. Chemical Basis of Biological Homochirality during the Abiotic Evolution Stages on Earth. Symmetry 2019, 11, 814. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Shushenachev, Y.V.; Zlotin, S.G. Chiral and Racemic Fields Concept for Understanding of the Homochirality Origin, Asymmetric Catalysis, Chiral Superstructure Formation from Achiral Molecules, and B-Z DNA Conformational Transition. Symmetry 2019, 11, 649. [Google Scholar] [CrossRef]
- Toxvaerd, S. The Role of Carbohydrates at the Origin of Homochirality in Biosystems. Orig. Life Evol. Biosph. 2013, 43, 391–409. [Google Scholar] [CrossRef] [PubMed]
- Toxvaerd, S. A Prerequisite for Life. J. Theor. Biol. 2019, 474, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Hein, J.E.; Blackmond, D.G. On the Origin of Single Chirality of Amino Acids and Sugars in Biogenesis. Acc. Chem. Res. 2012, 45, 2045–2054. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Klabunovskii, E.I. Homochirality Origin in Nature: Possible Versions. Curr. Org. Chem. 2014, 18, 93–114. [Google Scholar] [CrossRef]
- Avalos, M.; Babiano, R.; Cintas, P.; Jimenez, J.L.; Palacios, J.C. Homochirality and chemical evolution: New vistas and reflections on recent models. Tetrahedron Asymmetry 2010, 21, 1030–1040. [Google Scholar] [CrossRef]
- Suzuki, N.; Itabashi, Y. Possible Roles of Amphiphilic Molecules in the Origin of Biological Homochirality. Symmetry 2019, 11, 966. [Google Scholar] [CrossRef]
- Dressel, C.; Reppe, T.; Prehm, M.; Brautzsch, M.; Tschierske, C. Chiral self-sorting and amplification in isotropic liquids of achiral molecules. Nat. Chem. 2014, 6, 971–977. [Google Scholar] [CrossRef]
- Dressel, C.; Liu, F.; Prehm, M.; Zeng, X.B.; Ungar, G.; Tschierske, C. Dynamic Mirror-Symmetry Breaking in Bicontinuous Cubic Phases. Angew. Chem. Int. Ed. 2014, 53, 13115–13120. [Google Scholar] [CrossRef]
- Yashima, E.; Ousaka, N.; Taura, D.; Shimomura, K.; Ikai, T.; Maeda, K. Supramolecular helical systems: Helical assemblies of small molecules, foldamers, and polymers with chiral amplification and their functions. Chem. Rev. 2016, 116, 13752–13990. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, L.; Wang, T. Supramolecular chirality in self-assembled systems. Chem. Rev. 2015, 115, 7304–7397. [Google Scholar] [CrossRef]
- Barclay, T.G.; Constantopoulos, K.; Matisons, J. Nanotubes self-assembled from amphiphilic molecules via helical intermediates. Chem. Rev. 2014, 114, 10217–10291. [Google Scholar] [CrossRef]
- La, A.D.D.; Al Kobaisi, M.; Gupta, A.; Bhosale, S.V. Chiral Assembly of AIE-Active Achiral Molecules: An Odd Effect in Self-Assembly. Chem. Eur. J. 2017, 23, 3950–3956. [Google Scholar] [CrossRef]
- Sang, Y.; Liu, M. Symmetry Breaking in Self-Assembled Nanoassemblies. Symmetry 2019, 11, 950. [Google Scholar] [CrossRef]
- Changa, B.; Lib, X.; Suna, T. Self-assembled chiral materials from achiral components or racemates. Eur. Polym. J. 2019, 118, 365–381. [Google Scholar] [CrossRef]
- Hoeben, F.J.M.; Jonkheijm, P.; Meijer, E.W.; Schenning, A.P.H.J. About Supramolecular Assemblies of π-Conjugated Systems. Chem. Rev. 2005, 105, 1491–1546. [Google Scholar] [CrossRef]
- Palmans, A.R.A.; Meijer, E.W. Amplification of chirality in dynamic supramolecular Aggregates. Angew. Chem. Int. Ed. 2007, 46, 8948–8968. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, T.; Shen, Z.; Liu, M. Chiral Nanoarchitectonics: Towards the Design, Self-Assembly, and Function of Nanoscale Chiral Twists and Helices. Adv. Mater. 2016, 28, 1044–1059. [Google Scholar] [CrossRef]
- Pijper, D.; Feringa, B.L. Control of dynamic helicity at the macro- and supramolecular level. Soft Matter 2008, 4, 1349–1372. [Google Scholar] [CrossRef]
- Ariga, K.; Mori, T.; Kitao, T.; Uemura, T. Supramolecular Chiral Nanoarchitectonics. Adv. Mater. 2020, 1905657. [Google Scholar] [CrossRef]
- De Greef, T.F.A.; Smulders, M.M.J.; Wolffs, M.; Schenning, A.P.H.J.; Sijbesma, R.P.; Meijer, E.W. Supramolecular polymerization. Chem. Rev. 2009, 109, 5687–5754. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.; Alaasar, M.; Poppe, M.; Poppe, S.; Prehm, M.; Nagaraj, M.; Sreenilayam, S.P.; Panarin, J.P.; Vij, J.K.; Tschierske, C. Stereochemical Rules Govern the Soft Self-Assembly of Achiral Compounds: Understanding the Heliconical Liquid-Crystalline Phases of Bent-Core Mesogens. Chem. Eur. J. 2020, 26, 4714–4733. [Google Scholar] [CrossRef] [PubMed]
- Tschierske, C. Mirror symmetry breaking in liquids and liquid crystals. Liq. Cryst. 2018, 45, 2221–2252. [Google Scholar] [CrossRef]
- Le, K.V.; Takezoe, H.; Araoka, F. Chiral Superstructure mesophases of achiral bent-shaped molecules-hierarchical chirality amplification and physical properties. Adv. Mater. 2017, 29, 1602737. [Google Scholar] [CrossRef] [PubMed]
- Tschierske, C.; Ungar, G. Mirror Symmetry Breaking by Chirality Synchronisation in Liquids and Liquid Crystals of Achiral Molecules. ChemPhysChem 2016, 17, 9–26. [Google Scholar] [CrossRef]
- Dierking, I. Chiral Liquid Crystals: Structures, Phases, Effects. Symmetry 2014, 6, 444–472. [Google Scholar] [CrossRef]
- Nishyiama, I. Remarkable Effect of Pre-organization on the Self Assembly in Chiral Liquid Crystals. Chem. Rec. 2010, 9, 340–355. [Google Scholar] [CrossRef] [PubMed]
- Bisoyi, H.K.; Bunning, T.J.; Li, Q. Stimuli-Driven Control of the Helical Axis of Self-Organized Soft Helical Superstructures. Adv. Mater. 2018, 30, 1706512. [Google Scholar] [CrossRef]
- Eliel, E.L.; Wilen, S.H.; Mander, L.N. Stereochemistry of Organic Compounds; Wiley: New York, NY, USA, 1994. [Google Scholar]
- Jacques, J.; Collet, A.; Wilen, S.H. Enantiomers, Racemates and Resolutions; Wiley: New York, NY, USA, 1981. [Google Scholar]
- Mills, W.H. Some Aspects of Stereochemistry. Chem. Ind. 1932, 51, 750–759. [Google Scholar] [CrossRef]
- Morowitz, M. A mechanism for the amplification of fluctuations in racemic mixtures. J. Theor. Biol. 1969, 25, 491–494. [Google Scholar] [CrossRef]
- Siegel, J. Homochiral Imperative of Molecular Evolution. Chirality 1998, 10, 24–27. [Google Scholar] [CrossRef]
- Lente, G. The Role of Stochastic Models in Interpreting the Origins of Biological Chirality. Symmetry 2010, 2, 767–798. [Google Scholar] [CrossRef]
- Hochberg, D.; Zorzano, M.-P. Reaction-noise induced homochirality. Chem. Phys. Lett. 2006, 431, 185–189. [Google Scholar] [CrossRef]
- Kondepudi, D.K.; Aaskura, K. Chiral Autocatalysis, Spontaneous Symmetry Breaking, and Stochastic Behavior. Acc. Chem. Res. 2001, 34, 946–954. [Google Scholar] [CrossRef]
- Silva-Dias, L.; Lopez-Castillo, A. Stochastic chiral symmetry breaking process besides the deterministic one. Phys. Chem. Chem. Phys. 2017, 19, 29424–29428. [Google Scholar] [CrossRef] [PubMed]
- Frank, F.C. On spontaneous asymmetric synthesis. Biochim. Biophys. Acta 1953, 11, 459–464. [Google Scholar] [CrossRef]
- Weissbuch, I.; Leiserowitz, L.; Lahav, M. Stochastic “Mirror Symmetry Breaking” via Self-Assembly, Reactivity and Amplification of Chirality: Relevance to Abiotic Conditions. Top. Curr. Chem. 2005, 259, 123–165. [Google Scholar] [CrossRef]
- Soai, K.; Kawasaki, T.; Matsumoto, A. The Origins of Homochirality Examined by Using Asymmetric Autocatalysis. Chem. Rec. 2014, 14, 70–83. [Google Scholar] [CrossRef]
- Soai, K.; Kawasaki, T.; Matsumoto, A. Role of Asymmetric Autocatalysis in the Elucidation of Origins of Homochirality of Organic Compounds. Symmetry 2019, 11, 694. [Google Scholar] [CrossRef]
- Satyanarayana, T.; Abraham, S.; Kagan, H.B. Nonlinear Effects in Asymmetric Catalysis. Angew. Chem. Int. Ed. 2009, 48, 456–494. [Google Scholar] [CrossRef]
- Kawasaki, T.; Tanaka, H.; Tsutsumi, T.; Kasahara, T.; Sato, I.; Soai, K. Chiral Discrimination of Cryptochiral Saturated Quaternary and Tertiary Hydrocarbons by Asymmetric Autocatalysis. J. Am. Chem. Soc. 2006, 128, 6032–6033. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Okano, Y.; Suzuki, E.; Takano, S.; Oji, S.; Soai, K. Asymmetric Autocatalysis: Triggered by Chiral Isotopomer Arising from Oxygen Isotope Substitution. Angew. Chem. Int. Ed. 2011, 50, 8131–8133. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Kaimori, Y.; Uchida, M.; Omori, H.; Kawasaki, T.; Soai, K. Achiral Inorganic Gypsum Acts as an Origin of Chirality through Its Enantiotopic Surface in Conjunction with Asymmetric Autocatalysis. Angew. Chem. Int. Ed. 2017, 56, 545–548. [Google Scholar] [CrossRef]
- Soai, K.; Sato, I.; Shibata, T.; Komiya, S.; Hayashi, M.; Matsueda, Y.; Imamura, H.; Hayase, T.; Morioka, H.; Tabira, H.; et al. Asymmetric synthesis of pyrimidyl alkanol without adding chiral substances by the addition of diisopropylzinc to pyrimidine-5-carbaldehyde in conjunction with asymmetric autocatalysis. Tetrahedron Asymmetry 2003, 14, 185–188. [Google Scholar] [CrossRef]
- Singleton, D.A.; Vo, L.K. A Few Molecules Can Control the Enantiomeric Outcome. Evidence Supporting Absolute Asymmetric Synthesis Using the Soai Asymmetric Autocatalysis. Org. Lett. 2003, 5, 4337–4339. [Google Scholar] [CrossRef]
- Toxvaerd, S. Origin of homochirallity in biosystems. Int. J. Mol. Sci. 2009, 10, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Plasson, R.; Kondepudi, D.K.; Bersine, H.; Commeyras, A.; Asakura, K. Emergence of Homochirality in Far-From-Equilibrium Systems: Mechanisms and Role in Prebiotic Chemistry. Chirality 2007, 19, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Ribo, J.M.; Blanco, C.; Crusats, J.; El-Hachemi, Z.; Hochberg, D.; Moyano, A. Absolute Asymmetric Synthesis in Enantioselective Autocatalytic Reaction Networks: Theoretical Games, Speculations on Chemical Evolution and Perhaps a Synthetic Option. Chem. Eur. J. 2014, 20, 17250–17271. [Google Scholar] [CrossRef]
- Toxvaerd, S. Molecular Dynamics Simulations in Isomerization Kinetics in Condensed Fluids. Phys. Rev. Lett. 2000, 85, 4747–4750. [Google Scholar]
- Jafarpour, F.; Biancalani, T.; Goldenfeld, N. Noise-induced symmetry breaking far from equilibrium and the emergence of biological homochirality. Phys. Rev. E 2017, 95, 032407. [Google Scholar] [CrossRef]
- Sugimori, T.; Hyuga, H.; Saito, Y. Fluctuation Induced Homochirality. J. Phys. Soc. Jpn. 2008, 77, 064606. [Google Scholar] [CrossRef]
- Latinwo, F.; Stillinger, F.H.; Debenedetti, P.G. Molecular model for chirality phenomena. J. Chem. Phys. 2016, 145, 154503. [Google Scholar] [CrossRef]
- Brandenburg, A. The Limited Roles of Autocatalysis and Enantiomeric Cross-Inhibition in Achieving Homochirality in Dilute Systems. Orig. Life Evol. Biosph. 2019, 49, 49–60. [Google Scholar] [CrossRef]
- Kondepudi, D.K.; Kaufman, R.J.; Singh, N. Chiral Symmetry Breaking in Sodium Chlorate Crystallizaton. Science 1990, 250, 975–976. [Google Scholar] [CrossRef] [PubMed]
- Viedma, C. Chiral Symmetry Breaking During Crystallization: Complete Chiral PurityInduced by Nonlinear Autocatalysis and Recycling. Phys. Rev. Lett. 2005, 94, 065504. [Google Scholar] [CrossRef] [PubMed]
- Cintas, P.; Viedma, C. On the Physical Basis of Asymmetry and Homochirality. Chirality 2012, 24, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Blackmond, D.G. “Chiral Amnesia” as a Driving Force for Solid-Phase Homochirality. Chem. Eur. J. 2007, 13, 3290–3295. [Google Scholar] [CrossRef]
- Noorduin, W.L.; Vlieg, E.; Kellogg, R.M.; Kaptein, B. From Ostwald Ripening to Single Chirality. Angew. Chem. Int. Ed. 2009, 48, 9600–9606. [Google Scholar] [CrossRef]
- Amabilino, D.B.; Kellogg, R.M. Spontaneous Deracemization. Isr. J. Chem. 2011, 51, 1034–1040. [Google Scholar] [CrossRef]
- Uemura, N.; Sano, K.; Matsumoto, A.; Yoshida, Y.; Mino, T.; Sakamoto, M. Absolute Asymmetric Synthesis of an Aspartic Acid Derivative from Prochiral Maleic Acid and Pyridine under Achiral Conditions. Chem. Asian J. 2019, 14, 4150–4153. [Google Scholar] [CrossRef]
- Tsogoeva, S.B.; Wei, S.; Freund, M.; Mauksch, M. Generation of Highly Enantioenriched Crystalline Products in Reversible Asymmetric Reactions with Racemic or Achiral Catalysts. Angew. Chem. Int. Ed. 2009, 48, 590–594. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, D.T.; Thao Nguyen, T.P.; Mengnjo, L.; Bian, C.; Leung, Y.H.; Goodfellow, E.; Ramrup, P.; Woo, S.; Cuccia, L.A. Viedma Ripening of Conglomerate Crystals of Achiral Molecules Monitored Using Solid-State Circular Dichroism. Cryst. Growth Des. 2014, 14, 1067–1076. [Google Scholar] [CrossRef]
- Runnels, C.M.; Lanier, K.A.; Williams, J.K.; Bowman, J.C.; Petrov, A.S.; Hud, N.V.; Williams, L.D. Folding, Assembly, and Persistence: The Essential Nature and Origins of Biopolymers. J. Mol. Evol. 2018, 86, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Kenney, J.F.; Deiters, U.K. The evolution of multicomponent systems at high pressures Part IV. The genesis of optical activity in high-density, abiotic Ñuids. Phys. Chem. Chem. Phys. 2000, 2, 3163–3174. [Google Scholar] [CrossRef]
- Hochberg, D.; Cintas, P. Does Pressure Break Mirror-Image Symmetry? A Perspective and New Insights. ChemPhysChem 2020, 21, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Leporl, L.; Mengherl, M.; Molllca, V. Discriminating Interactions between Chiral Molecules in the Liquid Phase: Effect on Volumetric Properties. J. Phys. Chem. 1983, 87, 3520–3525. [Google Scholar] [CrossRef]
- Atik, Z.; Ewing, M.B.; McGlashan, M.L. Chiral Discrimination in Liquids. Excess Molar Volumes of (1 − x)A+ + xA, Where A Denotes Limonene, Fenchone, and a-Methylbenzylamine. J. Phys. Chem. 1981, 85, 3300–3303. [Google Scholar] [CrossRef]
- Dressel, C.; Weissflog, W.; Tschierske, C. Spontaneous mirror symmetry breaking in a re-entrant isotropic liquid. Chem. Commun. 2015, 51, 15850–15853. [Google Scholar] [CrossRef]
- Alaasar, M.; Poppe, S.; Dong, Q.; Liu, F.; Tschierske, C. Mirror symmetry breaking in cubic phases and isotropic liquids driven by hydrogen bonding. Chem. Commun. 2016, 52, 13869–13872. [Google Scholar] [CrossRef]
- Alaasar, M.; Prehm, M.; Cao, Y.; Liu, F.; Tschierske, C. Spontaneous Mirror-Symmetry Breaking in Isotropic Liquid Phases of Photoisomerizable Achiral Molecules. Angew. Chem. Int. Ed. 2016, 55, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Alaasar, M.; Poppe, S.; Dong, Q.; Liu, F.; Tschierske, C. Isothermal Chirality Switching in Liquid-Crystalline Azobenzene Compounds with Non Polarized Light. Angew. Chem. Int. Ed. 2017, 56, 10801–10805. [Google Scholar] [CrossRef]
- Goodby, J.W.; Collings, P.J.; Kato, T.; Tschierske, C.; Gleeson, H.F.; Raynes, P. Handbook of Liquid Crystals, 2nd ed; Wiley-VHC: Weinheim, Germany, 2014. [Google Scholar]
- Kato, T.; Uchida, J.; Ichikawa, T.; Sakamoto, T. Functional Liquid Crystals towards the Next Generation of Materials. Angew. Chem. Int. Ed. 2018, 57, 4355–4371. [Google Scholar] [CrossRef]
- Tschierske, C. Development of Structural Complexity by Liquid-Crystal Self-assembly. Angew. Chem. Int. Ed. 2013, 52, 8828–8878. [Google Scholar] [CrossRef]
- Mitov, M. Cholesteric liquid crystals in living matter. Soft Matter 2017, 13, 4176–4209. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.L.; Spicer, P.T. (Eds.) Bicontinuous Liquid Crystals; Surfactant Science Series 127; CRC Press—Taylor & Francis Group: Boca Raton, FL, USA, 2005. [Google Scholar]
- Seddon, J.M.; Templer, R.H. Polymorphism of Lipid-Water Systems. In Handbook of Biological Physics; Lipowsky, R., Sackmann, E., Eds.; Elsevier: Amsterdam, the Netherlands, 1995; Volume 1, pp. 97–160. [Google Scholar]
- Tschierske, C. Micro-segregation, molecular shape and molecular topology—Partners for the design of liquid crystalline materials with complex mesophase morphologies. J. Mater. Chem. 2001, 11, 2647–2671. [Google Scholar] [CrossRef]
- Borisch, K.; Diele, S.; Göring, P.; Kresse, H.; Tschierske, C. Tailoring Thermotropic Cubic Mesophases: Amphiphilic Polyhydroxy Derivatives. J. Mater. Chem. 1998, 8, 529–543. [Google Scholar] [CrossRef]
- Kutsumizu, S. Recent Progress in the Synthesis and Structural Clarification of Thermotropic Cubic Phases. Isr. J. Chem. 2012, 52, 844–853. [Google Scholar] [CrossRef]
- Ungar, G.; Liu, F.; Zeng, X. Cubic and 3D Thermotropic Liquid Crystal Phases and Quasicrystals. In Handbook of Liquid Crystals, 2nd ed; Goodby, J.W., Collings, P.J., Kato, T., Tschierske, C., Gleeson, H.F., Raynes, P., Eds.; Wiley-VHC: Weinheim, Germany, 2014; Volume 5, pp. 363–436. [Google Scholar]
- Meuler, A.J.; Hillmyer, M.A.; Bates, F.S. Ordered Network Mesostructures in Block Polymer Materials. Macromolecules 2009, 42, 7221–7250. [Google Scholar] [CrossRef]
- Hyde, S.; Andersson, S.; Larsson, K.; Blum, Z.; Landh, T.; Lidin, S.; Ninham, B.W. The Language of Shape, The Role of Curvature in Condensed Matter: Physics, Chemistry and Biology; Elsevier: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Deamer, D. Liquid crystalline nanostructures: Organizing matrices for non-enzymatic nucleic acid polymerization. Chem. Soc. Rev. 2012, 41, 5375–5379. [Google Scholar] [CrossRef] [PubMed]
- Hamley, I.W. Liquid crystal phase formation by biopolymers. Soft Matter 2010, 6, 1863–1871. [Google Scholar] [CrossRef]
- Jewell, S.A. Living systems and liquid crystals. Liq. Cryst. 2011, 38, 1699–1714. [Google Scholar] [CrossRef]
- Steward, G.T. Liquid crystals in biology II. Origins and processes of life. Liq. Cryst. 2004, 31, 443–471. [Google Scholar] [CrossRef]
- Rey, A.D. Liquid crystal models of biological materials and processes. Soft Matter 2010, 6, 3402–3429. [Google Scholar] [CrossRef]
- Giraud-Guille, M.M.; Belamie, E.; Mosser, G.; Helary, C.; Gobeaux, F.; Vigier, S. Liquid crystalline properties of type I collagen: Perspectives in tissue morphogenesis. Comptes Rendus Chim. 2008, 11, 245–252. [Google Scholar] [CrossRef]
- Lydon, J. Microtubules: Nature’s smartest mesogens—A liquid crystal model for cell division. Liq. Cryst. Today 2006, 15, 1–10. [Google Scholar] [CrossRef]
- Bouligand, Y. Geometry and topology of cell membranes. In Geometry in Condensed Matter Physics; Sadoc, J.F., Ed.; World Scientific: Singapore, 1990; pp. 193–231. [Google Scholar]
- Kulkarni, C.V. Lipid crystallization: From self-assembly to hierarchical and biological ordering. Nanoscale 2012, 4, 5779–5791. [Google Scholar] [CrossRef]
- Livolant, F.; Levelut, A.M.; Doucet, J.; Benoit, J.P. The highly concentrated liquid crystalline phase of DNA is columnar hexagonal. Nature 1989, 339, 724–726. [Google Scholar] [CrossRef]
- Leforestie, A.; Bertin, A.; Dubochet, J.; Richter, K.; Sartori Blanc, N.; Livolant, F. Expression of chirality in columnar hexagonal phases of DNA and nucleosomes. Comptes Rendus Chim. 2008, 11, 229–244. [Google Scholar] [CrossRef]
- Leal, C.; Ewert, K.K.; Bouxsein, N.F.; Shirazi, R.S.; Lic, Y.; Safinya, C.R. Stacking of short DNA induces the gyroid cubic-to inverted hexagonal phase transition in lipid–DNA complexes. Soft Matter 2013, 9, 795–804. [Google Scholar] [CrossRef]
- Zanchetta, G. Spontaneous self-assembly of nucleic acids: Liquid crystal condensation of complementary sequences in mixtures of DNA and RNA oligomers. Liq. Cryst. Today 2009, 18, 40–49. [Google Scholar] [CrossRef]
- Mahadevi, S.A.; Sastry, G.N. Cooperativity in Noncovalent Interactions. Chem. Rev. 2016, 116, 2775–2825. [Google Scholar] [CrossRef] [PubMed]
- Tschierske, C. Microsegregation: From Basic Concepts to Complexity in Liquid Crystal Self-Assembly. Isr. J. Chem. 2012, 52, 935–959. [Google Scholar] [CrossRef]
- Rest, C.; Kandanelli, R.; Fernandez, G. Strategies to create hierarchical self-assembled structures via cooperative non-covalent interactions. Chem. Soc. Rev. 2015, 44, 2543–2572. [Google Scholar] [CrossRef]
- Takezoe, H. Spontaneous Achiral Symmetry Breaking in Liquid Crystalline Phases. Top. Curr. Chem. 2012, 318, 303–330. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Kieffer, R.; Ebert, H.; Prehm, M.; Zhang, R.-B.; Zeng, X.; Liu, F.; Ungar, G.; Tschierske, C. Chirality Induction through Nano-Phase Separation: Alternating Network Gyroid Phase by Thermotropic Self-Assembly of X-Shaped Bolapolyphiles. Angew. Chem. Int. Ed. 2020, 59, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Poppe, S.; Lehmann, A.; Prehm, M.; Chen, C.; Liu, F.; Lu, H.; Ungar, G.; Tschierske, C. A Self-assembled Bicontinuous Cubic Phase with Single Diamond Network. Angew. Chem. Int. Ed. 2019, 131, 7453–7457. [Google Scholar] [CrossRef]
- Poppe, S.; Cheng, X.; Chen, C.; Zeng, X.; Zhang, R.-B.; Liu, F.; Ungar, G.; Tschierske, C. Liquid Organic Frameworks: The Single-Network “Plumber’s Nightmare” Bicontinuous Cubic Liquid Crystal. J. Am. Chem. Soc. 2020, 142, 3296–3300. [Google Scholar] [CrossRef]
- Zeng, X.B.; Ungar, G. Spontaneously chiral cubic liquid crystal: Three interpenetrating networks with a twist. J. Mater. Chem. C 2020, 8, 5389–5398. [Google Scholar] [CrossRef]
- Kauffman, S. At Home in the Universe; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Malthete, J.; Nguyen, H.T.; Destrade, C. Phasmids and polycatenar mesogens. Liq. Cryst. 1993, 13, 171–187. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Destrade, C.; Malthete, J. Phasmids and Polycatenar Mesogens. Adv. Mater. 1997, 9, 375–388. [Google Scholar] [CrossRef]
- Bruce, D.W. Calamitics, Cubics, and Columnars. Liquid-Crystalline Complexes of Silver (I). Acc. Chem. Res. 2000, 33, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Reppe, T.; Dressel, C.; Poppe, S.; Tschierske, C. Controlling spontaneous mirror symmetry breaking in cubic liquid crystalline phases by the cycloaliphatic ring size. Chem. Commun. 2020, 56, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Dressel, C.; Reppe, T.; Poppe, S.; Prehm, M.; Lu, H.; Zeng, X.; Ungar, G.; Tschierske, C. Helical networks of π-conjugated rods—A robust design concept for bicontinuous cubic liquid crystalline phases with achiral d and chiral I23 lattice. Adv. Funct. Mater. 2020, submitted. [Google Scholar]
- Reppe, T.; Poppe, S.; Cai, X.; Cao, Y.; Liu, F.; Tschierske, C. Spontaneous mirror symmetry breaking in benzil-based soft crystalline, cubic liquid crystalline and isotropic liquid phases. Chem. Sci. 2020, 11, 5902–5908. [Google Scholar] [CrossRef]
- Wolska, J.M.; Wilk, J.; Pociecha, D.; Mieczkowski, J.; Gorecka, E. Optically Active Cubic Liquid Crystalline Phase Made of Achiral Polycatenar Stilbene Derivatives. Chem. Eur. J. 2017, 23, 6853–6857. [Google Scholar] [CrossRef]
- Kutsumizu, S.; Yamada, Y.; Sugimoto, T.; Yamada, N.; Udagawa, T.; Miwa, Y. Systematic exploitation of thermotropic bicontinuous cubic phase families from 1,2-bis(aryloyl)hydrazine-based molecules. Phys. Chem. Chem. Phys. 2018, 20, 7953–7961. [Google Scholar] [CrossRef]
- Kutsumizu, S.; Miisako, S.; Miwa, Y.; Kitagawa, M.; Yamamura, Y.; Saito, K. Mirror symmetry breaking by mixing of equimolar amounts of two gyroid phase-forming achiral molecules. Phys. Chem. Chem. Phys. 2016, 18, 17341–17344. [Google Scholar] [CrossRef]
- Pescitelli, G.; Di Bari, L.; Berova, N. Application of electronic circular dichroism in the study of supramolecular systems. Chem. Soc. Rev. 2014, 43, 5211–5233. [Google Scholar] [CrossRef]
- Gottarelli, G.; Lena, S.; Masiero, S.; Pieraccini, S.; Spada, G.P. The Use of Circular Dichroism Spectroscopy for Studying the Chiral Molecular Self-Assembly: An Overview. Chirality 2008, 20, 471–485. [Google Scholar] [CrossRef]
- Jouvelet, B.; Isare, B.; Bouteiller, L.; von der Schoot, P. Direct probing of the free-energy penalty for helix reversal and chiral mismatches in chiral supramolecular polymers. Langmuir 2014, 30, 4570–4575. [Google Scholar] [CrossRef]
- Lu, H.; Zeng, X.; Ungar, G.; Dressel, C.; Tschierske, C. The Solution of the Puzzle of Smectic-Q: The Phase Structure and the Origin of Spontaneous Chirality. Angew. Chem. Int. Ed. 2018, 57, 2835–2840. [Google Scholar] [CrossRef] [PubMed]
- Brand, H.R.; Pleiner, H. Cubic and tetragonal liquid crystal phases composed of non-chiral molecules: Chirality and macroscopic properties. Eur. Phys. J. E 2019, 42, 142. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, T.; Kohmoto, S.; Yamamoto, M.; Kishikawa, K. Spontaneous Chiral Induction in a Cubic Phase. Chem. Mater. 2005, 17, 3812–3819. [Google Scholar] [CrossRef]
- Quack, M. How important is parity violation for molecular and biomolecular chirality? Angew. Chem. Int. Ed. 2002, 41, 4618–4630. [Google Scholar] [CrossRef]
- Chandrasekhar, S. Molecular homochirality and the parity-violating energy difference. A critique with new proposals. Chirality 2008, 20, 84–95. [Google Scholar] [CrossRef]
- Ha, A.; Cohen, I.; Zhao, X.L.; Lee, M.; Kivelson, D. Supercooled Liquids and Polyamorphism. J. Phys. Chem. 1996, 100, 1–4. [Google Scholar] [CrossRef]
- Anisimov, M.A.; Duška, M.; Caupin, F.; Amrhein, L.E.; Rosenbaum, A.; Sadus, R.J. Thermodynamics of Fluid Polyamorphism. Phys. Rev. X 2018, 8, 011004. [Google Scholar] [CrossRef]
- Goodby, J.W.; Dunmur, D.A.; Collings, J.P. Lattice melting at the clearing point in frustrated Systems. Liq. Cryst. 1995, 19, 703–709. [Google Scholar] [CrossRef]
- de las Heras, D.; Tavares, J.M.; Telo da Gama, M.M. Phase diagrams of binary mixtures of patchy colloids with distinct numbers of patches: The network fluid regime. Soft Matter 2011, 7, 5615–5626. [Google Scholar] [CrossRef]
- Zhuang, Y.; Charbonneau, P. Recent Advances in the Theory and Simulation of Model Colloidal Microphase Formers. J. Phys. Chem. B 2016, 120, 7775–7782. [Google Scholar] [CrossRef]
- Henrich, O.; Stratford, K.; Cates, M.E.; Marenduzzo, D. Structure of Blue Phase III of Cholesteric Liquid Crystals. Phys. Rev. Lett. 2011, 106, 107801. [Google Scholar] [CrossRef]
- Brand, H.R.; Pleiner, H. On the influence of a network on optically isotropic fluid phases with tetrahedral/octupolar order. Eur. Phys. J. E 2017, 40, 34. [Google Scholar] [CrossRef] [PubMed]
- Damer, B.; Deamer, D. The Hot Spring Hypothesis for an Origin of Life. Astrobiology 2020, 20, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Camprubí, E.; de Leeuw, J.W.; House, C.H.; Raulin, F.; Russell, M.J.; Spang, A.; Tirumalai, M.R.; Westall, F. The Emergence of Life. Space Sci. Rev. 2019, 215, 56. [Google Scholar] [CrossRef]
- Toxvaerd, S. The role of the peptides at the origin of life. J. Theor. Biol. 2017, 429, 164–169. [Google Scholar] [CrossRef]
- Gilbert, W. The RNA world. Nature 1986, 319, 618. [Google Scholar] [CrossRef]
- Pressman, A.; Blanco, C.; Chen, I.A. The RNA World as a Model System to Study the Origin of Life. Curr. Biol. 2015, 25, R953–R963. [Google Scholar] [CrossRef]
- Sutherland, J.D. The Origin of Life—Out of the Blue Angew. Chem. Int. Ed. 2016, 55, 104–121. [Google Scholar] [CrossRef]
- Cafferty, B.J.; Fialho, D.M.; Hud., N.V. Searching for Possible Ancestors of RNA: The Self-Assembly Hypothesis for the Origin of Proto-RNA. In Prebiotic Chemistry and Chemical Evolution of Nucleic Acids. Nucleic Acids and Molecular Biology; Menor-Salván, C., Nicholson, A.W., Eds.; Springer-Nature: Cham, Switzerland, 2018; Volume 35. [Google Scholar]
- Krishnamurthy, R. On the Emergence of RNA. Isr. J. Chem. 2015, 55, 837–850. [Google Scholar] [CrossRef]
- Xu, J.; Tsanakopoulou, M.; Magnani, C.R.; Szabla, R.; Šponer, J.E.; Šponer, J.; Góra, R.W.; Sutherland, J.D. A prebiotically plausible synthesis of pyrimidine β-ribonucleosides and their phosphate derivatives involving photoanomerization. Nat. Chem. 2017, 9, 303–309. [Google Scholar] [CrossRef]
- Nama, I.; Nama, H.G.; Zareb, R.N. Abiotic synthesis of purine and pyrimidine ribonucleosides in aqueous microdroplets. Proc. Natl. Acad. Sci. USA 2018, 115, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Feldmann, J.; Wiedemann, S.; Okamura, H.; Schneider, C.; Iwan, K.; Crisp, A.; Rossa, M.; Amatov, T.; Carell, T. Unified prebiotically plausible synthesis of pyrimidine and purine RNA ribonucleotides. Science 2019, 366, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Sanders, J.K.M. The Nature of Interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Martinez, C.R.; Iverson, B.L. Rethinking the term “pi-stacking”. Chem. Sci. 2012, 3, 2191–2201. [Google Scholar] [CrossRef]
- Krueger, A.T.; Kool, E.T. Model systems for understanding DNA base pairing. Curr. Opin. Chem. Biol. 2007, 11, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Joyce, G.F.; Schwartz, A.W.; Miller, S.L.; Orgel, L.E. The case for an ancestral genetic system involving simple analogues of the nucleotides. Proc. Natl. Acad. Sci. USA 1987, 84, 4398–4402. [Google Scholar] [CrossRef]
- Chen, Y.; Ma, W. The origin of biological homochirality along with the origin of life. PLoS Comput. Biol. 2020, 16, e1007592. [Google Scholar] [CrossRef]
- Sandars, P.G.H. A toy model for the generation of homochirality during polymerization. Orig. Life Evol. Biosph. 2003, 33, 575–587. [Google Scholar] [CrossRef]
- Wattis, J.A.D.; Coveney, P.V. Symmetry-breaking in chiral polymerisation. Orig. Life Evol. Biosph. 2005, 35, 243–273. [Google Scholar] [CrossRef]
- Jain, V.; Cheon, K.-S.; Tang, K.; Jha, S.; Green, M.M. Chiral Cooperativity in Helical Polymers. Isr. J. Chem. 2011, 51, 1067–1074. [Google Scholar] [CrossRef]
- Baumgarten, J.L. Ferrochirality: A simple theoretical model of interacting, dynamically invertible, helical polymers, 2†. Molecular field approach: Supports and the details. Macromol. Theory Simul. 1995, 4, 1–43. [Google Scholar] [CrossRef]
- Stich, M.; Hochberg, D. Mechanically Induced Homochirality in Nucleated Enantioselective Polymerization, Celia Blanco. J. Phys. Chem. B 2017, 121, 942–955. [Google Scholar] [CrossRef]
- Buchs, J.; Vogel, L.; Janietz, D.; Prehm, M.; Tschierske, C. Chirality Synchronization of Hydrogen-Bonded Complexes of Achiral N-Heterocycles. Angew. Chem. Int. Ed. 2017, 56, 280–284. [Google Scholar] [CrossRef]
- Karunakaran, S.C.; Cafferty, B.J.; Weigert-MuÇoz, A.; Schuster, G.B.; Hud, N.V. Spontaneous Symmetry Breaking in the Formation of Supramolecular Polymers: Implications for the Origin of Biological Homochirality. Angew. Chem. Int. Ed. 2019, 58, 1453–1457. [Google Scholar] [CrossRef]


















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tschierske, C.; Dressel, C. Mirror Symmetry Breaking in Liquids and Their Impact on the Development of Homochirality in Abiogenesis: Emerging Proto-RNA as Source of Biochirality? Symmetry 2020, 12, 1098. https://doi.org/10.3390/sym12071098
Tschierske C, Dressel C. Mirror Symmetry Breaking in Liquids and Their Impact on the Development of Homochirality in Abiogenesis: Emerging Proto-RNA as Source of Biochirality? Symmetry. 2020; 12(7):1098. https://doi.org/10.3390/sym12071098
Chicago/Turabian StyleTschierske, Carsten, and Christian Dressel. 2020. "Mirror Symmetry Breaking in Liquids and Their Impact on the Development of Homochirality in Abiogenesis: Emerging Proto-RNA as Source of Biochirality?" Symmetry 12, no. 7: 1098. https://doi.org/10.3390/sym12071098
APA StyleTschierske, C., & Dressel, C. (2020). Mirror Symmetry Breaking in Liquids and Their Impact on the Development of Homochirality in Abiogenesis: Emerging Proto-RNA as Source of Biochirality? Symmetry, 12(7), 1098. https://doi.org/10.3390/sym12071098