
Trends in the Development of Antibody-Drug Conjugates for Cancer Therapy


Abstract
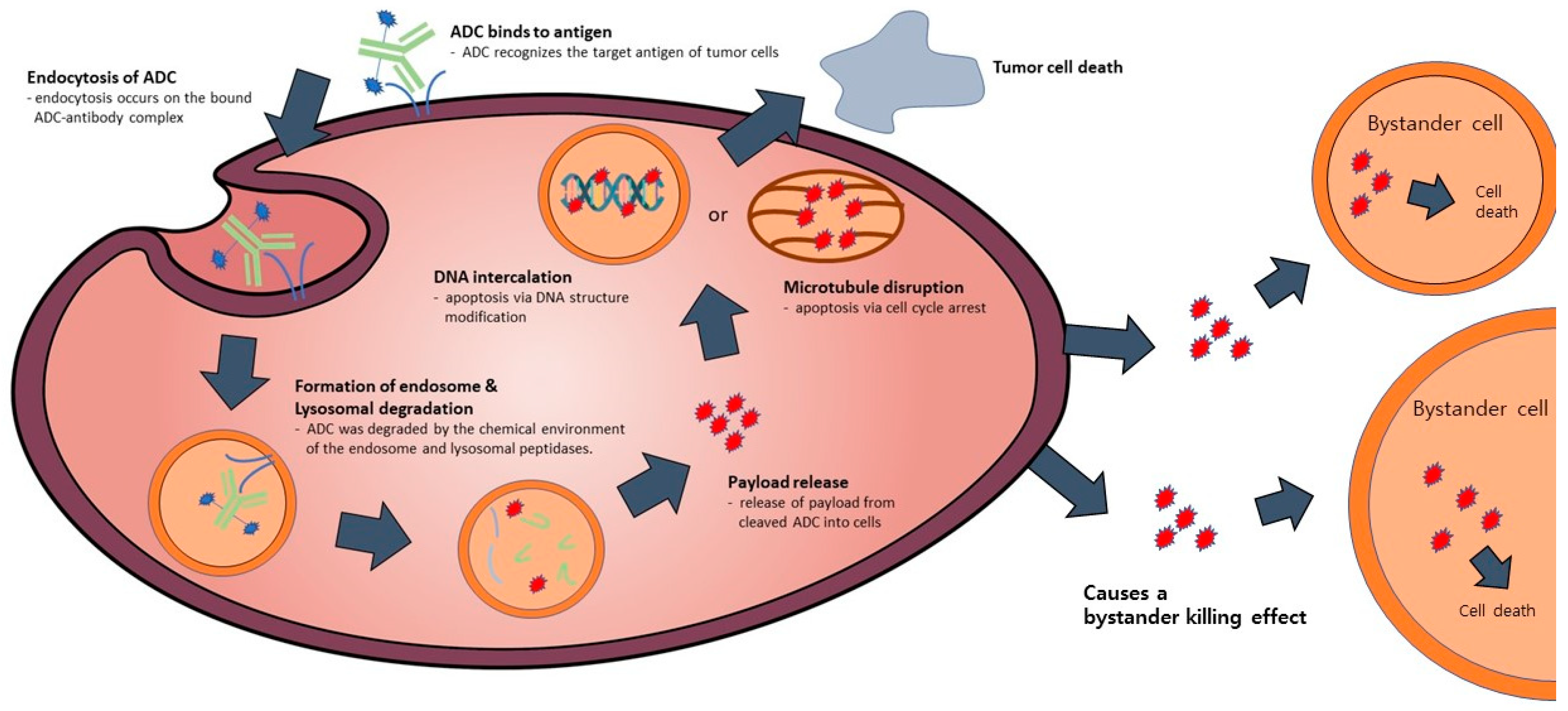
:1. Introduction
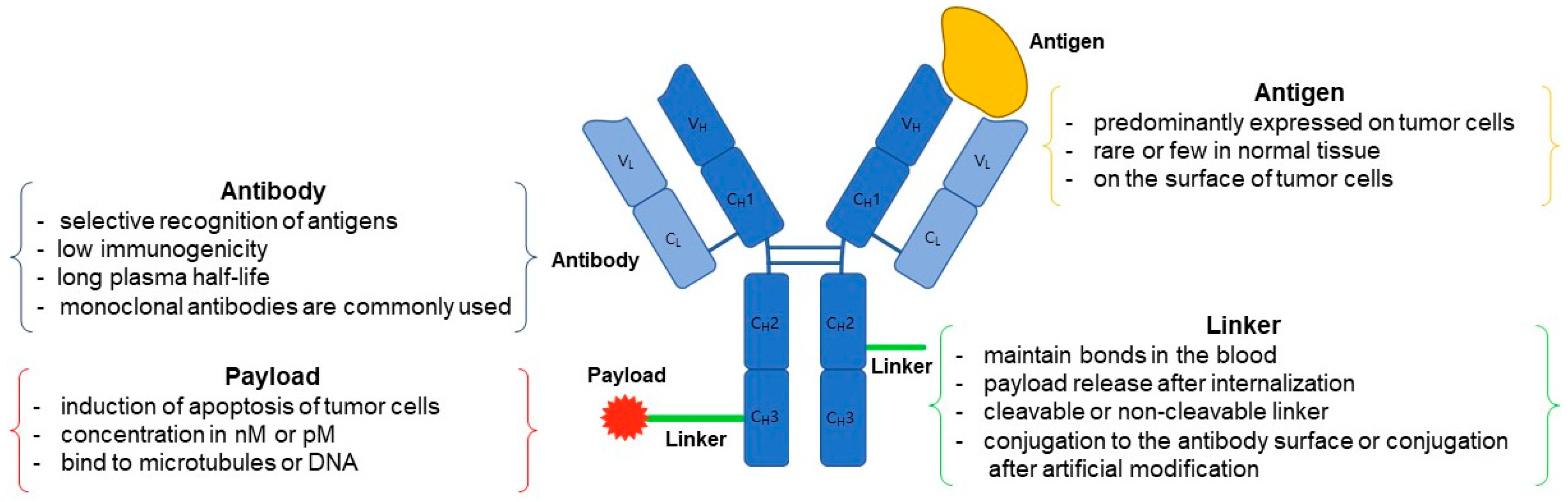
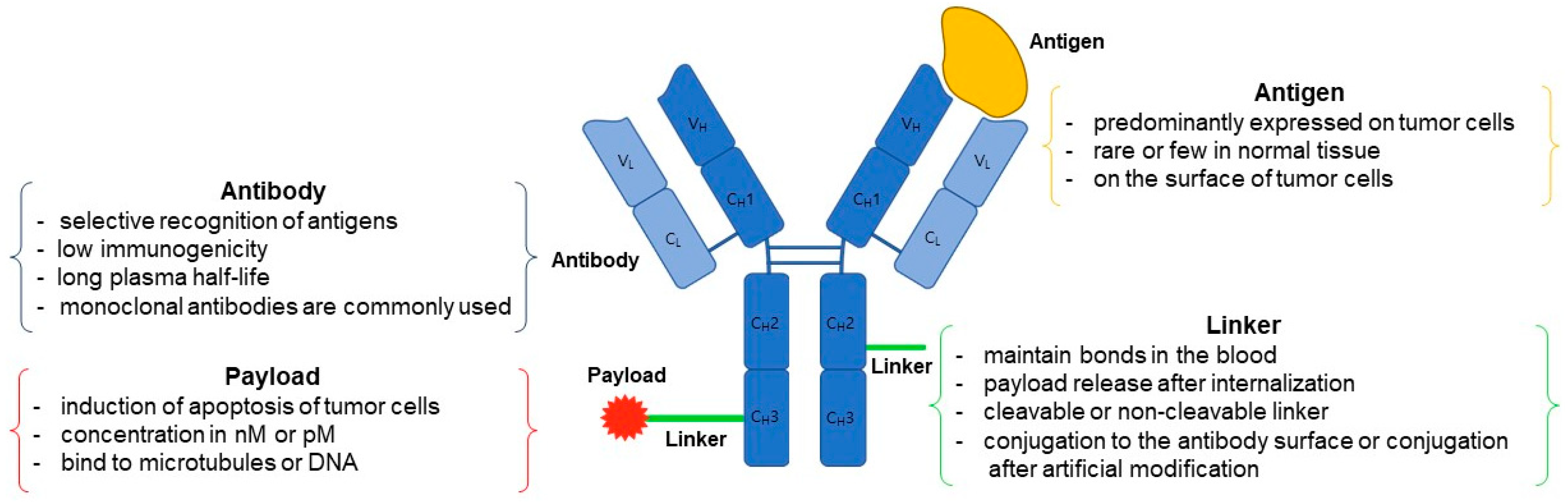
2. ADC Structure
2.1. Antibody
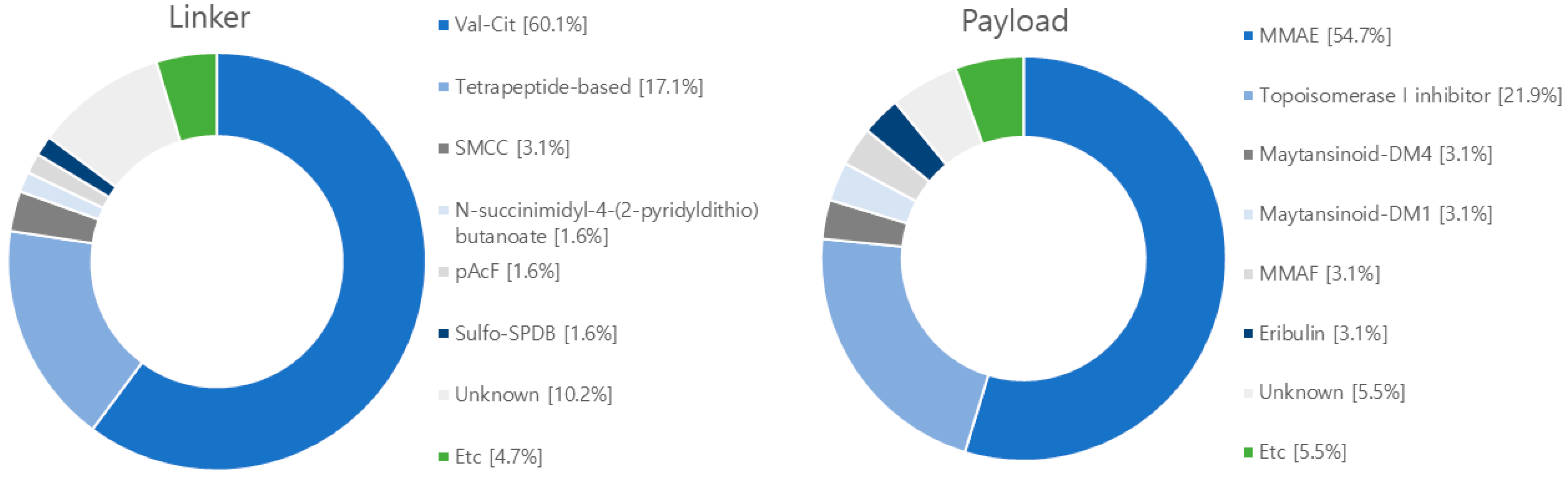
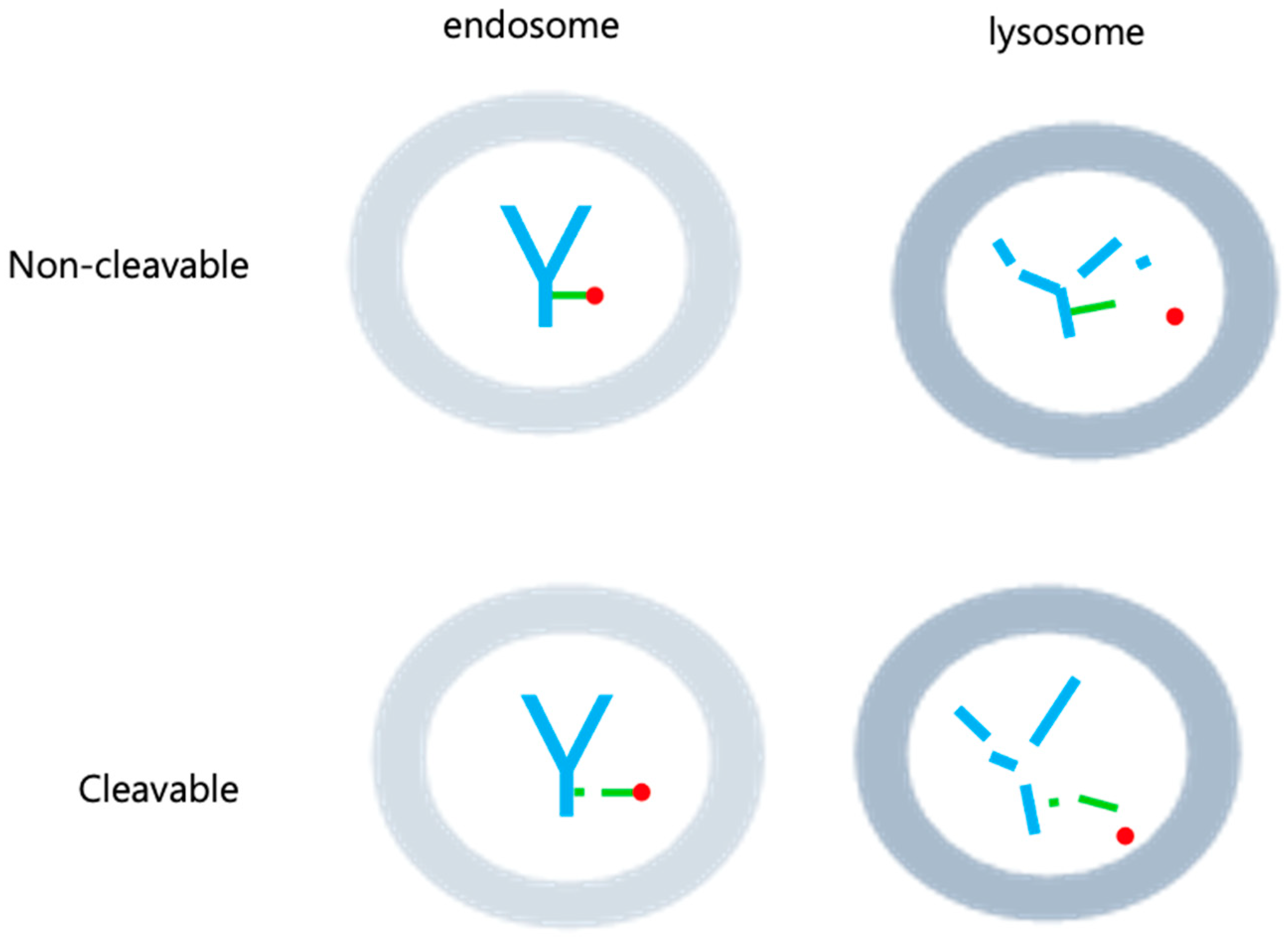
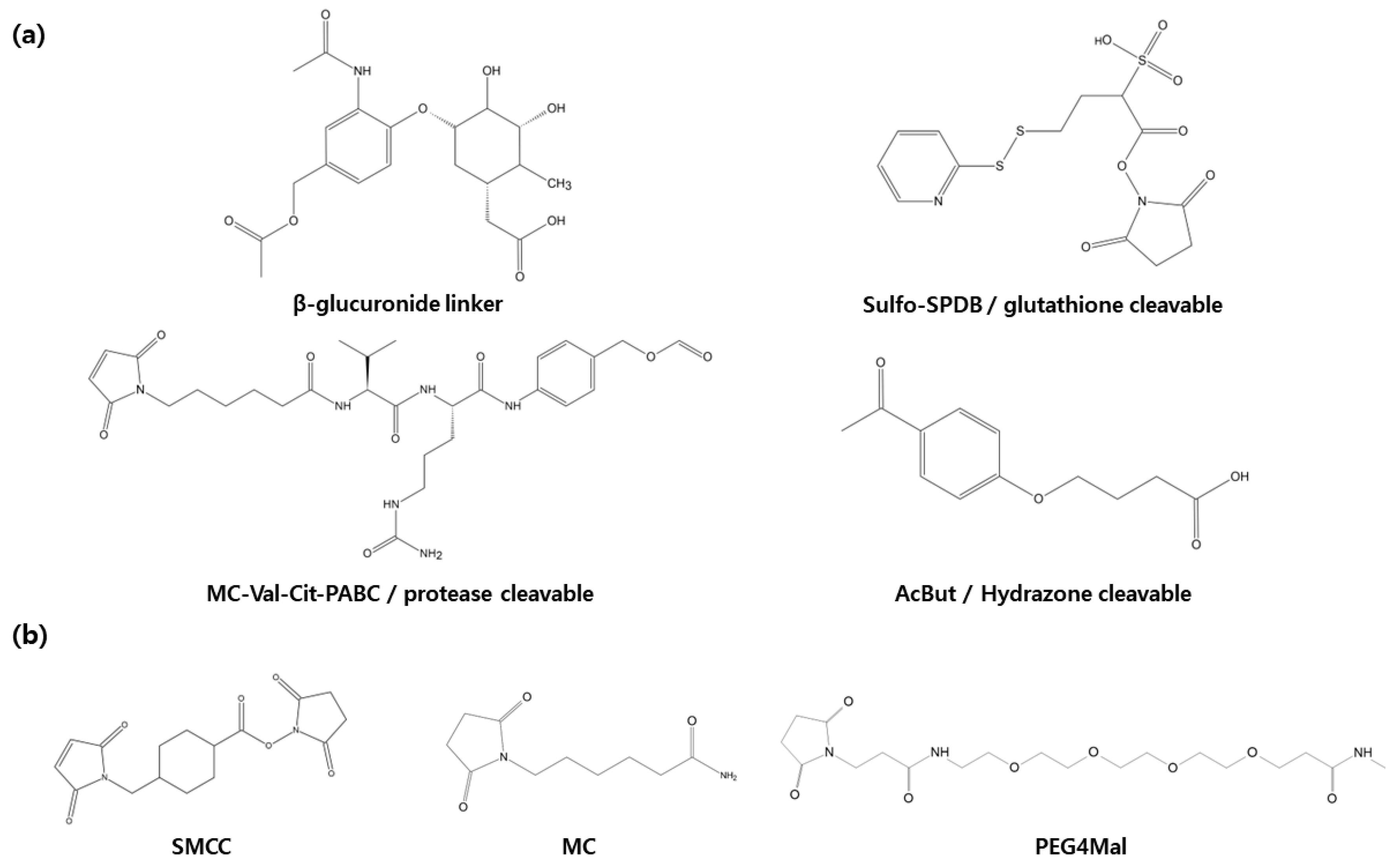
2.2. Linker
2.3. Payload
2.4. Conjugation
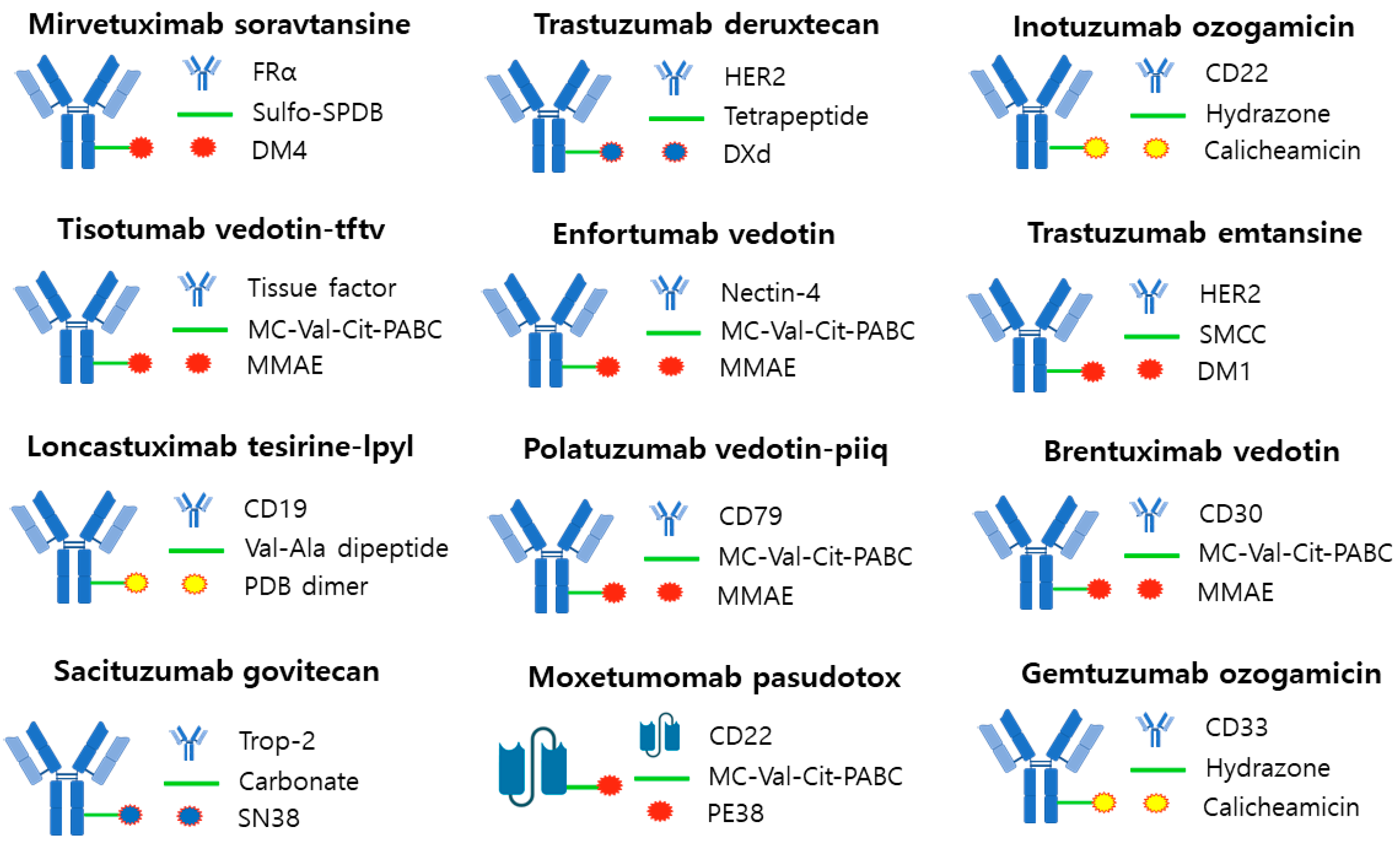
3. US FDA Approved ADC and Market Status
3.1. Mirvetuximab Soravtansine (ELAHERE®—ImmunoGen)
3.2. Tisotumab Vedotin-Tftv (Tivdak®—Seagen)
3.3. Loncastuximab Tesirine-Lpyl (Zynlonta®—ADC Therapeutics)
3.4. Belantamab Mafodotin-Blmf (Blenrep®—GlaxoSmithKline)
3.5. Sacituzumab Govitecan (Trodelvy®—Immunomedics)
3.6. Trastuzumab Deruxtecan (Enhertu®—Daiichi Sankyo/AstraZeneca)
3.7. Enfortumab Vedotin (Padcev®—Astellas/Seagen Genetics)
3.8. Polatuzumab Vedotin-Piiq (Polivy®—Genentech/Roche)
3.9. Inotuzumab Ozogamicin (Besponsa®—Pfizer/Wyeth)
3.10. Trastuzumab Emtansine (Kadcyla®—Roche)
3.11. Brentuximab Vedotin (Adcetris®—Seagen/Takeda)
3.12. Gemtuzumab Ozogamicin (Mylotarg®—Pfizer/Wyeth)
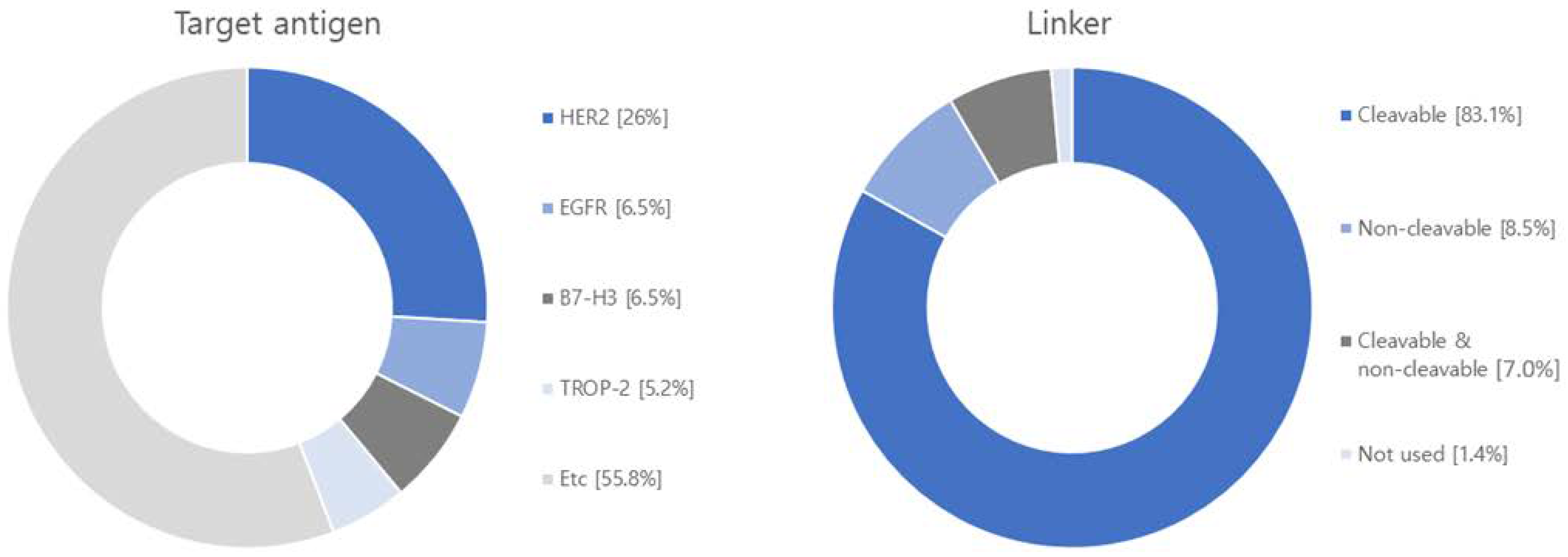
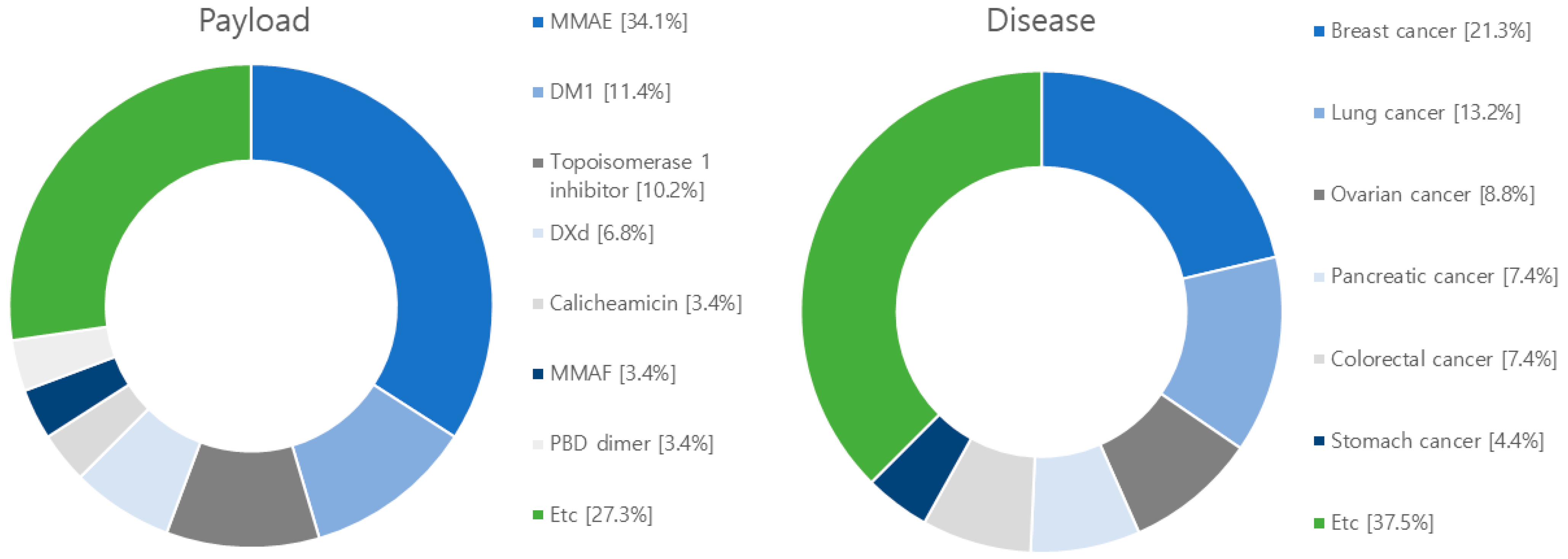
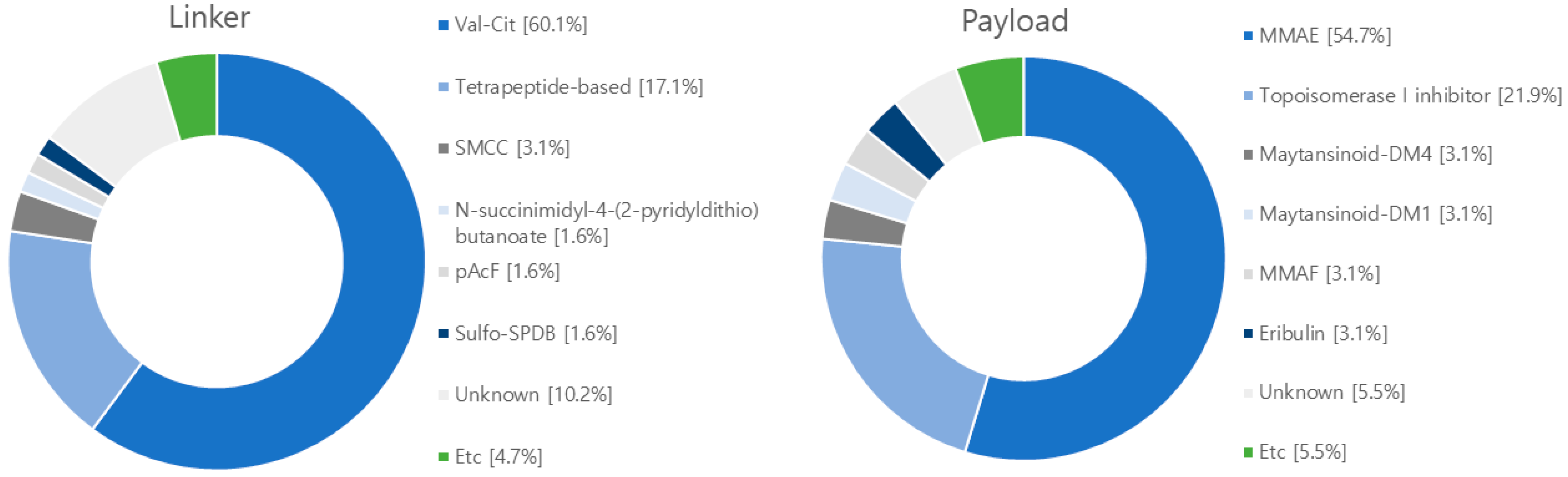
4. Research Trends in ADCs
5. Current Clinical Progress of ADCs
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kesik-Brodacka, M. Progress in biopharmaceutical development. Biotechnol. Appl. Biochem. 2018, 65, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Mongre, R.K.; Mishra, C.B.; Shukla, A.K.; Prakash, A.; Jung, S.; Ashraf-Uz-Zaman, M.; Lee, M.S. Emerging Importance of Tyrosine Kinase Inhibitors against Cancer: Quo Vadis to Cure? Int. J. Mol. Sci. 2021, 22, 11659. [Google Scholar] [CrossRef] [PubMed]
- Dempke, W.C.M.; Fenchel, K.; Uciechowski, P.; Dale, S.P. Second- and third-generation drugs for immuno-oncology treatment-The more the better? Eur. J. Cancer 2017, 74, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Tsao, L.C.; Force, J.; Hartman, Z.C. Mechanisms of Therapeutic Antitumor Monoclonal Antibodies. Cancer Res. 2021, 81, 4641–4651. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers 2023, 15, 713. [Google Scholar] [CrossRef]
- Lim, B.; Yoo, D.; Chun, Y.; Go, A.; Kim, J.Y.; Lee, H.Y.; Boohaker, R.J.; Cho, K.J.; Ahn, S.; Lee, J.S.; et al. Integrative Analyses Reveal the Anticancer Mechanisms and Sensitivity Markers of the Next-Generation Hypomethylating Agent NTX-301. Cancers 2023, 15, 1737. [Google Scholar] [CrossRef]
- Kwon, S.-I. Market Trend and Current Status of the Research and Development of Antibody-Drug Conjugates. Biomed. Sci. Lett. 2021, 27, 121–133. [Google Scholar] [CrossRef]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- PEG, B. FDA Approved Antibody-Drug Conjugates (ADCs) by 2023. Available online: https://www.biochempeg.com/article/74.html (accessed on 3 September 2023).
- Hong, Y.; Nam, S.M.; Moon, A. Antibody-drug conjugates and bispecific antibodies targeting cancers: Applications of click chemistry. Arch. Pharm. Res. 2023, 46, 131–148. [Google Scholar] [CrossRef]
- Drake, P.M.; Rabuka, D. Recent Developments in ADC Technology: Preclinical Studies Signal Future Clinical Trends. BioDrugs 2017, 31, 521–531. [Google Scholar] [CrossRef]
- Tai, Y.T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef] [PubMed]
- Birrer, M.J.; Moore, K.N.; Betella, I.; Bates, R.C. Antibody-Drug Conjugate-Based Therapeutics: State of the Science. J. Natl. Cancer Inst. 2019, 111, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Staudacher, A.H.; Brown, M.P. Antibody drug conjugates and bystander killing: Is antigen-dependent internalisation required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody-Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- De Cecco, M.; Galbraith, D.N.; McDermott, L.L. What makes a good antibody-drug conjugate? Expert Opin. Biol. Ther. 2021, 21, 841–847. [Google Scholar] [CrossRef]
- Lipman, N.S.; Jackson, L.R.; Trudel, L.J.; Weis-Garcia, F. Monoclonal versus polyclonal antibodies: Distinguishing characteristics, applications, and information resources. ILAR J. 2005, 46, 258–268. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; Ko, C.W.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist 2018, 23, 1103–1108. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Oh, D.Y.; Bang, Y.J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef]
- Hock, M.B.; Thudium, K.E.; Carrasco-Triguero, M.; Schwabe, N.F. Immunogenicity of antibody drug conjugates: Bioanalytical methods and monitoring strategy for a novel therapeutic modality. AAPS J. 2015, 17, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs 2013, 5, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016, 8, 659–671. [Google Scholar] [CrossRef]
- Zhang, J.; Woods, C.; He, F.; Han, M.; Treuheit, M.J.; Volkin, D.B. Structural Changes and Aggregation Mechanisms of Two Different Dimers of an IgG2 Monoclonal Antibody. Biochemistry 2018, 57, 5466–5479. [Google Scholar] [CrossRef]
- Mahalingaiah, P.K.; Ciurlionis, R.; Durbin, K.R.; Yeager, R.L.; Philip, B.K.; Bawa, B.; Mantena, S.R.; Enright, B.P.; Liguori, M.J.; Van Vleet, T.R. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol. Ther. 2019, 200, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Tsumura, R.; Manabe, S.; Takashima, H.; Koga, Y.; Yasunaga, M.; Matsumura, Y. Influence of the dissociation rate constant on the intra-tumor distribution of antibody-drug conjugate against tissue factor. J. Control. Release 2018, 284, 49–56. [Google Scholar] [CrossRef]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef]
- Bordeau, B.M.; Nguyen, T.D.; Polli, J.R.; Chen, P.; Balthasar, J.P. Payload-Binding Fab Fragments Increase the Therapeutic Index of MMAE Antibody-Drug Conjugates. Mol. Cancer Ther. 2023, 22, 459–470. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Q.; Kong, Y.; Wang, Z.; Lei, C.; Li, J.; Ding, L.; Wang, C.; Cheng, Y.; Wei, Y.; et al. A highly stable human single-domain antibody-drug conjugate exhibits superior penetration and treatment of solid tumors. Mol. Ther. 2022, 30, 2785–2799. [Google Scholar] [CrossRef]
- Saber, H.; Leighton, J.K. An FDA oncology analysis of antibody-drug conjugates. Regul. Toxicol. Pharmacol. 2015, 71, 444–452. [Google Scholar] [CrossRef]
- Su, D.; Kozak, K.R.; Sadowsky, J.; Yu, S.F.; Fourie-O’Donohue, A.; Nelson, C.; Vandlen, R.; Ohri, R.; Liu, L.; Ng, C.; et al. Modulating Antibody-Drug Conjugate Payload Metabolism by Conjugation Site and Linker Modification. Bioconjug. Chem. 2018, 29, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Delaria, K.; Foletti, D.; Witt, J.M.; Hasa-Moreno, A.; Poulsen, K.; Casas, M.G.; Dorywalska, M.; Farias, S.; Pios, A.; et al. Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nat. Biotechnol. 2015, 33, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, Y.; Li, W.; Jeanty, C.; Xiang, G.; Dong, Y. Recent advances of antibody drug conjugates for clinical applications. Acta Pharm. Sin. B 2020, 10, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Ducry, L.; Stump, B. Antibody-drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjug. Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Kayser, S.; Levis, M.J. The clinical impact of the molecular landscape of acute myeloid leukemia. Haematologica 2023, 108, 308–320. [Google Scholar] [CrossRef]
- Whiteman, K.R.; Johnson, H.A.; Mayo, M.F.; Audette, C.A.; Carrigan, C.N.; LaBelle, A.; Zukerberg, L.; Lambert, J.M.; Lutz, R.J. Lorvotuzumab mertansine, a CD56-targeting antibody-drug conjugate with potent antitumor activity against small cell lung cancer in human xenograft models. MAbs 2014, 6, 556–566. [Google Scholar] [CrossRef]
- Desai, A.; Abdayem, P.; Adjei, A.A.; Planchard, D. Antibody-drug conjugates: A promising novel therapeutic approach in lung cancer. Lung Cancer 2022, 163, 96–106. [Google Scholar] [CrossRef]
- Moskowitz, C.H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M.H.; Chen, A.I.; Stiff, P.; Gianni, A.M.; Carella, A.; et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862. [Google Scholar] [CrossRef]
- Bargh, J.D.; Walsh, S.J.; Isidro-Llobet, A.; Omarjee, S.; Carroll, J.S.; Spring, D.R. Sulfatase-cleavable linkers for antibody-drug conjugates. Chem. Sci. 2020, 11, 2375–2380. [Google Scholar] [CrossRef]
- Jackson, C.P.; Fang, S.; Benjamin, S.R.; Alayi, T.; Hathout, Y.; Gillen, S.M.; Handel, J.P.; Brems, B.M.; Howe, J.M.; Tumey, L.N. Evaluation of an ester-linked immunosuppressive payload: A case study in understanding the stability and cleavability of ester-containing ADC linkers. Bioorg. Med. Chem. Lett. 2022, 75, 128953. [Google Scholar] [CrossRef]
- Bargh, J.D.; Walsh, S.J.; Ashman, N.; Isidro-Llobet, A.; Carroll, J.S.; Spring, D.R. A dual-enzyme cleavable linker for antibody-drug conjugates. Chem. Commun. 2021, 57, 3457–3460. [Google Scholar] [CrossRef] [PubMed]
- Lopez Rivas, P.; Muller, C.; Breunig, C.; Hechler, T.; Pahl, A.; Arosio, D.; Belvisi, L.; Pignataro, L.; Dal Corso, A.; Gennari, C. beta-Glucuronidase triggers extracellular MMAE release from an integrin-targeted conjugate. Org. Biomol. Chem. 2019, 17, 4705–4710. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, C.; Wu, Y.; Dong, L.; Chen, F.; Dong, K.; Song, H. Simple and Rapid LC-MS/MS Methods for Quantifying Catabolites of Antibody-Drug Conjugates with SMCC Linker. J. Chromatogr. Sci. 2021, 59, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kim, M.T.; Zheng, L.; Deperalta, G.; Jacobson, F. Structural Characterization of Cross-Linked Species in Trastuzumab Emtansine (Kadcyla). Bioconjug Chem. 2016, 27, 2037–2047. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Dahan, A. Computational Simulations to Guide Enzyme-Mediated Prodrug Activation. Int. J. Mol. Sci. 2020, 21, 3621. [Google Scholar] [CrossRef]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef]
- Liu, J.; Yu, Y.; Liu, C.; Gao, C.; Zhuang, J.; Liu, L.; Wu, Q.; Ma, W.; Zhang, Q.; Sun, C. Combinatorial regimens of chemotherapeutic agents: A new perspective on raising the heat of the tumor immune microenvironment. Front. Pharmacol. 2022, 13, 1035954. [Google Scholar] [CrossRef]
- Yu, B.; Jiang, T.; Liu, D. BCMA-targeted immunotherapy for multiple myeloma. J. Hematol. Oncol. 2020, 13, 125. [Google Scholar] [CrossRef]
- Yaghoubi, S.; Karimi, M.H.; Lotfinia, M.; Gharibi, T.; Mahi-Birjand, M.; Kavi, E.; Hosseini, F.; Sineh Sepehr, K.; Khatami, M.; Bagheri, N.; et al. Potential drugs used in the antibody-drug conjugate (ADC) architecture for cancer therapy. J. Cell. Physiol. 2020, 235, 31–64. [Google Scholar] [CrossRef]
- Mantaj, J.; Jackson, P.J.; Rahman, K.M.; Thurston, D.E. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody-Drug Conjugates (ADCs). Angew. Chem. Int. Ed. Engl. 2017, 56, 462–488. [Google Scholar] [CrossRef]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates--an emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Best, R.L.; LaPointe, N.E.; Azarenko, O.; Miller, H.; Genualdi, C.; Chih, S.; Shen, B.Q.; Jordan, M.A.; Wilson, L.; Feinstein, S.C.; et al. Microtubule and tubulin binding and regulation of microtubule dynamics by the antibody drug conjugate (ADC) payload, monomethyl auristatin E (MMAE): Mechanistic insights into MMAE ADC peripheral neuropathy. Toxicol. Appl. Pharmacol. 2021, 421, 115534. [Google Scholar] [CrossRef] [PubMed]
- Yudong, H.; Kyoung-gon, K.; Jae-Youn, J.; Suk-Jin, H.; Kwang, L. Antibody-Drug Conjugates: Development and Advances. KSBB J. 2021, 36, 87–98. [Google Scholar]
- Nittoli, T.; Kelly, M.P.; Delfino, F.; Rudge, J.; Kunz, A.; Markotan, T.; Spink, J.; Chen, Z.; Shan, J.; Navarro, E.; et al. Antibody drug conjugates of cleavable amino-alkyl and aryl maytansinoids. Bioorg. Med. Chem. 2018, 26, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Goldmacher, V.S.; Kovtun, Y.V. Antibody-drug conjugates: Using monoclonal antibodies for delivery of cytotoxic payloads to cancer cells. Ther. Deliv. 2011, 2, 397–416. [Google Scholar] [CrossRef] [PubMed]
- Hobson, A.D.; McPherson, M.J.; Waegell, W.; Goess, C.A.; Stoffel, R.H.; Li, X.; Zhou, J.; Wang, Z.; Yu, Y.; Hernandez, A., Jr.; et al. Design and Development of Glucocorticoid Receptor Modulators as Immunology Antibody-Drug Conjugate Payloads. J. Med. Chem. 2022, 65, 4500–4533. [Google Scholar] [CrossRef]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef]
- Zhijia Wang, H.L.; Gou, L.; Li, W.; Wang, Y. Antibody-drug conjugates: Recent advances in payloads. Acta Pharm. Sin. B 2023, 13, 4025–4059. [Google Scholar] [CrossRef]
- Almaliti, J.; Miller, B.; Pietraszkiewicz, H.; Glukhov, E.; Naman, C.B.; Kline, T.; Hanson, J.; Li, X.; Zhou, S.; Valeriote, F.A.; et al. Exploration of the carmaphycins as payloads in antibody drug conjugate anticancer agents. Eur. J. Med. Chem. 2019, 161, 416–432. [Google Scholar] [CrossRef]
- Puthenveetil, S.; Loganzo, F.; He, H.; Dirico, K.; Green, M.; Teske, J.; Musto, S.; Clark, T.; Rago, B.; Koehn, F.; et al. Natural Product Splicing Inhibitors: A New Class of Antibody-Drug Conjugate (ADC) Payloads. Bioconjug. Chem. 2016, 27, 1880–1888. [Google Scholar] [CrossRef]
- Burke, P.J.; Toki, B.E.; Meyer, D.W.; Miyamoto, J.B.; Kissler, K.M.; Anderson, M.; Senter, P.D.; Jeffrey, S.C. Novel immunoconjugates comprised of streptonigrin and 17-amino-geldanamycin attached via a dipeptide-p-aminobenzyl-amine linker system. Bioorg. Med. Chem. Lett. 2009, 19, 2650–2653. [Google Scholar] [CrossRef]
- Yao, H.P.; Zhao, H.; Hudson, R.; Tong, X.M.; Wang, M.H. Duocarmycin-based antibody-drug conjugates as an emerging biotherapeutic entity for targeted cancer therapy: Pharmaceutical strategy and clinical progress. Drug Discov. Today 2021, 26, 1857–1874. [Google Scholar] [CrossRef]
- Hartley, J.A.; Flynn, M.J.; Bingham, J.P.; Corbett, S.; Reinert, H.; Tiberghien, A.; Masterson, L.A.; Antonow, D.; Adams, L.; Chowdhury, S.; et al. Pre-clinical pharmacology and mechanism of action of SG3199, the pyrrolobenzodiazepine (PBD) dimer warhead component of antibody-drug conjugate (ADC) payload tesirine. Sci. Rep. 2018, 8, 10479. [Google Scholar] [CrossRef]
- Szot, C.; Saha, S.; Zhang, X.M.; Zhu, Z.; Hilton, M.B.; Morris, K.; Seaman, S.; Dunleavey, J.M.; Hsu, K.S.; Yu, G.J.; et al. Tumor stroma-targeted antibody-drug conjugate triggers localized anticancer drug release. J. Clin. Investig. 2018, 128, 2927–2943. [Google Scholar] [CrossRef] [PubMed]
- Mckertish, C.M.; Kayser, V. A Novel Dual-Payload ADC for the Treatment of HER2+ Breast and Colon Cancer. Pharmaceutics 2023, 15, 2020. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of Drug-Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody-Maytansinoid Conjugates. Bioconjug. Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Lhospice, F.; Bregeon, D.; Belmant, C.; Dennler, P.; Chiotellis, A.; Fischer, E.; Gauthier, L.; Boedec, A.; Rispaud, H.; Savard-Chambard, S.; et al. Site-Specific Conjugation of Monomethyl Auristatin E to Anti-CD30 Antibodies Improves Their Pharmacokinetics and Therapeutic Index in Rodent Models. Mol. Pharm. 2015, 12, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Chih, H.W.; Gikanga, B.; Yang, Y.; Zhang, B. Identification of amino acid residues responsible for the release of free drug from an antibody-drug conjugate utilizing lysine-succinimidyl ester chemistry. J. Pharm. Sci. 2011, 100, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Madler, S.; Bich, C.; Touboul, D.; Zenobi, R. Chemical cross-linking with NHS esters: A systematic study on amino acid reactivities. J. Mass Spectrom. 2009, 44, 694–706. [Google Scholar] [CrossRef]
- Wang, L.; Amphlett, G.; Blattler, W.A.; Lambert, J.M.; Zhang, W. Structural characterization of the maytansinoid-monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci. 2005, 14, 2436–2446. [Google Scholar] [CrossRef] [PubMed]
- Wakankar, A.; Chen, Y.; Gokarn, Y.; Jacobson, F.S. Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs 2011, 3, 161–172. [Google Scholar] [CrossRef]
- Gordon, M.R.; Canakci, M.; Li, L.; Zhuang, J.; Osborne, B.; Thayumanavan, S. Field Guide to Challenges and Opportunities in Antibody-Drug Conjugates for Chemists. Bioconjug. Chem. 2015, 26, 2198–2215. [Google Scholar] [CrossRef] [PubMed]
- Sadowsky, J.D.; Pillow, T.H.; Chen, J.; Fan, F.; He, C.; Wang, Y.; Yan, G.; Yao, H.; Xu, Z.; Martin, S.; et al. Development of Efficient Chemistry to Generate Site-Specific Disulfide-Linked Protein- and Peptide-Payload Conjugates: Application to THIOMAB Antibody-Drug Conjugates. Bioconjug. Chem. 2017, 28, 2086–2098. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Kyazike, J.; Boudanova, E.; Drzyzga, M.; Honey, D.; Cost, R.; Hou, L.; Duffieux, F.; Brun, M.P.; Park, A.; et al. Site-Specific Antibody Conjugation to Engineered Double Cysteine Residues. Pharmaceuticals 2021, 14, 672. [Google Scholar] [CrossRef]
- Li, X.; Patterson, J.T.; Sarkar, M.; Pedzisa, L.; Kodadek, T.; Roush, W.R.; Rader, C. Site-Specific Dual Antibody Conjugation via Engineered Cysteine and Selenocysteine Residues. Bioconjug. Chem. 2015, 26, 2243–2248. [Google Scholar] [CrossRef]
- Badescu, G.; Bryant, P.; Bird, M.; Henseleit, K.; Swierkosz, J.; Parekh, V.; Tommasi, R.; Pawlisz, E.; Jurlewicz, K.; Farys, M.; et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug. Chem. 2014, 25, 1124–1136. [Google Scholar] [CrossRef]
- Rosen, C.B.; Francis, M.B. Targeting the N terminus for site-selective protein modification. Nat. Chem. Biol. 2017, 13, 697–705. [Google Scholar] [CrossRef]
- York, D.; Baker, J.; Holder, P.G.; Jones, L.C.; Drake, P.M.; Barfield, R.M.; Bleck, G.T.; Rabuka, D. Generating aldehyde-tagged antibodies with high titers and high formylglycine yields by supplementing culture media with copper(II). BMC Biotechnol. 2016, 16, 23. [Google Scholar] [CrossRef]
- Chin, J.W. Expanding and reprogramming the genetic code. Nature 2017, 550, 53–60. [Google Scholar] [CrossRef]
- Young, T.S.; Schultz, P.G. Beyond the canonical 20 amino acids: Expanding the genetic lexicon. J. Biol. Chem. 2010, 285, 11039–11044. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-selective modification strategies in antibody-drug conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef] [PubMed]
- Jeger, S.; Zimmermann, K.; Blanc, A.; Grunberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew. Chem. Int. Ed. Engl. 2010, 49, 9995–9997. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Deweid, L.; Avrutina, O.; Kolmar, H. Recent progress in transglutaminase-mediated assembly of antibody-drug conjugates. Anal. Biochem. 2020, 595, 113615. [Google Scholar] [CrossRef]
- van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody-Drug Conjugates. Bioconjug. Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef]
- Ramakrishnan, B.; Qasba, P.K. Structure-based design of beta 1,4-galactosyltransferase I (beta 4Gal-T1) with equally efficient N-acetylgalactosaminyltransferase activity: Point mutation broadens beta 4Gal-T1 donor specificity. J. Biol. Chem. 2002, 277, 20833–20839. [Google Scholar] [CrossRef]
- Matsuda, Y.; Kliman, M.; Mendelsohn, B.A. Application of Native Ion Exchange Mass Spectrometry to Intact and Subunit Analysis of Site-Specific Antibody-Drug Conjugates Produced by AJICAP First Generation Technology. J. Am. Soc. Mass Spectrom. 2020, 31, 1706–1712. [Google Scholar] [CrossRef]
- Fujii, T.; Matsuda, Y.; Seki, T.; Shikida, N.; Iwai, Y.; Ooba, Y.; Takahashi, K.; Isokawa, M.; Kawaguchi, S.; Hatada, N.; et al. AJICAP Second Generation: Improved Chemical Site-Specific Conjugation Technology for Antibody-Drug Conjugate Production. Bioconjug. Chem. 2023, 34, 728–738. [Google Scholar] [CrossRef]
- Martins, A.C.; Albericio, F.; de la Torre, B.G. FDA Approvals of Biologics in 2022. Biomedicines 2023, 11, 1434. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal. Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Rassy, E.; Rached, L.; Pistilli, B. Antibody drug conjugates targeting HER2: Clinical development in metastatic breast cancer. Breast 2022, 66, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Ellis-Taitt, L. ADCs Coming of Age: Deals, Targets and Catalysts; CITELINE: London, UK, 2023; Available online: https://invivo.citeline.com/IV147692/ADCs-Coming-Of-Age-Deals-Targets-And-Catalysts (accessed on 3 September 2023).
- Company, T.B.R. Antibody Drug Conjugates Global Market Report 2023. Available online: https://hk.finance.yahoo.com/news/antibody-drug-conjugates-global-market-170800766.html (accessed on 9 February 2023).
- Porter, R.L.; Matulonis, U.A. Mirvetuximab soravtansine for platinum-resistant epithelial ovarian cancer. Expert Rev. Anticancer Ther. 2023, 23, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Lorusso, D.; Oaknin, A.; Pignata, S.; Dean, A.; Denys, H.; Colombo, N.; Van Gorp, T.; Konner, J.A.; Marin, M.R.; et al. Efficacy and Safety of Mirvetuximab Soravtansine in Patients with Platinum-Resistant Ovarian Cancer with High Folate Receptor Alpha Expression: Results From the SORAYA Study. J. Clin. Oncol. 2023, 41, 2436–2445. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Li, R.; Wang, H.; Song, P.; Guo, W.; Chen, Z.S. Tisotumab vedotin for the treatment of cervical carcinoma. Drugs Today 2022, 58, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Lorusso, D.; Gennigens, C.; Gonzalez-Martin, A.; Randall, L.; Cibula, D.; Lund, B.; Woelber, L.; Pignata, S.; Forget, F.; et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG-3023/ENGOT-cx6): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021, 22, 609–619. [Google Scholar] [CrossRef]
- Xu, B. Loncastuximab tesirine: An effective therapy for relapsed or refractory diffuse large B-cell lymphoma. Eur. J. Clin. Pharmacol. 2022, 78, 707–719. [Google Scholar] [CrossRef]
- Ketchum, E.B.; Clarke, A.; Clemmons, A.B. Belantamab Mafodotin-blmf: A Novel Antibody-Drug Conjugate for Treatment of Patients With Relapsed/Refractory Multiple Myeloma. J. Adv. Pr. Oncol. 2022, 13, 77–85. [Google Scholar] [CrossRef]
- Baines, A.C.; Ershler, R.; Kanapuru, B.; Xu, Q.; Shen, G.; Li, L.; Ma, L.; Okusanya, O.O.; Simpson, N.E.; Nguyen, W.; et al. FDA Approval Summary: Belantamab Mafodotin for Patients with Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2022, 28, 4629–4633. [Google Scholar] [CrossRef]
- Wahby, S.; Fashoyin-Aje, L.; Osgood, C.L.; Cheng, J.; Fiero, M.H.; Zhang, L.; Tang, S.; Hamed, S.S.; Song, P.; Charlab, R.; et al. FDA Approval Summary: Accelerated Approval of Sacituzumab Govitecan-hziy for Third-line Treatment of Metastatic Triple-negative Breast Cancer. Clin. Cancer Res. 2021, 27, 1850–1854. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Sharkey, R.M. Antibody-drug conjugates targeting TROP-2 and incorporating SN-38: A case study of anti-TROP-2 sacituzumab govitecan. MAbs 2019, 11, 987–995. [Google Scholar] [CrossRef]
- Keam, S.J. Trastuzumab Deruxtecan: First Approval. Drugs 2020, 80, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Mantia, C.M.; Sonpavde, G. Enfortumab vedotin-ejfv for the treatment of advanced urothelial carcinoma. Expert Rev. Anticancer Ther. 2022, 22, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Heath, E.I.; Rosenberg, J.E. The biology and rationale of targeting nectin-4 in urothelial carcinoma. Nat. Rev. Urol. 2021, 18, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Polatuzumab Vedotin: First Global Approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gokbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Zein, N.; Sinha, A.M.; McGahren, W.J.; Ellestad, G.A. Calicheamicin gamma 1I: An antitumor antibiotic that cleaves double-stranded DNA site specifically. Science 1988, 240, 1198–1201. [Google Scholar] [CrossRef]
- Garcia-Alonso, S.; Ocana, A.; Pandiella, A. Trastuzumab Emtansine: Mechanisms of Action and Resistance, Clinical Progress, and Beyond. Trends Cancer 2020, 6, 130–146. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef]
- Castellino, S.M.; Pei, Q.; Parsons, S.K.; Hodgson, D.; McCarten, K.; Horton, T.; Cho, S.; Wu, Y.; Punnett, A.; Dave, H.; et al. Brentuximab Vedotin with Chemotherapy in Pediatric High-Risk Hodgkin’s Lymphoma. N. Engl. J. Med. 2022, 387, 1649–1660. [Google Scholar] [CrossRef]
- van de Donk, N.W.; Dhimolea, E. Brentuximab vedotin. MAbs 2012, 4, 458–465. [Google Scholar] [CrossRef]
- Gandullo-Sanchez, L.; Pandiella, A. An anti-EGFR antibody-drug conjugate overcomes resistance to HER2-targeted drugs. Cancer Lett. 2023, 554, 216024. [Google Scholar] [CrossRef] [PubMed]
- Ashman, N.; Bargh, J.D.; Walsh, S.J.; Greenwood, R.D.; Tiberghien, A.; Carroll, J.S.; Spring, D.R. Peroxide-cleavable linkers for antibody-drug conjugates. Chem. Commun. 2023, 59, 1841–1844. [Google Scholar] [CrossRef] [PubMed]
- Indini, A.; Rijavec, E.; Grossi, F. Trastuzumab Deruxtecan: Changing the Destiny of HER2 Expressing Solid Tumors. Int. J. Mol. Sci. 2021, 22, 4774. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Kim, S.B.; Chung, W.P.; Im, S.A.; Park, Y.H.; Hegg, R.; Kim, M.H.; Tseng, L.M.; Petry, V.; Chung, C.F.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. N. Engl. J. Med. 2022, 386, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Saraswat, N.; Sood, A.; Kumar, D.; Verma, R.; Sushil, K. Clinical Profile of Cutaneous Adverse Effects of Epidermal Growth Factor Receptor Inhibitors: A Prospective Observational Study of 76 Cases. Indian Dermatol. Online J. 2019, 10, 251–255. [Google Scholar] [CrossRef]
- Yu, J.; Fang, T.; Yun, C.; Liu, X.; Cai, X. Antibody-Drug Conjugates Targeting the Human Epidermal Growth Factor Receptor Family in Cancers. Front. Mol. Biosci. 2022, 9, 847835. [Google Scholar] [CrossRef]
- von Achenbach, C.; Silginer, M.; Blot, V.; Weiss, W.A.; Weller, M. Depatuxizumab Mafodotin (ABT-414)-induced Glioblastoma Cell Death Requires EGFR Overexpression, but not EGFR(Y1068) Phosphorylation. Mol. Cancer Ther. 2020, 19, 1328–1339. [Google Scholar] [CrossRef]
- Yamato, M.; Hasegawa, J.; Maejima, T.; Hattori, C.; Kumagai, K.; Watanabe, A.; Nishiya, Y.; Shibutani, T.; Aida, T.; Hayakawa, I.; et al. DS-7300a, a DNA Topoisomerase I Inhibitor, DXd-Based Antibody-Drug Conjugate Targeting B7-H3, Exerts Potent Antitumor Activities in Preclinical Models. Mol. Cancer Ther. 2022, 21, 635–646. [Google Scholar] [CrossRef]
- Guo, C.; Figueiredo, I.; Gurel, B.; Neeb, A.; Seed, G.; Crespo, M.; Carreira, S.; Rekowski, J.; Buroni, L.; Welti, J.; et al. B7-H3 as a Therapeutic Target in Advanced Prostate Cancer. Eur. Urol. 2023, 83, 224–238. [Google Scholar] [CrossRef]
- Okajima, D.; Yasuda, S.; Maejima, T.; Karibe, T.; Sakurai, K.; Aida, T.; Toki, T.; Yamaguchi, J.; Kitamura, M.; Kamei, R.; et al. Datopotamab Deruxtecan, a Novel TROP2-directed Antibody-drug Conjugate, Demonstrates Potent Antitumor Activity by Efficient Drug Delivery to Tumor Cells. Mol. Cancer Ther. 2021, 20, 2329–2340. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sacituzumab Govitecan: First Approval. Drugs 2020, 80, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Koyama, K.; Ishikawa, H.; Abe, M.; Shiose, Y.; Ueno, S.; Qiu, Y.; Nakamaru, K.; Murakami, M. Patritumab deruxtecan (HER3-DXd), a novel HER3 directed antibody drug conjugate, exhibits in vitro activity against breast cancer cells expressing HER3 mutations with and without HER2 overexpression. PLoS ONE 2022, 17, e0267027. [Google Scholar] [CrossRef] [PubMed]
- Gymnopoulos, M.; Betancourt, O.; Blot, V.; Fujita, R.; Galvan, D.; Lieuw, V.; Nguyen, S.; Snedden, J.; Stewart, C.; Villicana, J.; et al. TR1801-ADC: A highly potent cMet antibody-drug conjugate with high activity in patient-derived xenograft models of solid tumors. Mol. Oncol. 2020, 14, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.M.; Crescioli, S.; Mele, S.; Sachouli, E.; Cheung, A.; Chui, C.K.; Andriollo, P.; Jackson, P.J.M.; Lacy, K.E.; Spicer, J.F.; et al. A Novel Antibody-Drug Conjugate (ADC) Delivering a DNA Mono-Alkylating Payload to Chondroitin Sulfate Proteoglycan (CSPG4)-Expressing Melanoma. Cancers 2020, 12, 1029. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Thomas, A. New Life of Topoisomerase I Inhibitors as Antibody-Drug Conjugate Warheads. Clin. Cancer Res. 2023, 29, 991–993. [Google Scholar] [CrossRef]
- Tumey, L.N.; Leverett, C.A.; Vetelino, B.; Li, F.; Rago, B.; Han, X.; Loganzo, F.; Musto, S.; Bai, G.; Sukuru, S.C.; et al. Optimization of Tubulysin Antibody-Drug Conjugates: A Case Study in Addressing ADC Metabolism. ACS Med. Chem. Lett. 2016, 7, 977–982. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADC | Registered Trademark | Company | Disease | Antigen | Linker | Payload | Approved Year |
|---|---|---|---|---|---|---|---|
| Mirvetuximab soravtansine | ELAHERE | ImmunoGen (Waltham, MA, USA) | Platinum-resistant epithelial ovarian | FRα | Sulfo-SPDB | DM4 | 2022 |
| Tisotumab vedotin-tftv | Tivdak | Seagen Inc (Copenhagen, Denmark) | Recurrent or metastatic cervical cancer | Tissue factor | MC-Val-Cit-PABC | MMAE | 2021 |
| Loncastuximab tesirine-lpyl | Zynlonta | ADC Therapeutics (Epalinges, Switzerland) | Diffuse large B-cell lymphoma | CD19 | Val-Ala dipeptide | PDB dimer | 2021 |
| Belantamab mafodotin-blmf | Blenrep | GlaxoSmithKline (London, UK) | Relapsed or refractory multiple myeloma | BCMA | MC | MMAF | 2020; withdrawn 2022 |
| Sacituzumab govitecan | Trodelvy | Immunomedics (Foster, CA, USA) | Metastatic triple-negative breast cancer | Trop-2 | Carbonate | SN38 | 2020 |
| Trastuzumab deruxtecan | Enhertu | AstraZeneca/Daiichi Sankyo (Cambridge, UK) | Unresectable or metastatic HER2-positive breast cancer | HER2 | Tetrapeptide | DXd | 2019 |
| Enfortumab vedotin | Padcev | Astellas/Seagen Genetics (Tokyo, Japan) | Advanced or metastatic urothelial carcinoma | Nectin-4 | MC-Val-Cit-PABC | MMAE | 2019 |
| Polatuzumab vedotin-piiq | Polivy | Genentech, Roche (Basel, Switzerland) | Relapsed or refractory diffuse large B-cell lymphoma | CD79 | MC-Val-Cit-PABC | MMAE | 2019 |
| Moxetumomab pasudotox | Lumoxiti | Astrazeneca (Cambridge, UK) | Relapsed or refractory hairy cell leukemia | CD22 | MC-Val-Cit-PABC | PE38 | 2018 |
| Inotuzumab ozogamicin | Besponsa | Pfizer/Wyeth (New York, NY, USA) | B-cell acute lymphocytic leukemia | CD22 | Hydrazone | N-acetyl-γ calicheamicin | 2017 |
| Trastuzumab emtansine | Kadcyla | Genentech, Roche (Basel, Switzerland) | HER2-positive breast cancer | HER2 | SMCC | DM1 | 2013 |
| Brentuximab vedotin | Adcetris | Seagen Genetics, Millennium/Takeda (Tokyo, Japan) | Anaplastic large-cell lymphoma | CD30 | MC-Val-Cit-PABC | MMAE | 2011 |
| Gemtuzumab ozogamicin | Mylotarg | Pfizer/Wyeth (New York, NY, USA) | Acute myeloid leukemia | CD33 | Hydrazone | N-acetyl-γ calicheamicin | 2000; reapproved 2017 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, C.H.; Jeong, M.; In, H.; Kim, J.H.; Lin, C.-W.; Han, K.H. Trends in the Development of Antibody-Drug Conjugates for Cancer Therapy. Antibodies 2023, 12, 72. https://doi.org/10.3390/antib12040072
Song CH, Jeong M, In H, Kim JH, Lin C-W, Han KH. Trends in the Development of Antibody-Drug Conjugates for Cancer Therapy. Antibodies. 2023; 12(4):72. https://doi.org/10.3390/antib12040072
Chicago/Turabian StyleSong, Chi Hun, Minchan Jeong, Hyukmin In, Ji Hoe Kim, Chih-Wei Lin, and Kyung Ho Han. 2023. "Trends in the Development of Antibody-Drug Conjugates for Cancer Therapy" Antibodies 12, no. 4: 72. https://doi.org/10.3390/antib12040072
APA StyleSong, C. H., Jeong, M., In, H., Kim, J. H., Lin, C.-W., & Han, K. H. (2023). Trends in the Development of Antibody-Drug Conjugates for Cancer Therapy. Antibodies, 12(4), 72. https://doi.org/10.3390/antib12040072