
Evaluation of Two Chemoenzymatic Glycan Remodeling Approaches to Generate Site-Specific Antibody–Drug Conjugates

 , and
, and
Abstract
:1. Introduction
2. Results
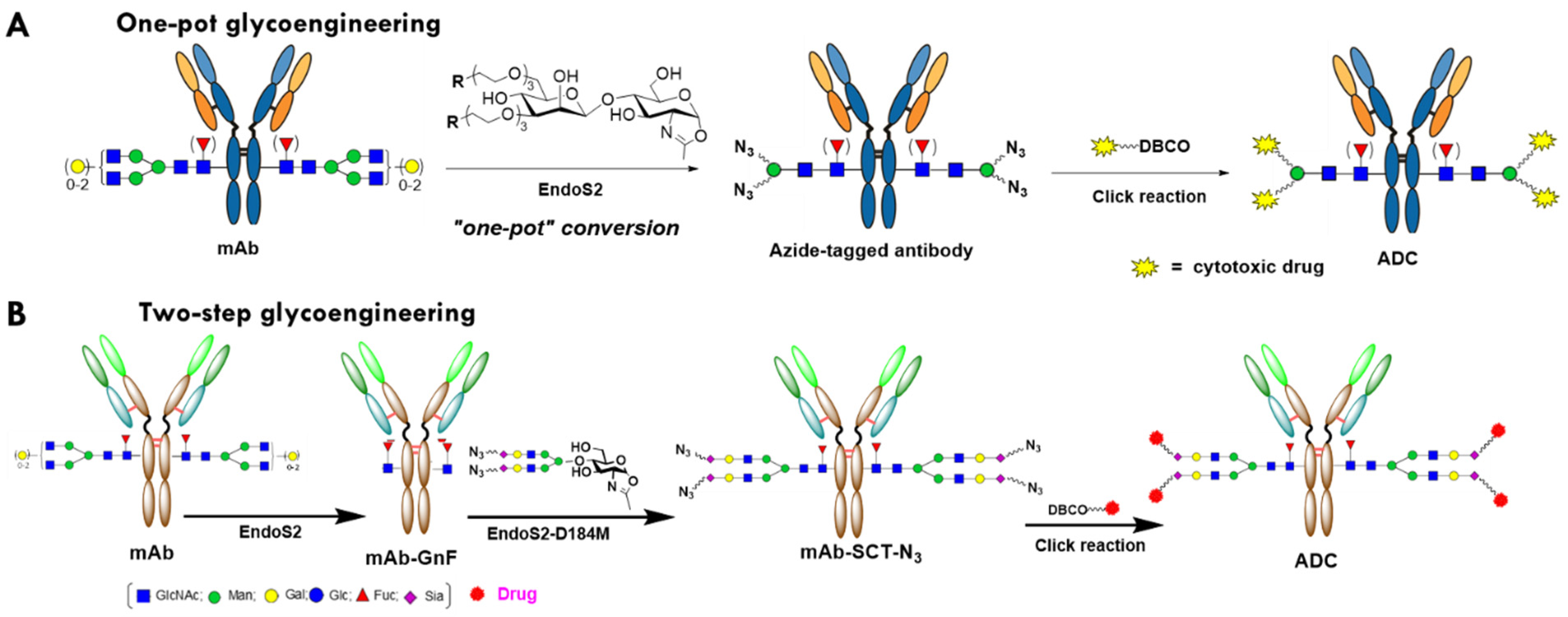
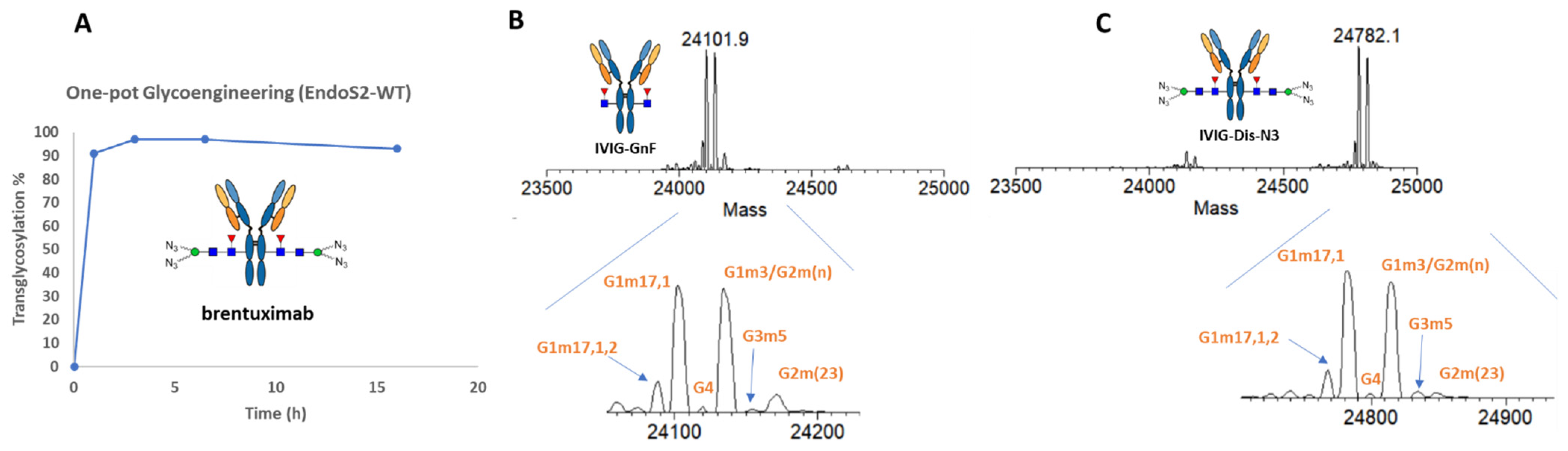
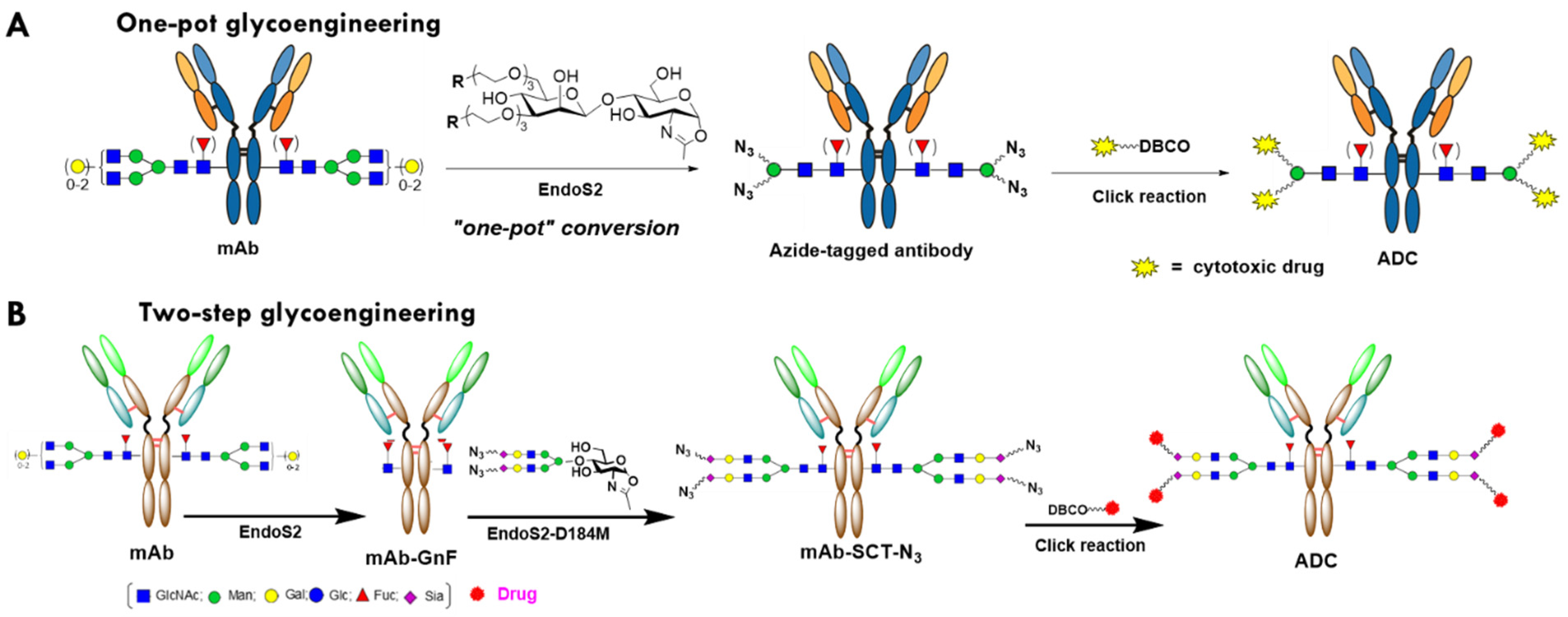
2.1. Evaluation of the One-Pot Glycan-Remodeling Strategy for Different IgGs
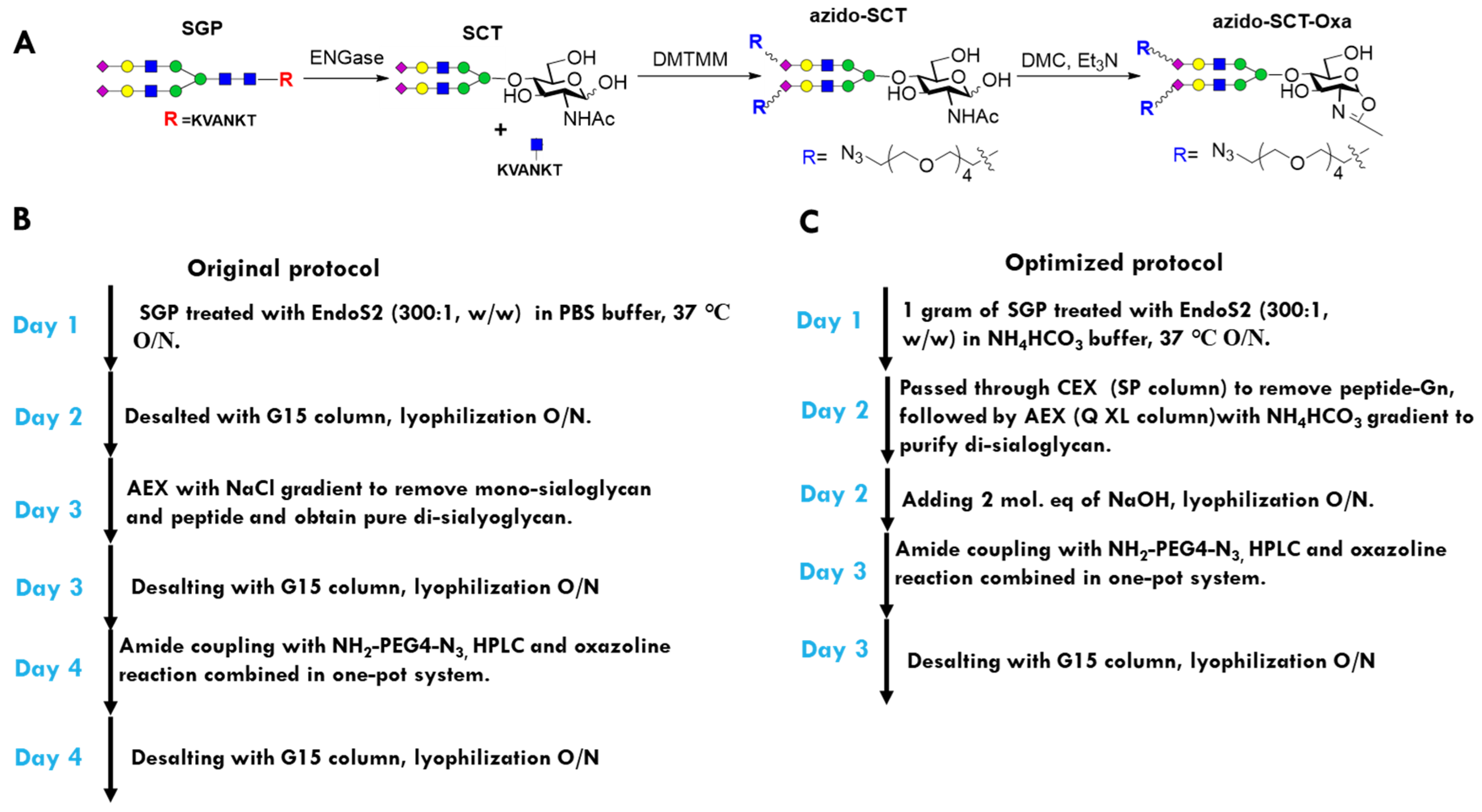
2.2. Optimization of the Synthetic Routes for Azido SCT Oxazoline
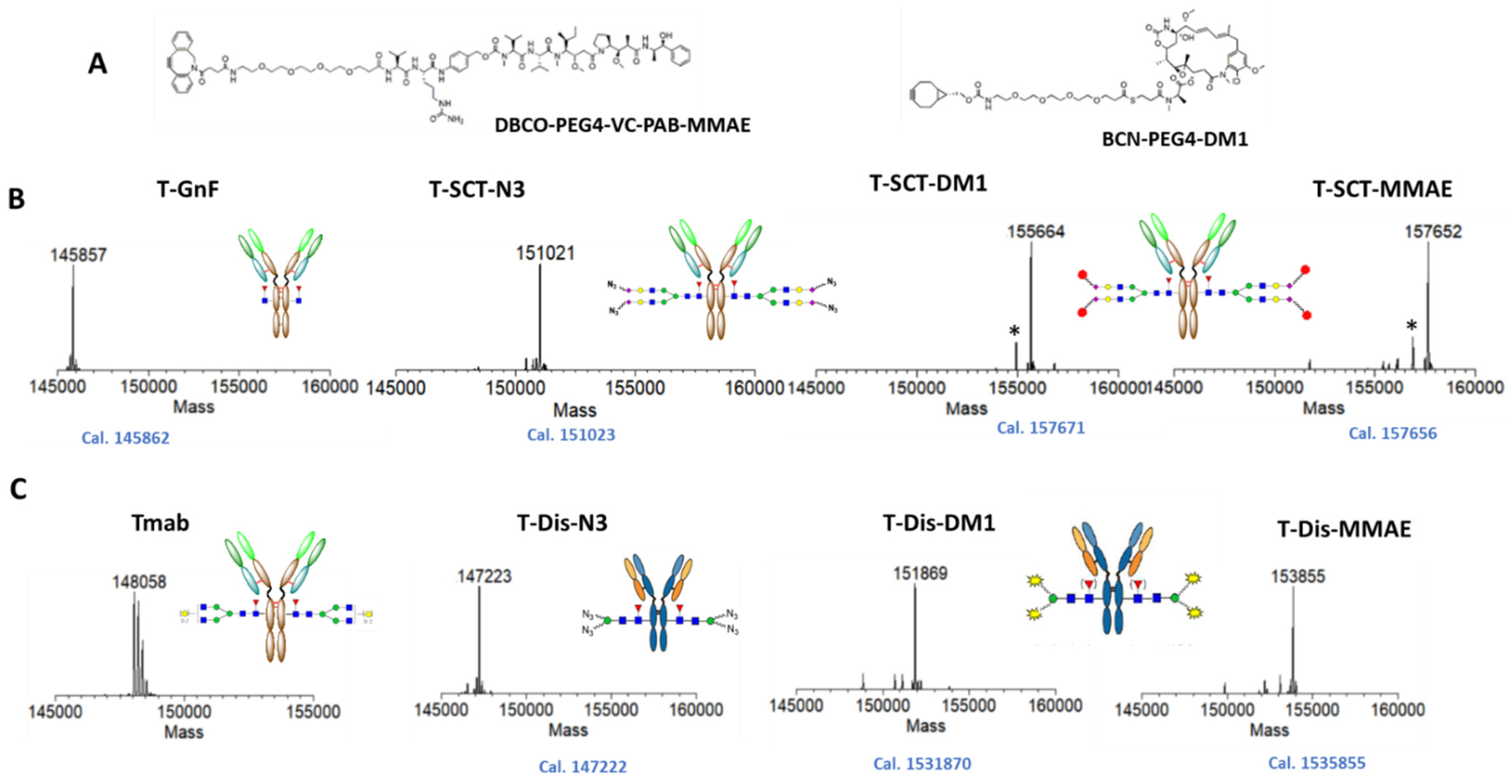
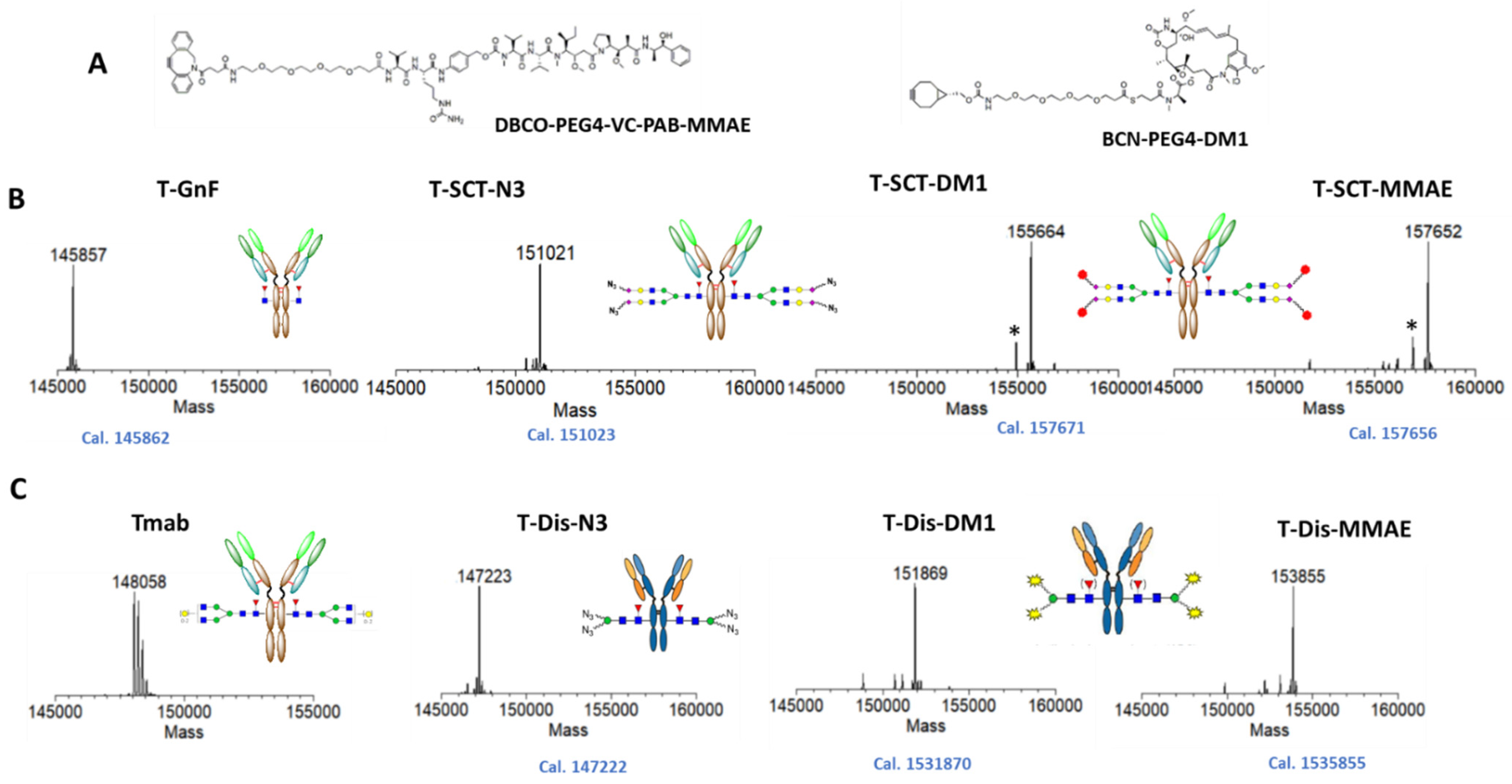
2.3. Preparation of Trastuzumab ADCs with Different Linker-Payload
2.4. In Vitro Characterization of the ADCs with Different Payloads
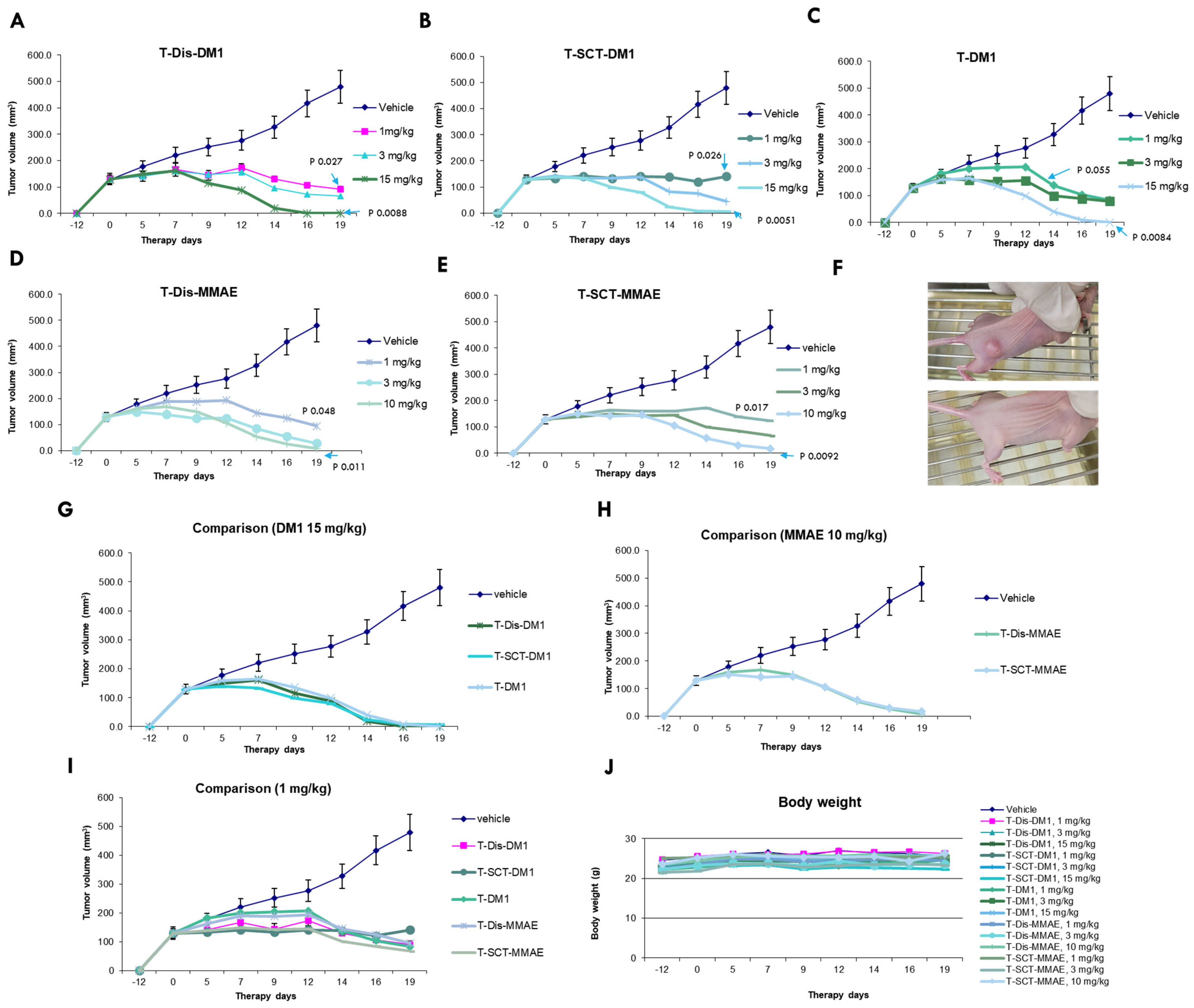
2.5. In Vivo Characterization of Glycosite-Specific ADCs with Different Payloads
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Expression and Purification of Enzymes
4.3. Liquid Chromatography Electrospray Mass Spectrometry (LC-ESI-MS)
4.4. Synthesis of Azido SCT Oxazoline
4.5. One-Pot Glycoengineering of Brentuximab and IVIG
4.6. Preparation of Trastuzumab gADCs with Different Linker-Payloads
4.7. SEC Analysis
4.8. Rat Serum Stability Test
4.9. Affinity to FcRn Receptors
4.10. Cancer Cell Killing Assay
4.11. Xenograft Mouse Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vira, S.; Mekhedov, E.; Humphrey, G.; Blank, P.S. Fluorescent-labeled antibodies: Balancing functionality and degree of labeling. Anal. Biochem. 2010, 402, 146–150. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug. Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Birrer, M.J.; Moore, K.N.; Betella, I.; Bates, R.C. Antibody-drug conjugate-based therapeutics: State of the science. J. Natl. Cancer Inst. 2019, 111, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Wolska-Washer, A.; Robak, T. Safety and tolerability of antibody-drug conjugates in cancer. Drug. Saf. 2019, 42, 295–314. [Google Scholar] [CrossRef]
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody-drug conjugates for cancer therapy. Lancet Oncol. 2016, 17, e254–e262. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell. 2018, 9, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Bertozzi, C.R. Site-specific antibody-drug conjugates: The nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug. Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Morisaki, J.H.; et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]
- Peck, M.; Rothenberg, M.E.; Deng, R.; Lewin-Koh, N.; She, G.; Kamath, A.V.; Carrasco-Triguero, M.; Saad, O.; Castro, A.; Teufel, L.; et al. A phase 1, randomized, single-ascending-dose study to investigate the safety, tolerability, and pharmacokinetics of dsta4637s, an anti-staphylococcus aureus thiomab antibody-antibiotic conjugate, in healthy volunteers. Antimicrob. Agents Chemother. 2019, 63, e02518–e02588. [Google Scholar] [CrossRef]
- Zhou, Q. Site-specific antibody conjugation with payloads beyond cytotoxins. Molecules 2023, 28, 917. [Google Scholar] [CrossRef]
- Ahn, G.; Banik, S.M.; Bertozzi, C.R. Degradation from the outside in: Targeting extracellular and membrane proteins for degradation through the endolysosomal pathway. Cell. Chem. Biol. 2021, 28, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q. Site-specific antibody conjugation for adc and beyond. Biomedicines 2017, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-selective modification strategies in antibody-drug conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef]
- Chudasama, V.; Maruani, A.; Caddick, S. Recent advances in the construction of antibody-drug conjugates. Nat. Chem. 2016, 8, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Delaria, K.; Foletti, D.; Witt, J.M.; Hasa-Moreno, A.; Poulsen, K.; Casas, M.G.; Dorywalska, M.; Farias, S.; Pios, A.; et al. Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nat. Biotechnol. 2015, 33, 694–696. [Google Scholar] [CrossRef]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Dorywalska, M.; Strop, P.; Melton-Witt, J.A.; Hasa-Moreno, A.; Farias, S.E.; Galindo Casas, M.; Delaria, K.; Lui, V.; Poulsen, K.; Loo, C.; et al. Effect of attachment site on stability of cleavable antibody drug conjugates. Bioconjug. Chem. 2015, 26, 650–659. [Google Scholar] [CrossRef]
- van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic conjugation of toxic payloads to the globally conserved n-glycan of native mabs provides homogeneous and highly efficacious antibody-drug conjugates. Bioconjug. Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef]
- Benjamin, S.R.; Jackson, C.P.; Fang, S.; Carlson, D.P.; Guo, Z.; Tumey, L.N. Thiolation of q295: Site-specific conjugation of hydrophobic payloads without the need for genetic engineering. Mol. Pharm. 2019, 16, 2795–2807. [Google Scholar] [CrossRef]
- Kaempffe, A.; Dickgiesser, S.; Rasche, N.; Paoletti, A.; Bertotti, E.; De Salve, I.; Sirtori, F.R.; Kellner, R.; Konning, D.; Hecht, S.; et al. Effect of conjugation site and technique on the stability and pharmacokinetics of antibody-drug conjugates. J. Pharm. Sci. 2021, 110, 3776–3785. [Google Scholar] [CrossRef]
- Shen, B.Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef]
- Ponte, J.F.; Sun, X.; Yoder, N.C.; Fishkin, N.; Laleau, R.; Coccia, J.; Lanieri, L.; Bogalhas, M.; Wang, L.; Wilhelm, S.; et al. Understanding how the stability of the thiol-maleimide linkage impacts the pharmacokinetics of lysine-linked antibody-maytansinoid conjugates. Bioconjug. Chem. 2016, 27, 1588–1598. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.A.; Stengl, A.; Ochtrop, P.; Gerlach, M.; Stoschek, T.; Schumacher, D.; Helma, J.; Penkert, M.; Krause, E.; Leonhardt, H.; et al. Ethynylphosphonamidates for the rapid and cysteine-selective generation of efficacious antibody-drug conjugates. Angew. Chem. Int. Ed. Engl. 2019, 58, 11631–11636. [Google Scholar] [CrossRef] [PubMed]
- Zuberbuhler, K.; Casi, G.; Bernardes, G.J.; Neri, D. Fucose-specific conjugation of hydrazide derivatives to a vascular-targeting monoclonal antibody in igg format. Chem. Commun. 2012, 48, 7100–7102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Stefano, J.E.; Manning, C.; Kyazike, J.; Chen, B.; Gianolio, D.A.; Park, A.; Busch, M.; Bird, J.; Zheng, X.; et al. Site-specific antibody-drug conjugation through glycoengineering. Bioconjug. Chem. 2014, 25, 510–520. [Google Scholar] [CrossRef]
- Wijdeven, M.A.; van Geel, R.; Hoogenboom, J.H.; Verkade, J.M.M.; Janssen, B.M.G.; Hurkmans, I.; de Bever, L.; van Berkel, S.S.; van Delft, F.L. Enzymatic glycan remodeling-metal free click (glycoconnect) provides homogenous antibody-drug conjugates with improved stability and therapeutic index without sequence engineering. mAbs 2022, 14, 2078466. [Google Scholar] [CrossRef]
- Zhang, X.; Ou, C.; Liu, H.; Prabhu, S.K.; Li, C.; Yang, Q.; Wang, L.X. General and robust chemoenzymatic method for glycan-mediated site-specific labeling and conjugation of antibodies: Facile synthesis of homogeneous antibody-drug conjugates. ACS Chem. Biol. 2021, 16, 2502–2514. [Google Scholar] [CrossRef]
- Zhang, X.; Ou, C.; Liu, H.; Wang, L.X. Synthesis and evaluation of three azide-modified disaccharide oxazolines as enzyme substrates for single-step fc glycan-mediated antibody-drug conjugation. Bioconjug. Chem. 2022, 33, 1179–1191. [Google Scholar] [CrossRef]
- Shi, W.; Li, W.; Zhang, J.; Li, T.; Song, Y.; Zeng, Y.; Dong, Q.; Lin, Z.; Gong, L.; Fan, S.; et al. One-step synthesis of site-specific antibody-drug conjugates by reprograming igg glycoengineering with lacnac-based substrates. Acta Pharm. Sin. B 2022, 12, 2417–2428. [Google Scholar] [CrossRef]
- Wang, L.X.; Lomino, J.V. Emerging technologies for making glycan-defined glycoproteins. ACS Chem. Biol. 2012, 7, 110–122. [Google Scholar] [CrossRef]
- Li, C.; Wang, L.X. Chemoenzymatic methods for the synthesis of glycoproteins. Chem. Rev. 2018, 118, 8359–8413. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.X.; Tong, X.; Li, C.; Giddens, J.P.; Li, T. Glycoengineering of antibodies for modulating functions. Annu. Rev. Biochem. 2019, 88, 433–459. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, A.J. Chemoenzymatic synthesis of glycoproteins. Curr. Opin. Chem. Biol. 2019, 53, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Yang, Y.; Tang, Y.; Tang, S.; Yang, L.; Sun, B.; Jiang, B.; Dong, J.; Liu, H.; Huang, M.; et al. One-pot n-glycosylation remodeling of igg with non-natural sialylglycopeptides enables glycosite-specific and dual-payload antibody-drug conjugates. Org. Biomol. Chem. 2016, 14, 9501–9518. [Google Scholar] [CrossRef]
- Ou, C.; Li, C.; Zhang, R.; Yang, Q.; Zong, G.; Dai, Y.; Francis, R.L.; Bournazos, S.; Ravetch, J.V.; Wang, L.X. One-pot conversion of free sialoglycans to functionalized glycan oxazolines and efficient synthesis of homogeneous antibody-drug conjugates through site-specific chemoenzymatic glycan remodeling. Bioconjug. Chem. 2021, 32, 1888–1897. [Google Scholar] [CrossRef]
- Beerli, R.R.; Hell, T.; Merkel, A.S.; Grawunder, U. Sortase enzyme-mediated generation of site-specifically conjugated antibody drug conjugates with high in vitro and in vivo potency. PLoS ONE 2015, 10, e0131177. [Google Scholar] [CrossRef]
- Jeger, S.; Zimmermann, K.; Blanc, A.; Grunberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew. Chem. Int. Ed. Engl. 2010, 49, 9995–9997. [Google Scholar] [CrossRef]
- Dickgiesser, S.; Rieker, M.; Mueller-Pompalla, D.; Schroter, C.; Tonillo, J.; Warszawski, S.; Raab-Westphal, S.; Kuhn, S.; Knehans, T.; Konning, D.; et al. Site-specific conjugation of native antibodies using engineered microbial transglutaminases. Bioconjug. Chem. 2020, 31, 1070–1076. [Google Scholar] [CrossRef]
- Lee, J.J.; Choi, H.J.; Yun, M.; Kang, Y.; Jung, J.E.; Ryu, Y.; Kim, T.Y.; Cha, Y.J.; Cho, H.S.; Min, J.J.; et al. Enzymatic prenylation and oxime ligation for the synthesis of stable and homogeneous protein-drug conjugates for targeted therapy. Angew. Chem. Int. Ed. Engl. 2015, 54, 12020–12024. [Google Scholar] [CrossRef]
- Toda, N.; Ota, Y.; DOI, F.; Meguro, M.; Hayakawa, I.; Ashida, S.; Masuda, T.; Nakada, T.; Iwamoto, M.; Harada, N.; et al. Antibody-Pyrrolobenzodiazepine Derivative Conjugate. US Patent Applicaiton US11583590, 21 February 2023. [Google Scholar]
- Sankyo, D. A Study of ds-9606a in Patients with Advanced Solid Tumors. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05394675 (accessed on 2 November 2023).
- Leblanc, Y.; Romanin, M.; Bihoreau, N.; Chevreux, G. Lc-ms analysis of polyclonal iggs using ides enzymatic proteolysis for oxidation monitoring. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2014, 961, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Seko, A.; Koketsu, M.; Nishizono, M.; Enoki, Y.; Ibrahim, H.R.; Juneja, L.R.; Kim, M.; Yamamoto, T. Occurence of a sialylglycopeptide and free sialylglycans in hen’s egg yolk. Biochim. Biophys. Acta 1997, 1335, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, T.; Boons, G.J. Preparation of well-defined antibody-drug conjugates through glycan remodeling and strain-promoted azide-alkyne cycloadditions. Angew. Chem. Int. Ed. Engl. 2014, 53, 7179–7182. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Zhang, J.; Liu, L.; Li, W.; Liu, Z.; Ren, A.; Wang, J.; Tang, C.; Yang, Y.; Xu, D.; et al. Hiding payload inside the igg fc cavity significantly enhances the therapeutic index of antibody-drug conjugates. J. Med. Chem. 2023, 66, 1011–1026. [Google Scholar] [CrossRef] [PubMed]
- Conilh, L.; Sadilkova, L.; Viricel, W.; Dumontet, C. Payload diversification: A key step in the development of antibody-drug conjugates. J. Hematol. Oncol. 2023, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Tong, X.; Yang, Q.; Giddens, J.P.; Wang, L.X. Glycosynthase mutants of endoglycosidase s2 show potent transglycosylation activity and remarkably relaxed substrate specificity for antibody glycosylation remodeling. J. Biol. Chem. 2016, 291, 16508–16518. [Google Scholar] [CrossRef] [PubMed]
- Giddens, J.P.; Lomino, J.V.; DiLillo, D.J.; Ravetch, J.V.; Wang, L.X. Site-selective chemoenzymatic glycoengineering of fab and fc glycans of a therapeutic antibody. Proc. Natl. Acad. Sci. USA 2018, 115, 12023–12027. [Google Scholar] [CrossRef]
- Liu, L.; Prudden, A.R.; Bosman, G.P.; Boons, G.J. Improved isolation and characterization procedure of sialylglycopeptide from egg yolk powder. Carbohydr. Res. 2017, 452, 122–128. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Company/Partner (Trade Mark) | Common Name | INN | Indications | Approval | Target | Antibody Conjugation | DAR | Linker | Payload |
|---|---|---|---|---|---|---|---|---|---|
| Pfizer (Mylotarg) | CDP-771 | Gemtuzumab ozogamicin | acute myeloid leukaemia (AML) | 2000/2017 (USA) | CD33 | IgG4 (Lysine) | 2~3 | Cleavable hydrazone-disulfide | Calicheamicin |
| Seagen/Takeda (Adcetris) | SGN-35 | Brentuximab vedotin | Hodgkin’s Lymphoma (HL) and anaplastic large-cell lymphoma (ALCL) | 2011 (USA) | CD30 | IgG1 (Cysteine) | ~4 | Cleavable Val-Cit | MMAE |
| Genetech (Kadcyla) | T-DM1 | Trastuzumab emtansine | HER2 + metastatic breast cancer | 2013 (USA) | HER2 | IgG1 (Lysine) | 3.5 | Non-cleavable SMCC | DM-1 |
| Pfizer (Besponsa) | CMC-544 | Inotuzumab ozogamicin | acute lymphoblastic leukaemia (ALL) | 2017 (USA) | CD22 | IgG4 (Lysine) | ~4 | Cleavable hydrazone-disulfide | Calicheamicin |
| Astrazeneca (Lumoxiti) | CAT-8015 | Moxetumomab pasudotox | hairy cell leukemia (HCL) | 2018 (USA) | CD22 | Fc/2 Ab | 1 | Fusion protein | PE38 |
| Genetech (Polivy) | DCDS4501 /RG7596 | Polatuzumab vedotin | diffuse large-B-cell lymphoma (DLBCL) | 2019 (USA) | CD79b | IgG1 (Cysteine) | 3.5 | Cleavable Val-Cit | MMAE |
| Daiichi Sankyo (Enhertu) | DS-8201/T-DXD | Trastuzumab deruxtecan | HER2+ metastatic breast cancer | 2019 (USA) | HER2 | IgG1 (Cysteine) | 7.6 | Cleavable GGFG | DXD |
| Seagen/Astella (Padcev) | ASG-22ME | Enfortumab vedotin | Metastatic urothelial cancer | 2019 (USA) | Nectin-4 | IgG1 (Cysteine) | 3.8 | Cleavable Val-Cit | MMAE |
| Immunomedics/Gilead (Trodelvy) | IMMU-132 | Sacituzumab govitecan | Triple negative breast cancer | 2020 (USA) | TROP2 | IgG (Cysteine) | 7.6 | Cleavable CL2A | SN38 |
| GSK (Blenrep) | GSK2857916 | belantamab mafodotin | Multiple myeloma | 2020 (USA) * | BCMA | IgG1, (Cysteine) | ~4 | Non-cleavable mc | MMAF |
| Rakuten (Akalux) | RM-1929 | Cetuximab sarotalocan | Head and neck squamous cell carcinomas (HNSCC) | 2020 (Japan) | EGFR | IgG1 (lycsine) | ~3 | Non-cleavable | IR700 |
| ADCT (Zynlonta) | ADCT-402 | Loncastuximab- tesirine | Diffuse Large B-cell lymphoma | 2021 (USA) | CD19 | IgG1 (Cysteine) | 2.3 | Cleavable Val-Ala | PBD dimer |
| Seagen (Tivdak) | TF-011-MMAE | Tisotumab vedotin | Recurrent or metastatic cervical cancer | 2021 (USA) | Tissue factor | IgG1 (Cysteine) | 4 | Cleavable Val-Cit | MMAE |
| RemeGen (Aidixi) | RC48 | Disitamab vedotin | gastric cancer | 2021 (China) | HER2 | IgG1 (Cysteine) | 4 | Cleavable Val-Cit | MMAE |
| Immunogen (ELAHERE) | IMGN-853 | Mirvetuximab soravtansine | Platinum-Resistant Ovarian Cancer | 2022 (USA) | FRa | IgG1 (lysine) | 3.4 | Cleavable Sulfo-SPDB | DM4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Chen, H.; Ou, C.; Zheng, Z.; Zhang, X.; Liu, Y.; Zong, G.; Wang, L.-X. Evaluation of Two Chemoenzymatic Glycan Remodeling Approaches to Generate Site-Specific Antibody–Drug Conjugates. Antibodies 2023, 12, 71. https://doi.org/10.3390/antib12040071
Yang Q, Chen H, Ou C, Zheng Z, Zhang X, Liu Y, Zong G, Wang L-X. Evaluation of Two Chemoenzymatic Glycan Remodeling Approaches to Generate Site-Specific Antibody–Drug Conjugates. Antibodies. 2023; 12(4):71. https://doi.org/10.3390/antib12040071
Chicago/Turabian StyleYang, Qiang, He Chen, Chong Ou, Zhihao Zheng, Xiao Zhang, Yunpeng Liu, Guanghui Zong, and Lai-Xi Wang. 2023. "Evaluation of Two Chemoenzymatic Glycan Remodeling Approaches to Generate Site-Specific Antibody–Drug Conjugates" Antibodies 12, no. 4: 71. https://doi.org/10.3390/antib12040071
APA StyleYang, Q., Chen, H., Ou, C., Zheng, Z., Zhang, X., Liu, Y., Zong, G., & Wang, L.-X. (2023). Evaluation of Two Chemoenzymatic Glycan Remodeling Approaches to Generate Site-Specific Antibody–Drug Conjugates. Antibodies, 12(4), 71. https://doi.org/10.3390/antib12040071