An Immunoregulatory Role for Complement Receptors in Murine Models of Breast Cancer

 , , and
, , and
Abstract
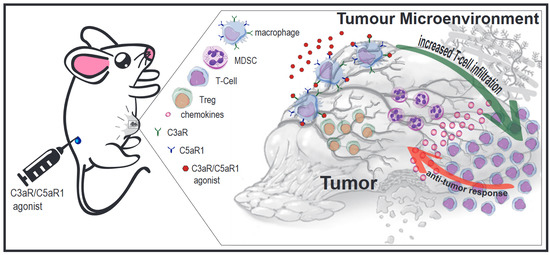
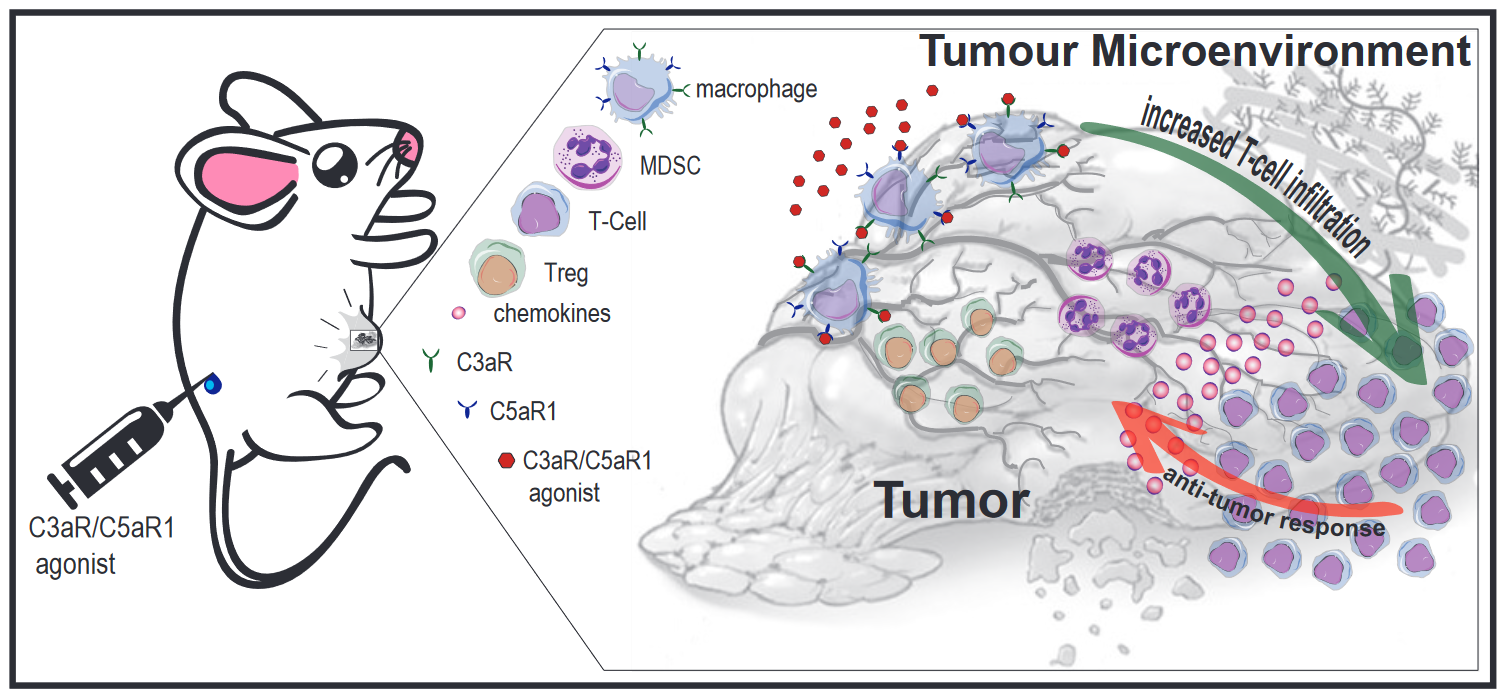{kind=link}
{kind=link}
{kind=link}
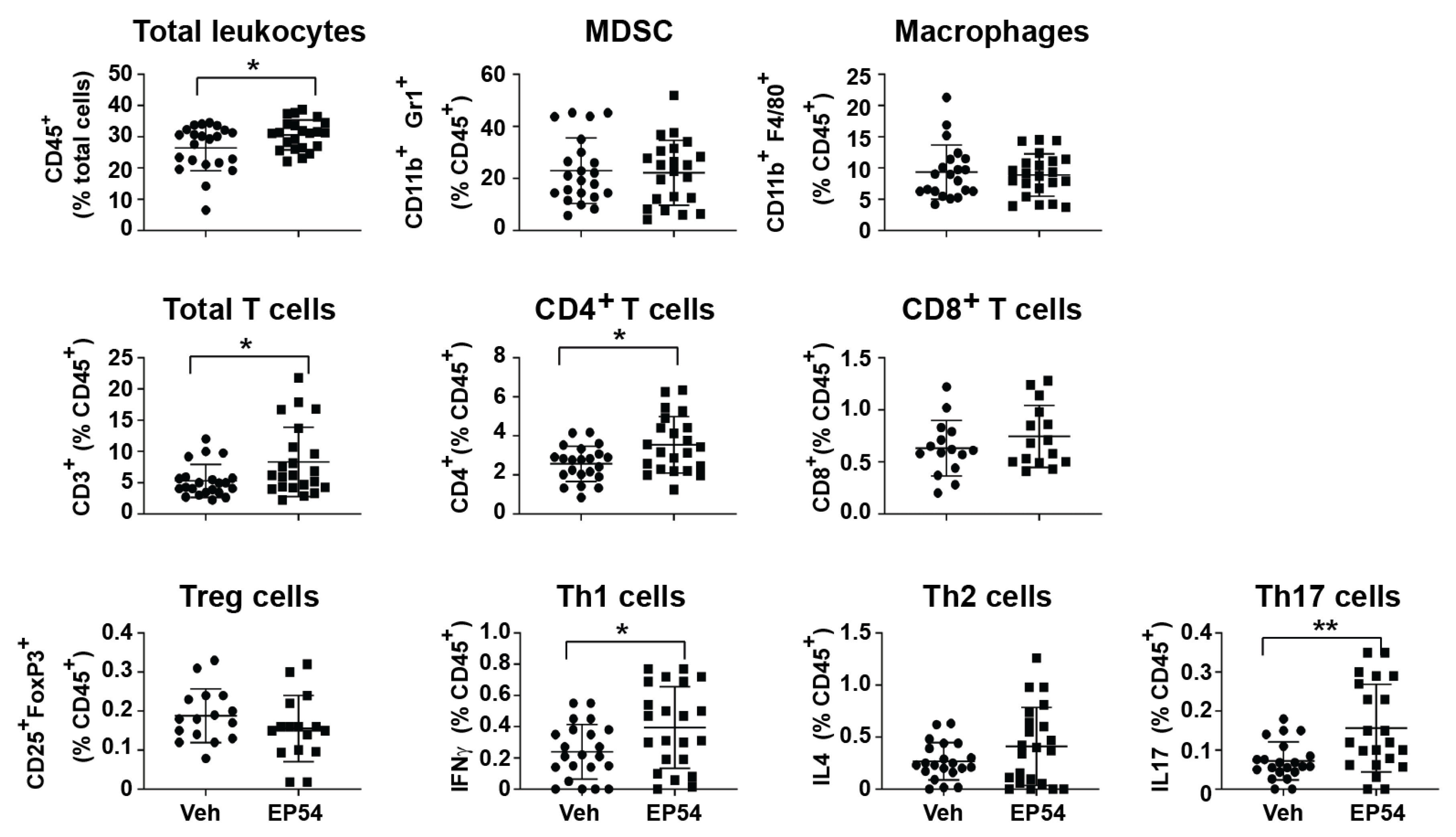{kind=link}
1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Cell Culture
2.3. Animals
Tumor Cell Injections and Drug Treatments
2.4. RNA Extraction and Quantitative Polymerase Chain Reaction (qPCR)
2.5. Calcium Mobilization Assay
2.6. Flow Cytometric Analysis
2.7. Statistical Analysis
3. Results
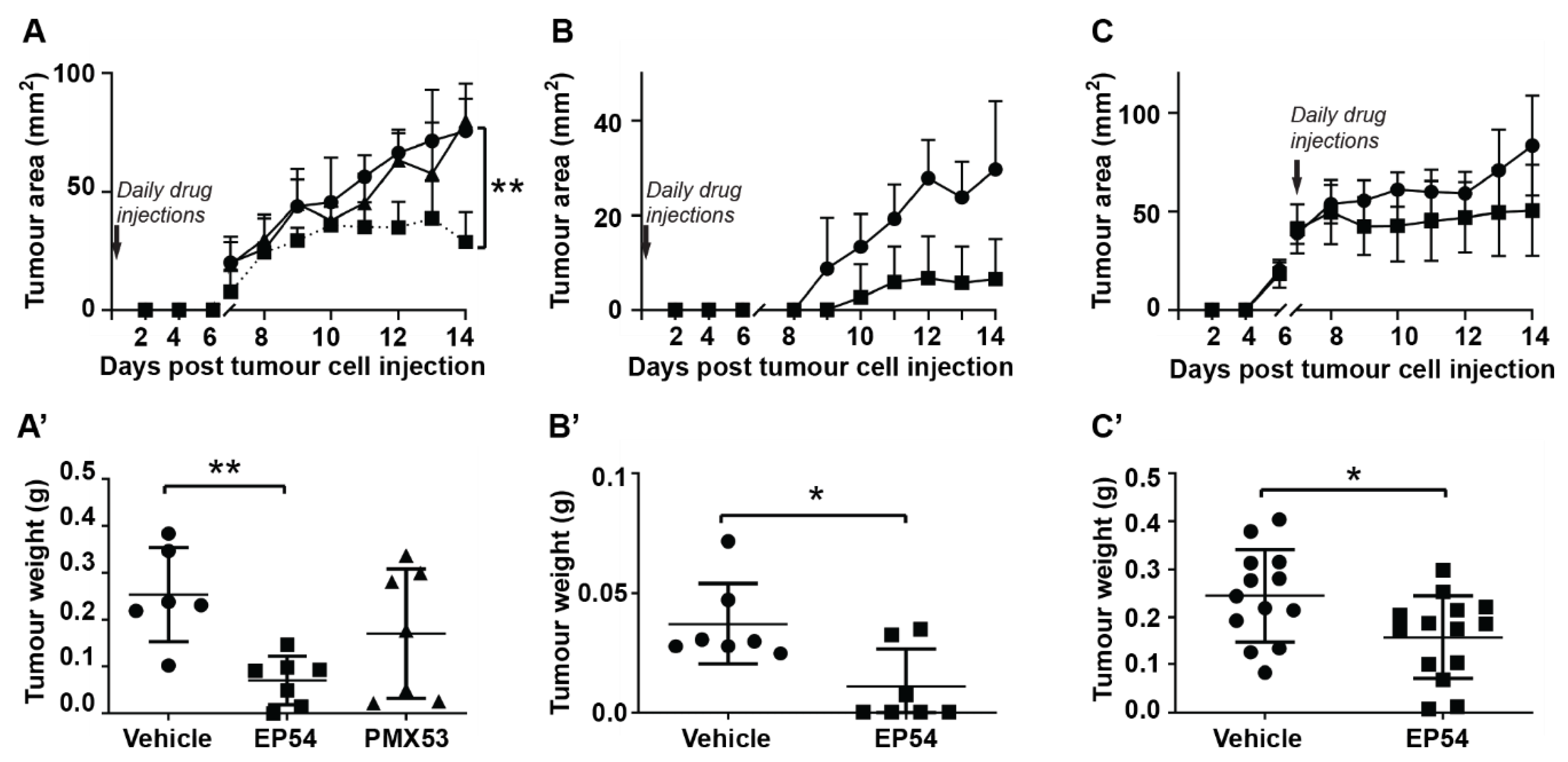
3.1. Effects of Pharmacological Modulation of C3aR/C5aR1 Signaling on Mammary Carcinoma Growth in Mice
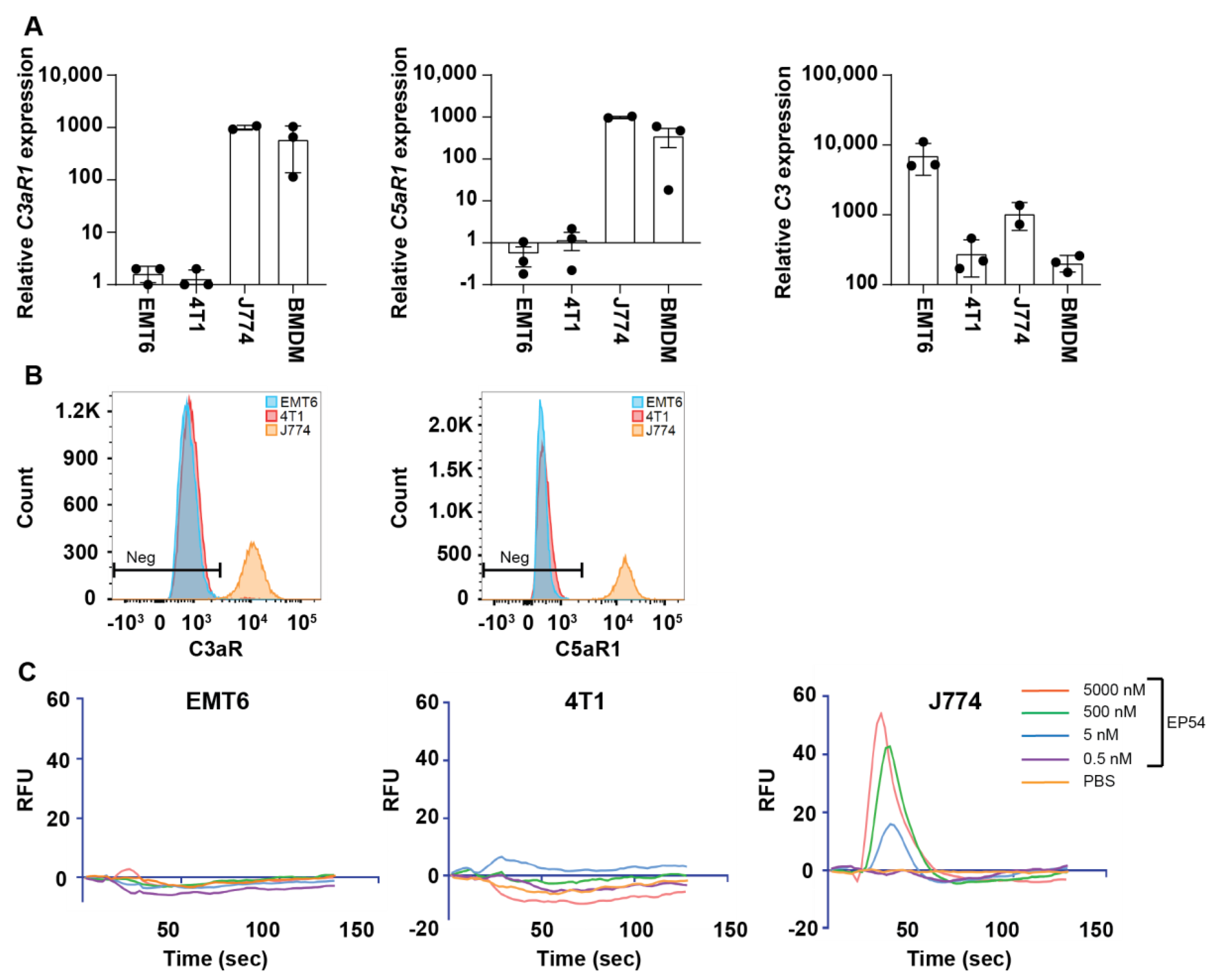
3.2. Expression of Complement Receptors C5aR1 (CD88) and C3aR by EMT6 and 4T1 Mammary Carcinoma Cell Lines
3.3. Leukocyte Response to Pharmacological Modulation of C3aR/C5aR1 in Tumor-Bearing Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Tang, Y.; Hua, S. Immunological Approaches towards Cancer and Inflammation: A Cross Talk. Front. Immunol. 2018, 9, 563. [Google Scholar] [CrossRef]
- Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Mantovani, A.; Lambris, J.D. Complement in cancer: Untangling an intricate relationship. Nat. Rev. Immunol. 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Niculescu, F.; Rus, H.G.; Retegan, M.; Vlaicu, R. Persistent complement activation on tumor cells in breast cancer. Am. J. Pathol. 1992, 140, 1039–1043. [Google Scholar]
- Chung, L.; Moore, K.; Phillips, L.; Boyle, F.M.; Marsh, D.J.; Baxter, R.C. Novel serum protein biomarker panel revealed by mass spectrometry and its prognostic value in breast cancer. Breast Cancer Res. 2014, 16, R63. [Google Scholar] [CrossRef]
- Imamura, T.; Yamamoto-Ibusuki, M.; Sueta, A.; Kubo, T.; Irie, A.; Kikuchi, K.; Kariu, T.; Iwase, H. Influence of the C5a-C5a receptor system on breast cancer progression and patient prognosis. Breast Cancer 2016, 23, 876–885. [Google Scholar] [CrossRef]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S. Cancer and complement. Nat. Biotechnol. 2008, 26, 1348–1349. [Google Scholar] [CrossRef]
- Gelderman, K.A.; Tomlinson, S.; Ross, G.D.; Gorter, A. Complement function in mAb-mediated cancer immunotherapy. Trends Immunol. 2004, 25, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Geller, A.; Yan, J. The Role of Membrane Bound Complement Regulatory Proteins in Tumor Development and Cancer Immunotherapy. Front. Immunol. 2019, 10, 1074. [Google Scholar] [CrossRef]
- Ouyang, Q.; Zhang, L.; Jiang, Y.; Ni, X.; Chen, S.; Ye, F.; Du, Y.; Huang, L.; Ding, P.; Wang, N.; et al. The membrane complement regulatory protein CD59 promotes tumor growth and predicts poor prognosis in breast cancer. Int. J. Oncol. 2016, 48, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Maciejczyk, A.; Szelachowska, J.; Szynglarewicz, B.; Szulc, R.; Szulc, A.; Wysocka, T.; Jagoda, E.; Lage, H.; Surowiak, P. CD46 Expression is an unfavorable prognostic factor in breast cancer cases. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Klos, A.; Wende, E.; Wareham, K.J.; Monk, P.N. International Union of Pharmacology. LXXXVII. Complement peptide C5a, C4a, and C3a receptors. Pharmacol. Rev. 2013, 65, 500–543. [Google Scholar] [CrossRef]
- Lo, M.W.; Woodruff, T.M. Complement: Bridging the innate and adaptive immune systems in sterile inflammation. J. Leukoc. Biol. 2020, 108, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Lee, J.D.; Kemper, C.; Woodruff, T.M. The complement receptor C5aR2: A powerful modulator of innate and adaptive immunity. J. Immunol. 2019, 202, 3339–3348. [Google Scholar] [CrossRef]
- Pandey, S.; Maharana, J.; Li, X.X.; Woodruff, T.M.; Shukla, A.K. Emerging Insights into the Structure and Function of Complement C5a Receptors. Trends Biochem. Sci. 2020, 45, 693–705. [Google Scholar] [CrossRef]
- Wu, M.C.; Brennan, F.H.; Lynch, J.P.; Mantovani, S.; Phipps, S.; Wetsel, R.A.; Ruitenberg, M.J.; Taylor, S.M.; Woodruff, T.M. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc. Natl. Acad. Sci. USA 2013, 110, 9439–9444. [Google Scholar] [CrossRef]
- Coulthard, L.G.; Woodruff, T.M. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J. Immunol. 2015, 194, 3542–3548. [Google Scholar] [CrossRef]
- Kim, D.Y.; Martin, C.B.; Lee, S.N.; Martin, B.K. Expression of complement protein C5a in a murine mammary cancer model: Tumor regression by interference with the cell cycle. Cancer Immunol. Immunother. 2005, 54, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Bandini, S.; Curcio, C.; Macagno, M.; Quaglino, E.; Arigoni, M.; Lanzardo, S.; Hysi, A.; Barutello, G.; Consolino, L.; Longo, D.L.; et al. Early onset and enhanced growth of autochthonous mammary carcinomas in C3-deficient Her2/neu transgenic mice. Oncoimmunology 2013, 2, e26137. [Google Scholar] [CrossRef] [PubMed]
- Vadrevu, S.K.; Chintala, N.K.; Sharma, S.K.; Sharma, P.; Cleveland, C.; Riediger, L.; Manne, S.; Fairlie, D.P.; Gorczyca, W.; Almanza, O.; et al. Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res. 2014, 74, 3454–3465. [Google Scholar] [CrossRef]
- Markiewski, M.M.; DeAngelis, R.A.; Benencia, F.; Ricklin-Lichtsteiner, S.K.; Koutoulaki, A.; Gerard, C.; Coukos, G.; Lambris, J.D. Modulation of the antitumor immune response by complement. Nat. Immunol. 2008, 9, 1225–1235. [Google Scholar] [CrossRef]
- Corrales, L.; Ajona, D.; Rafail, S.; Lasarte, J.J.; Riezu-Boj, J.I.; Lambris, J.D.; Rouzaut, A.; Pajares, M.J.; Montuenga, L.M.; Pio, R. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J. Immunol. 2012, 189, 4674–4683. [Google Scholar] [CrossRef]
- Nunez-Cruz, S.; Gimotty, P.A.; Guerra, M.W.; Connolly, D.C.; Wu, Y.Q.; DeAngelis, R.A.; Lambris, J.D.; Coukos, G.; Scholler, N. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplasia 2012, 14, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Nabizadeh, J.A.; Manthey, H.D.; Panagides, N.; Steyn, F.J.; Lee, J.D.; Li, X.X.; Akhir, F.N.M.; Chen, W.; Boyle, G.M.; Taylor, S.M.; et al. C5a receptors C5aR1 and C5aR2 mediate opposing pathologies in a mouse model of melanoma. FASEB J. 2019, 33, 11060–11071. [Google Scholar] [CrossRef] [PubMed]
- Nabizadeh, J.A.; Manthey, H.D.; Steyn, F.J.; Chen, W.; Widiapradja, A.; Md Akhir, F.N.; Boyle, G.M.; Taylor, S.M.; Woodruff, T.M.; Rolfe, B.E. The Complement C3a Receptor Contributes to Melanoma Tumorigenesis by Inhibiting Neutrophil and CD4+ T Cell Responses. J. Immunol. 2016, 196, 4783–4792. [Google Scholar] [CrossRef] [PubMed]
- Guglietta, S.; Chiavelli, A.; Zagato, E.; Krieg, C.; Gandini, S.; Ravenda, P.S.; Bazolli, B.; Lu, B.; Penna, G.; Rescigno, M. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat. Commun. 2016, 7, 11037. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.W.; Laskowski, J.; Li, H.Y.; McSharry, M.V.; Sippel, T.R.; Bullock, B.L.; Johnson, A.M.; Poczobutt, J.M.; Neuwelt, A.J.; Malkoski, S.P.; et al. Complement Activation via a C3a Receptor Pathway Alters CD4(+) T Lymphocytes and Mediates Lung Cancer Progression. Cancer Res. 2018, 78, 143–156. [Google Scholar] [CrossRef]
- Cho, M.S.; Vasquez, H.G.; Rupaimoole, R.; Pradeep, S.; Wu, S.; Zand, B.; Han, H.D.; Rodriguez-Aguayo, C.; Bottsford-Miller, J.; Huang, J.; et al. Autocrine effects of tumor-derived complement. Cell Rep. 2014, 6, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Boire, A.; Zou, Y.; Shieh, J.; Macalinao, D.G.; Pentsova, E.; Massague, J. Complement Component 3 Adapts the Cerebrospinal Fluid for Leptomeningeal Metastasis. Cell 2017, 168, 1101–1113.e1113. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, S.C.; Kallman, R.F.; Fajardo, L.F. Characteristics of a serially transplanted mouse mammary tumor and its tissue-culture-adapted derivative. J. Natl. Cancer Inst. 1972, 49, 735–749. [Google Scholar] [PubMed]
- Pulaski, B.A.; Ostrand Rosenberg, S. Mouse 4T1 Breast Tumor Model. Curr. Protoc. Immunol. 2001, 20, 20.2.1–20.2.16. [Google Scholar] [CrossRef] [PubMed]
- Finch, A.M.; Wong, A.K.; Paczkowski, N.J.; Wadi, S.K.; Craik, D.J.; Fairlie, D.P.; Taylor, S.M. Low-molecular-weight peptidic and cyclic antagonists of the receptor for the complement factor C5a. J. Med. Chem. 1999, 42, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.C.L.; Lee, J.D.; Ruitenberg, M.J.; Woodruff, T.M. Absence of the C5a Receptor C5aR2 Worsens Ischemic Tissue Injury by Increasing C5aR1-Mediated Neutrophil Infiltration. J. Immunol. 2020, 205, 2834–2839. [Google Scholar] [CrossRef]
- Proctor, L.M.; Arumugam, T.V.; Shiels, I.; Reid, R.C.; Fairlie, D.P.; Taylor, S.M. Comparative anti-inflammatory activities of antagonists to C3a and C5a receptors in a rat model of intestinal ischaemia/reperfusion injury. Br. J. Pharm. 2004, 142, 756–764. [Google Scholar] [CrossRef]
- Manthey, H.D.; Thomas, A.C.; Shiels, I.A.; Zernecke, A.; Woodruff, T.M.; Rolfe, B.; Taylor, S.M. Complement C5a inhibition reduces atherosclerosis in ApoE-/- mice. FASEB J. 2011, 25, 2447–2455. [Google Scholar] [CrossRef]
- Campbell, W.D.; Lazoura, E.; Okada, N.; Okada, H. Inactivation of C3a and C5a octapeptides by carboxypeptidase R and carboxypeptidase N. Microbiol. Immunol. 2002, 46, 131–134. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Strachan, A.J.; Sanderson, S.D.; Monk, P.N.; Wong, A.K.; Fairlie, D.P.; Taylor, S.M. Species dependence for binding of small molecule agonist and antagonists to the C5a receptor on polymorphonuclear leukocytes. Inflammation 2001, 25, 171–177. [Google Scholar] [CrossRef]
- Finch, A.M.; Vogen, S.M.; Sherman, S.A.; Kirnarsky, L.; Taylor, S.M.; Sanderson, S.D. Biologically active conformer of the effector region of human C5a and modulatory effects of N-terminal receptor binding determinants on activity. J. Med. Chem. 1997, 40, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Lee, J.D.; Massey, N.L.; Guan, C.; Robertson, A.A.B.; Clark, R.J.; Woodruff, T.M. Pharmacological characterisation of small molecule C5aR1 inhibitors in human cells reveals biased activities for signalling and function. Biochem. Pharm. 2020, 180, 114156. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Lee, J.D.; Clark, R.J.; Noakes, P.G.; Taylor, S.M.; Woodruff, T.M. Preclinical Pharmacokinetics of Complement C5a Receptor Antagonists PMX53 and PMX205 in Mice. ACS Omega 2020, 5, 2345–2354. [Google Scholar] [CrossRef] [PubMed]
- Hegde, G.V.; Meyers-Clark, E.; Joshi, S.S.; Sanderson, S.D. A conformationally-biased, response-selective agonist of C5a acts as a molecular adjuvant by modulating antigen processing and presentation activities of human dendritic cells. Int. Immunopharmacol. 2008, 8, 819–827. [Google Scholar] [CrossRef]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharm. 1989, 24, 148–154. [Google Scholar] [CrossRef]
- Spang-Thomsen, M.; Nielsen, A.; Visfeldt, J. Growth curves of three human malignant tumors transplanted to nude mice. Exp. Cell Biol. 1980, 48, 138–154. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Klos, A.; Tenner, A.J.; Johswich, K.O.; Ager, R.R.; Reis, E.S.; Kohl, J. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef]
- Bailey, S.R.; Nelson, M.H.; Himes, R.A.; Li, Z.; Mehrotra, S.; Paulos, C.M. Th17 cells in cancer: The ultimate identity crisis. Front. Immunol. 2014, 5, 276. [Google Scholar] [CrossRef]
- Faucheux, L.; Grandclaudon, M.; Perrot-Dockes, M.; Sirven, P.; Berger, F.; Hamy, A.S.; Fourchotte, V.; Vincent-Salomon, A.; Mechta-Grigoriou, F.; Reyal, F.; et al. A multivariate Th17 metagene for prognostic stratification in T cell non-inflamed triple negative breast cancer. Oncoimmunology 2019, 8, e1624130. [Google Scholar] [CrossRef]
- Markiewski, M.M.; Vadrevu, S.K.; Sharma, S.K.; Chintala, N.K.; Ghouse, S.; Cho, J.H.; Fairlie, D.P.; Paterson, Y.; Astrinidis, A.; Karbowniczek, M. The Ribosomal Protein S19 Suppresses Antitumor Immune Responses via the Complement C5a Receptor 1. J. Immunol. 2017, 198, 2989–2999. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Bhattacharya, S.; Yanamandra, N.; Kilian, D.; Shi, H.; Yadavilli, S.; Katlinskaya, Y.; Kaczynski, H.; Conner, M.; Benson, W.; et al. Tumor-immune profiling of murine syngeneic tumor models as a framework to guide mechanistic studies and predict therapy response in distinct tumor microenvironments. PLoS ONE 2018, 13, e0206223. [Google Scholar] [CrossRef] [PubMed]
- Ajona, D.; Zandueta, C.; Corrales, L.; Moreno, H.; Pajares, M.J.; Ortiz-Espinosa, S.; Martinez-Terroba, E.; Perurena, N.; de Miguel, F.J.; Jantus-Lewintre, E.; et al. Blockade of the Complement C5a/C5aR1 Axis Impairs Lung Cancer Bone Metastasis by CXCL16-mediated Effects. Am. J. Respir. Crit. Care Med. 2018, 197, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Mosely, S.I.; Prime, J.E.; Sainson, R.C.; Koopmann, J.O.; Wang, D.Y.; Greenawalt, D.M.; Ahdesmaki, M.J.; Leyland, R.; Mullins, S.; Pacelli, L.; et al. Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery. Cancer Immunol. Res. 2017, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Myers, J.S.; Wang, F.; Wang, K.; Lucas, J.; Rosfjord, E.; Lucas, J.; Hooper, A.T.; Yang, S.; Lemon, L.A.; et al. Comparison of the molecular and cellular phenotypes of common mouse syngeneic models with human tumors. BMC Genom. 2020, 21, 2. [Google Scholar] [CrossRef]
- Devaud, C.; Westwood, J.A.; John, L.B.; Flynn, J.K.; Paquet-Fifield, S.; Duong, C.P.; Yong, C.S.; Pegram, H.J.; Stacker, S.A.; Achen, M.G.; et al. Tissues in different anatomical sites can sculpt and vary the tumor microenvironment to affect responses to therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 18–27. [Google Scholar] [CrossRef]
- Gunn, L.; Ding, C.; Liu, M.; Ma, Y.; Qi, C.; Cai, Y.; Hu, X.; Aggarwal, D.; Zhang, H.G.; Yan, J. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J. Immunol. 2012, 189, 2985–2994. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Sellers, R.S.; Clifford, C.B.; Treuting, P.M.; Brayton, C. Immunological variation between inbred laboratory mouse strains: Points to consider in phenotyping genetically immunomodified mice. Vet. Pathol. 2012, 49, 32–43. [Google Scholar] [CrossRef]
- Pio, R.; Ajona, D.; Lambris, J.D. Complement inhibition in cancer therapy. Semin. Immunol. 2013, 25, 54–64. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhir, F.N.M.; Noor, M.H.M.; Leong, K.W.K.; Nabizadeh, J.A.; Manthey, H.D.; Sonderegger, S.E.; Fung, J.N.T.; McGirr, C.E.; Shiels, I.A.; Mills, P.C.; et al. An Immunoregulatory Role for Complement Receptors in Murine Models of Breast Cancer. Antibodies 2021, 10, 2. https://doi.org/10.3390/antib10010002
Akhir FNM, Noor MHM, Leong KWK, Nabizadeh JA, Manthey HD, Sonderegger SE, Fung JNT, McGirr CE, Shiels IA, Mills PC, et al. An Immunoregulatory Role for Complement Receptors in Murine Models of Breast Cancer. Antibodies. 2021; 10(1):2. https://doi.org/10.3390/antib10010002
Chicago/Turabian StyleAkhir, Fazrena Nadia Md, Mohd Hezmee Mohd Noor, Keith Weng Kit Leong, Jamileh A. Nabizadeh, Helga D. Manthey, Stefan E. Sonderegger, Jenny Nga Ting Fung, Crystal E. McGirr, Ian A. Shiels, Paul C. Mills, and et al. 2021. "An Immunoregulatory Role for Complement Receptors in Murine Models of Breast Cancer" Antibodies 10, no. 1: 2. https://doi.org/10.3390/antib10010002
APA StyleAkhir, F. N. M., Noor, M. H. M., Leong, K. W. K., Nabizadeh, J. A., Manthey, H. D., Sonderegger, S. E., Fung, J. N. T., McGirr, C. E., Shiels, I. A., Mills, P. C., Woodruff, T. M., & Rolfe, B. E. (2021). An Immunoregulatory Role for Complement Receptors in Murine Models of Breast Cancer. Antibodies, 10(1), 2. https://doi.org/10.3390/antib10010002