Viroinformatics-Based Analysis of SARS-CoV-2 Core Proteins for Potential Therapeutic Targets


 ,
,  and
and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Investigation of Conservative Motifs of SARS-CoV-2 Spike Protein and Phylogenetic Analysis
2.2. In Silico Protein-Protein Interaction
2.3. Finding the Most Interacting Motifs of Spike Protein with Antiviral Drugs and Heat-Map Representation of Motifs
2.4. Docking of Antivirals, Antibiotics, Antiparasitics, Flavonoids and Vitamins with Spike and RdRp Protein of SARS-CoV-2
3. Results
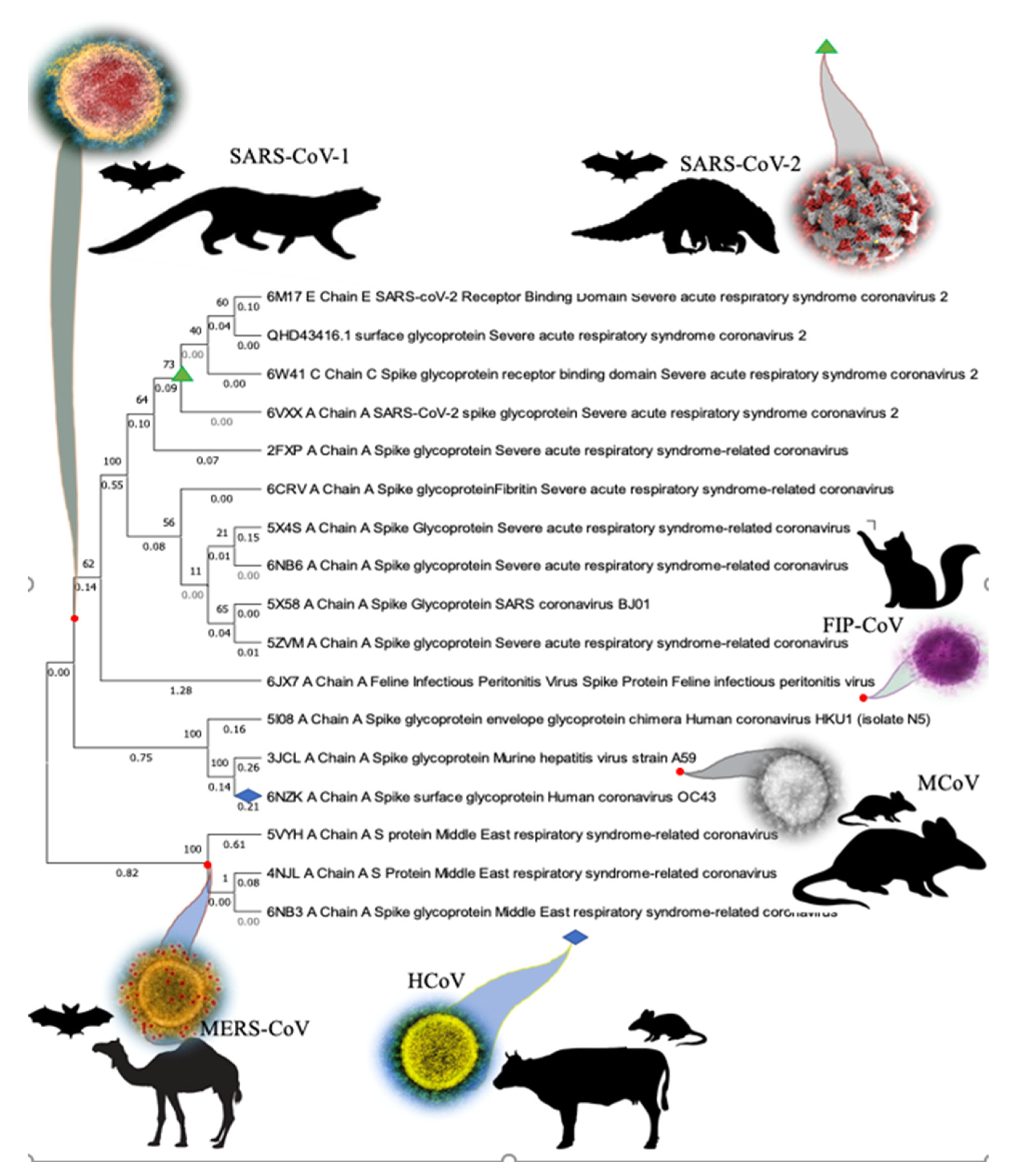
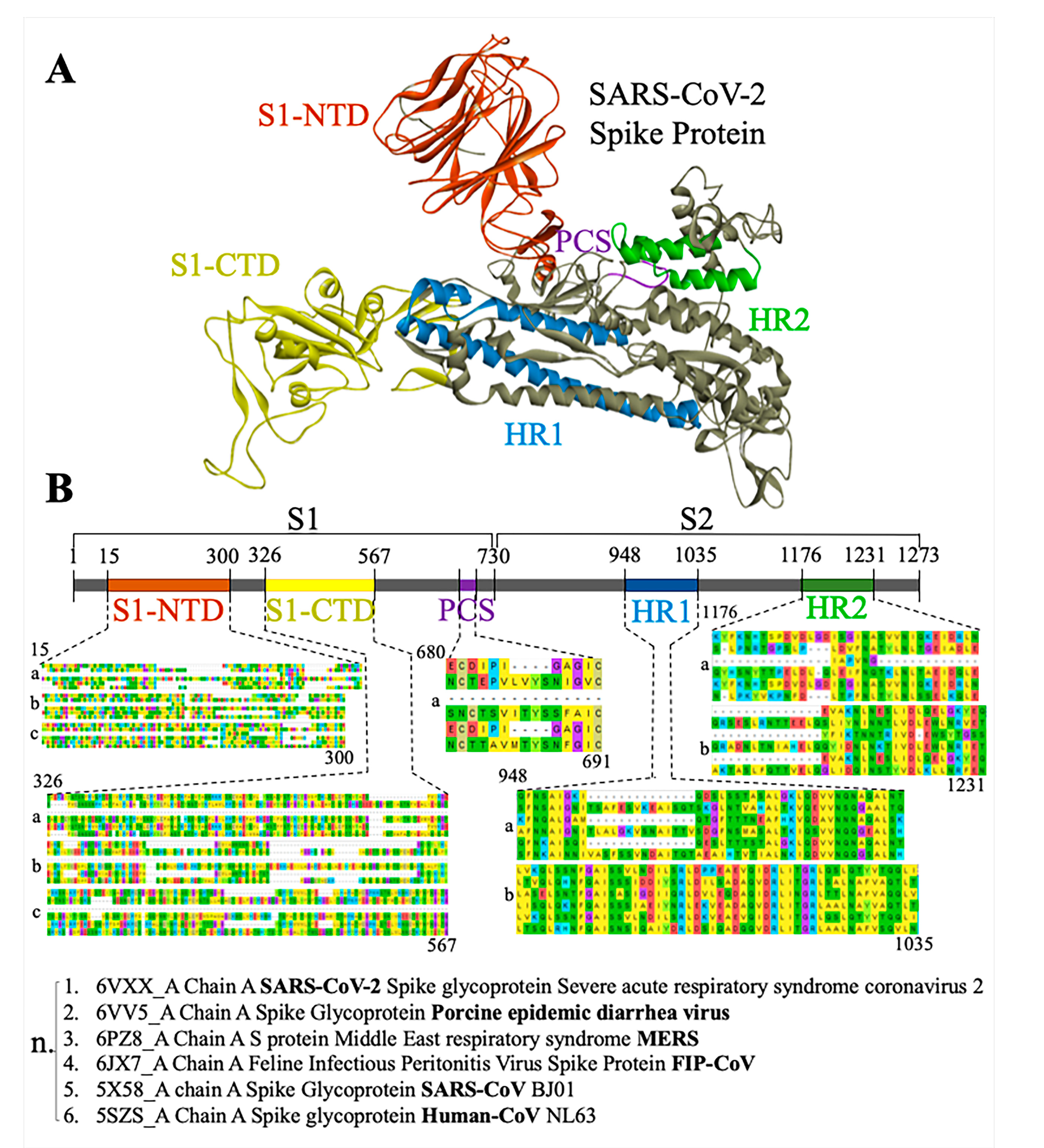
3.1. Phylogenetic Analysis of SARS-CoV-2 Spike Protein and Retrieval of Therapeutically Important Conservative Motifs of Evolutionary Significance
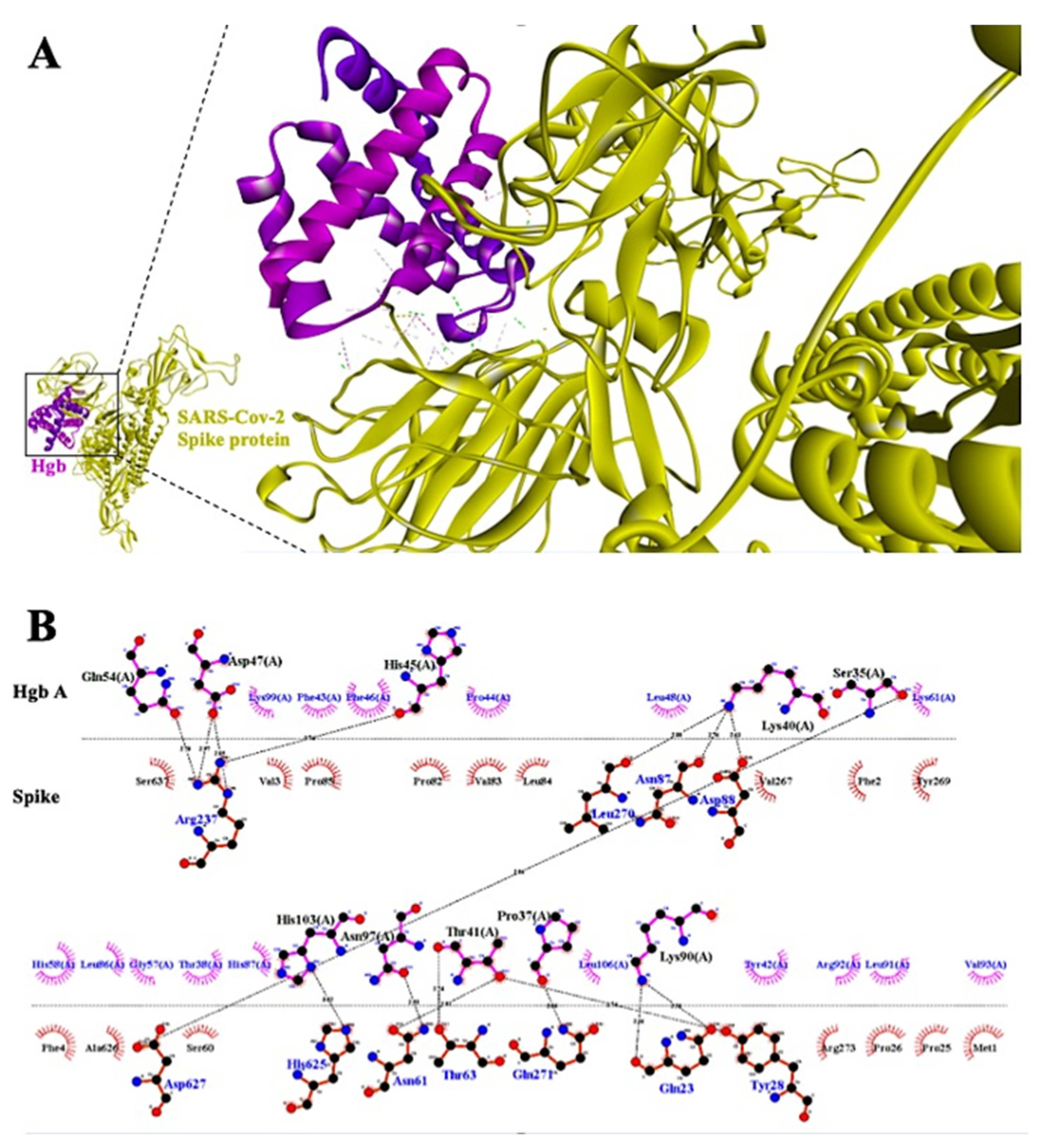
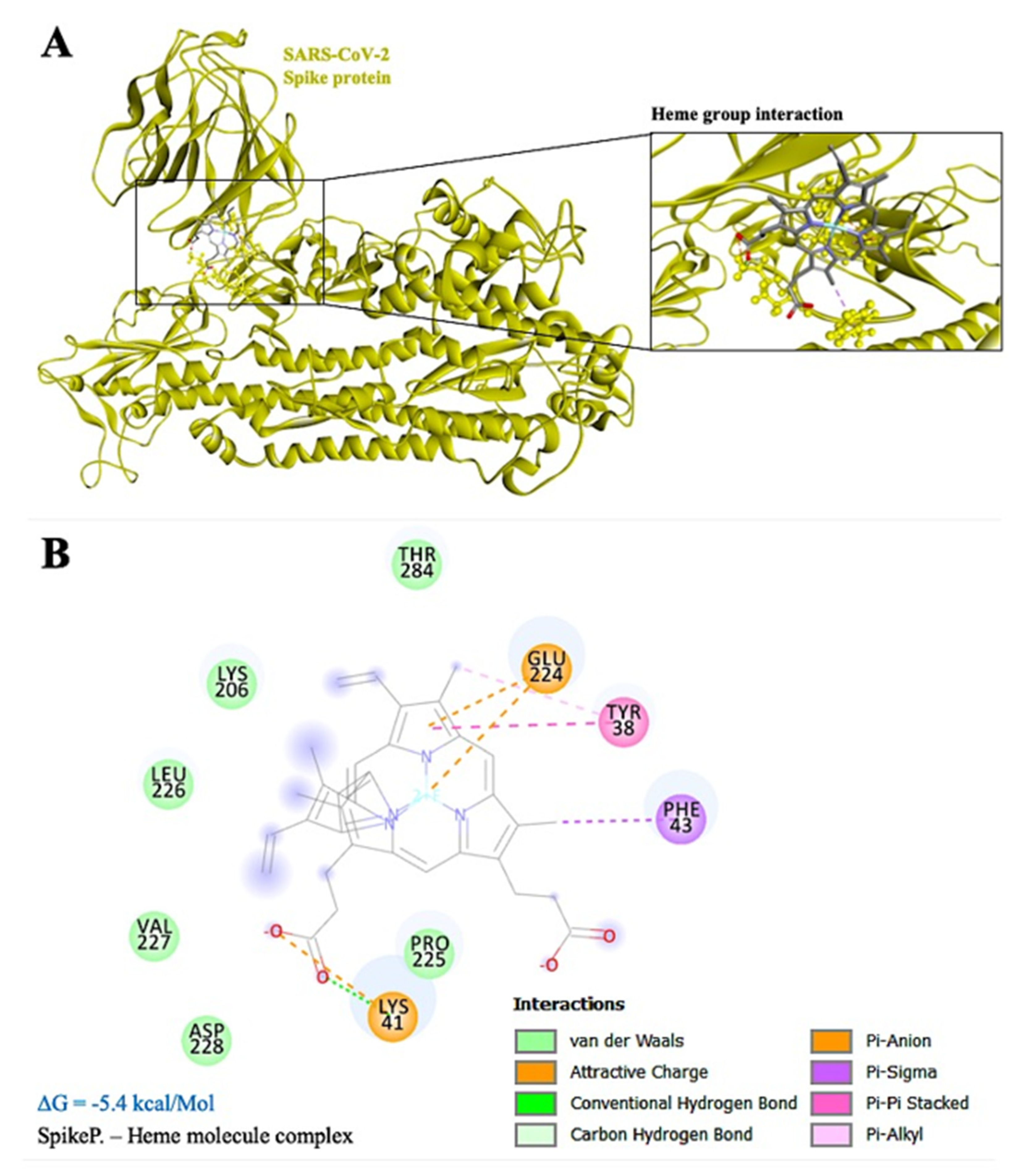
3.2. Mechanism of Hypoxia Caused by SARS-CoV-2 Spike Protein
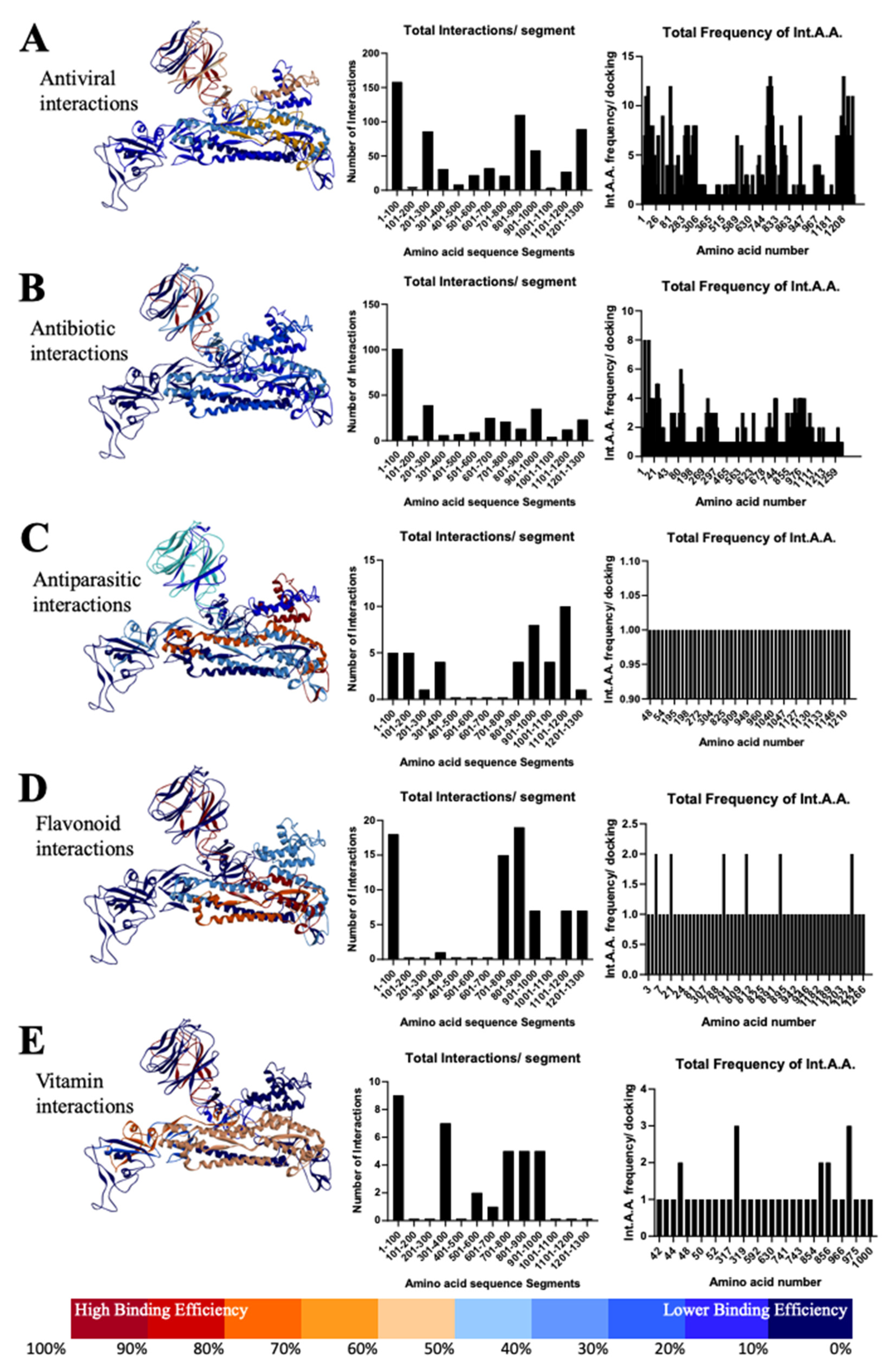
3.3. Most Important Conservative Motifs of SARS-CoV-2 Spike Protein as a Target for the Development of Therapeutic Drugs
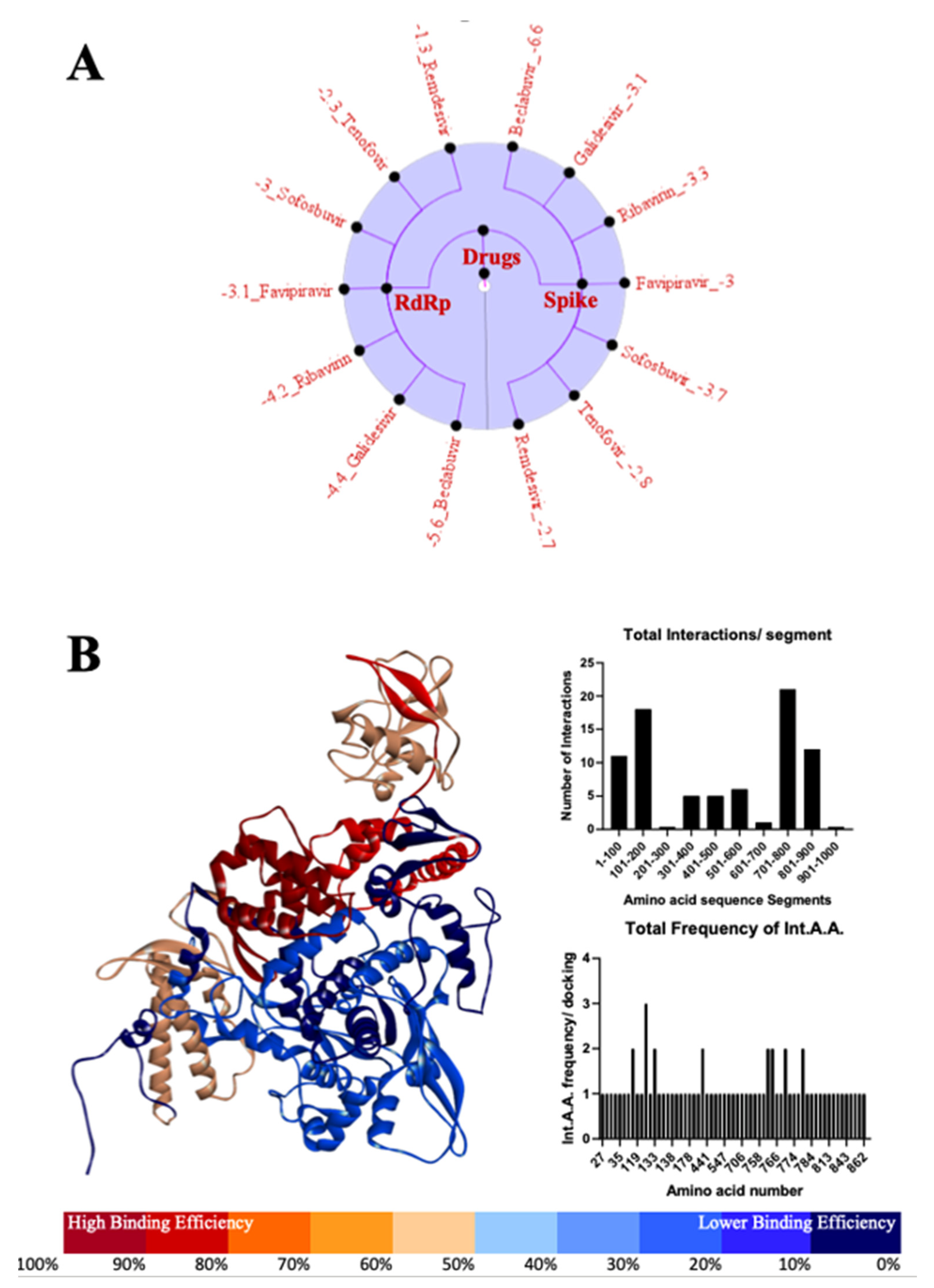
3.4. Most Important Motifs of RdRp Protein as a Target for Therapeutic Drugs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Shereen, M.A.; Khan, S.; Kazmi, A.; Bashir, N.; Siddique, R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020, 24, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; the Northwell, C.-R.C. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef] [PubMed]
- Baldi, E.; Sechi, G.M.; Mare, C.; Canevari, F.; Brancaglione, A.; Primi, R.; Klersy, C.; Palo, A.; Contri, E.; Ronchi, V.; et al. Out-of-Hospital Cardiac Arrest during the Covid-19 Outbreak in Italy. N. Engl. J. Med. 2020, 383, 496–498. [Google Scholar] [CrossRef] [PubMed]
- Avula, A.; Nalleballe, K.; Narula, N.; Sapozhnikov, S.; Dandu, V.; Toom, S.; Glaser, A.; Elsayegh, D. COVID-19 presenting as stroke. Brain Behav. Immun. 2020, 87, 115–119. [Google Scholar] [CrossRef]
- Li, X.; Ma, X. Acute respiratory failure in COVID-19: Is it “typical” ARDS? Crit. Care 2020, 24, 198. [Google Scholar] [CrossRef]
- Boopathi, S.; Poma, A.B.; Kolandaivel, P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Vinson, V. Structure of the nCoV trimeric spike. Science 2020, 367, 1207. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Kashani, K.B. Hypoxia in COVID-19: Sign of Severity or Cause for Poor Outcomes. Mayo Clin. Proc. 2020, 95, 1094–1096. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Covassin, N.; Fan, Z.; Singh, P.; Gao, W.; Li, G.; Kara, T.; Somers, V.K. Association Between Hypoxemia and Mortality in Patients With COVID-19. Mayo Clin. Proc. 2020, 95, 1138–1147. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Cavezzi, A.; Troiani, E.; Corrao, S. COVID-19: Hemoglobin, iron, and hypoxia beyond inflammation. A narrative review. Clin. Pract. 2020, 10, 1271. [Google Scholar] [CrossRef]
- Liu, W.; Li, H. COVID-19:Attacks the 1-Beta Chain of Hemoglobin and Captures the Porphyrin to Inhibit Human Heme Metabolism. ChemRxiv 2020. [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. CABIOS 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. Protein Structure and Function Prediction Using I-TASSER. In Curr Protoc Bioinformatics; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2015; Volume 52, pp. 1–15. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Vijayan, R. Dynamics of the ACE2–SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci. Rep. 2020, 10, 14214. [Google Scholar] [CrossRef]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef]
- Chakraborti, S.; Prabakaran, P.; Xiao, X.; Dimitrov, D.S. The SARS Coronavirus S Glycoprotein Receptor Binding Domain: Fine Mapping and Functional Characterization. Virol. J. 2005, 2, 73. [Google Scholar] [CrossRef]
- Xia, S.; Yan, L.; Xu, W.; Agrawal, A.S.; Algaissi, A.; Tseng, C.-T.K.; Wang, Q.; Du, L.; Tan, W.; Wilson, I.A.; et al. A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 2019, 5, eaav4580. [Google Scholar] [CrossRef]
- Menachery, V.D.; Dinnon, K.H., 3rd; Yount, B.L., Jr.; McAnarney, E.T.; Gralinski, L.E.; Hale, A.; Graham, R.L.; Scobey, T.; Anthony, S.J.; Wang, L.; et al. Trypsin Treatment Unlocks Barrier for Zoonotic Bat Coronavirus Infection. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Pastick, K.A.; Okafor, E.C.; Wang, F.; Lofgren, S.M.; Skipper, C.P.; Nicol, M.R.; Pullen, M.F.; Rajasingham, R.; McDonald, E.G.; Lee, T.C.; et al. Review: Hydroxychloroquine and Chloroquine for Treatment of SARS-CoV-2 (COVID-19). Op. Forum Infect. Dis. 2020, 7, 1–9. [Google Scholar] [CrossRef]
- Iyamu, E.; Perdew, H.; Woods, G. Growth inhibitory and differentiation effects of chloroquine and its analogue on human leukemic cells potentiate fetal hemoglobin production by targeting the polyamine pathway. Biochem. Pharmacol. 2009, 77, 1021–1028. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Gautret, P.; Lagier, J.-C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020, 56, 105949. [Google Scholar] [CrossRef] [PubMed]
- Kalra, R.S.; Tomar, D.; Meena, A.S.; Kandimalla, R. SARS-CoV-2, ACE2, and Hydroxychloroquine: Cardiovascular Complications, Therapeutics, and Clinical Readouts in the Current Settings. Pathogens 2020, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Heidary, F.; Gharebaghi, R. Ivermectin: A systematic review from antiviral effects to COVID-19 complementary regimen. J. Antibiot. 2020, 73, 593–602. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Roschek, B., Jr.; Fink, R.C.; McMichael, M.D.; Li, D.; Alberte, R.S. Elderberry flavonoids bind to and prevent H1N1 infection in vitro. Phytochemistry 2009, 70, 1255–1261. [Google Scholar] [CrossRef]
- Chen, C.; Zuckerman, D.M.; Brantley, S.; Sharpe, M.; Childress, K.; Hoiczyk, E.; Pendleton, A.R. Sambucus nigra extracts inhibit infectious bronchitis virus at an early point during replication. BMC Vet. Res. 2014, 10, 24. [Google Scholar] [CrossRef]
- Chen, Z.; Zheng, S.; Li, L.; Jiang, H. Metabolism of flavonoids in human: A comprehensive review. Curr. Drug Metab. 2014, 15, 48–61. [Google Scholar] [CrossRef]
- Jo, S.; Kim, S.; Shin, D.H.; Kim, M.-S. Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzym. Inhib. Med. Chem. 2020, 35, 145–151. [Google Scholar] [CrossRef]
- Jitendra Subhash, R.; Aroni, C.; Abhijeet, K.; Shashikant, R. Targeting SARS-CoV-2 Spike Protein of COVID-19 with Naturally Occurring Phytochemicals: An in Silco Study for Drug Development. ChemRxiv 2020. [CrossRef]
- Hemilä, H. Vitamin C and SARS coronavirus. J. Antimicrob. Chemother. 2003, 52, 1049–1050. [Google Scholar] [CrossRef] [PubMed]
- Ginde, A.A.; Mansbach, J.M.; Camargo, C.A., Jr. Association between serum 25-hydroxyvitamin D level and upper respiratory tract infection in the Third National Health and Nutrition Examination Survey. Archiv. Intern. Med. 2009, 169, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Lv, Q.; Chen, F.; Wang, D.; Liu, Y.; Shi, W. Identification of Recent Trends in Research on Vitamin D: A Quantitative and Co-Word Analysis. Med. Sci. Monit. 2019, 25, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Ilie, P.C.; Stefanescu, S.; Smith, L. The role of vitamin D in the prevention of coronavirus disease 2019 infection and mortality. Aging Clin. Exp. Res. 2020, 32, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Yang, M.; Liu, D.; Chen, J.; Shu, D.; Xia, J.; Liao, X.; Gu, Y.; Cai, Q.; Yang, Y.; et al. Experimental Treatment with Favipiravir for COVID-19: An Open-Label Control Study. Engineering 2020, 6, 1192–1198. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agrawal, L.; Poullikkas, T.; Eisenhower, S.; Monsanto, C.; Bakku, R.K.; Chen, M.-H.; Kalra, R.S. Viroinformatics-Based Analysis of SARS-CoV-2 Core Proteins for Potential Therapeutic Targets. Antibodies 2021, 10, 3. https://doi.org/10.3390/antib10010003
Agrawal L, Poullikkas T, Eisenhower S, Monsanto C, Bakku RK, Chen M-H, Kalra RS. Viroinformatics-Based Analysis of SARS-CoV-2 Core Proteins for Potential Therapeutic Targets. Antibodies. 2021; 10(1):3. https://doi.org/10.3390/antib10010003
Chicago/Turabian StyleAgrawal, Lokesh, Thanasis Poullikkas, Scott Eisenhower, Carlo Monsanto, Ranjith Kumar Bakku, Min-Hua Chen, and Rajkumar Singh Kalra. 2021. "Viroinformatics-Based Analysis of SARS-CoV-2 Core Proteins for Potential Therapeutic Targets" Antibodies 10, no. 1: 3. https://doi.org/10.3390/antib10010003
APA StyleAgrawal, L., Poullikkas, T., Eisenhower, S., Monsanto, C., Bakku, R. K., Chen, M.-H., & Kalra, R. S. (2021). Viroinformatics-Based Analysis of SARS-CoV-2 Core Proteins for Potential Therapeutic Targets. Antibodies, 10(1), 3. https://doi.org/10.3390/antib10010003