Genome Sequencing and Assembly by Long Reads in Plants


Abstract
1. Introduction
2. Sanger Sequencing: A Milestone in Plant Genomics
3. Next Generation Sequencing Enables Unprecedented Development in Plant Genomics
4. Long-Read Sequencing Opens a New Era of Solving Complex Plant Genomes
4.1. TruSeq Synthetic Long-Read
4.2. Nanopore Sequencing
4.3. PacBio Sequencing
5. Application and Consideration of PacBio Sequencing
5.1. Application of PacBio Sequencing
5.1.1. De Novo Assembly
5.1.2. Scaffolding
5.1.3. Filling the Gaps
5.2. Consideration of PacBio Sequencing
5.2.1. Sample Preparation
5.2.2. Sequencing Strategy
5.2.3. Bioinformatics Analysis
6. Long-Range Scaffolding Technologies Improve Assembly
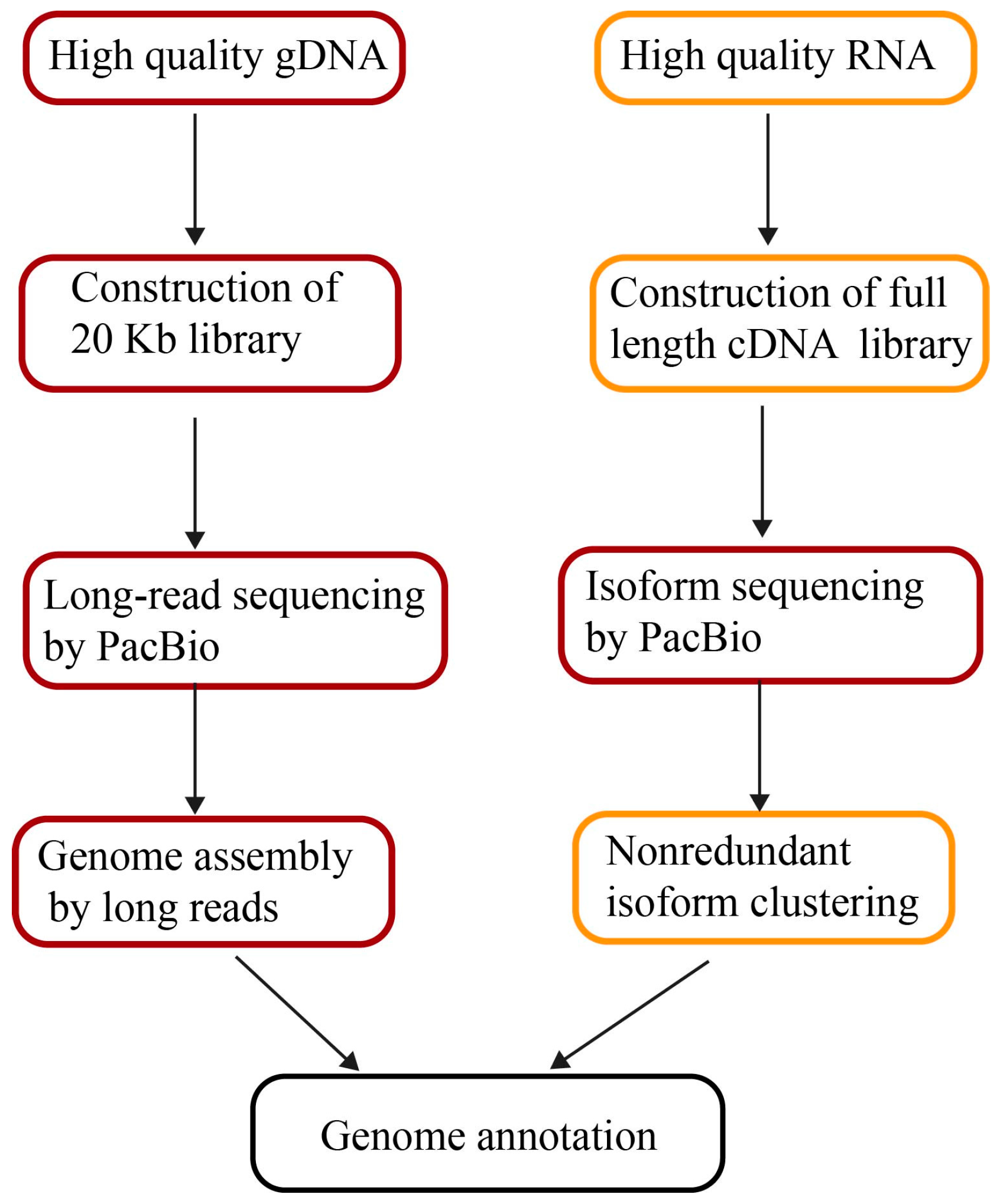
7. Genome Annotations by Long Reads
8. Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Yuan, Y.; Bayer, P.E.; Batley, J.; Edwards, D. Improvements in genomic technologies: Application to crop genomics. Trends Biotechnol. 2017, 35, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Argout, X.; Salse, J.; Aury, J.M.; Guiltinan, M.J.; Droc, G.; Gouzy, J.; Allegre, M.; Chaparro, C.; Legavre, T.; Maximova, S.N.; et al. The genome of Theobroma cacao. Nat. Genet. 2011, 43, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Feng, Y.; Kumar, D.; Zhang, W.; Zhu, T.; Luo, M.-C.; Messing, J. Analysis of tandem gene copies in maize chromosomal regions reconstructed from long sequence reads. Proc. Natl. Acad. Sci. USA 2016, 113, 7949–7956. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Mackay, J.; Dean, J.F.; Plomion, C.; Peterson, D.G.; Canovas, F.M.; Pavy, N.; Ingvarsson, P.K.; Savolainen, O.; Guevara, M.A.; Fluch, S.; et al. Towards decoding the conifer giga-genome. Plant Mol. Biol. 2012, 80, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.; Stevens, K.A.; Crepeau, M.W.; Holtz-Morris, A.; Koriabine, M.; Marçais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.L.; de Jong, P.J.; et al. Sequencing and assembly of the 22-Gb Loblolly pine genome. Genetics 2014, 196, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Michael, T.P.; VanBuren, R. Progress, challenges and the future of crop genomes. Curr. Opin. Plant Biol. 2015, 24, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Jiao, W.B.; Schneeberger, K. The impact of third generation genomic technologies on plant genome assembly. Curr. Opin. Plant Biol. 2017, 36, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.-S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Zhang, Y.; Hao, P.; Wang, S.; Fu, G.; Huang, Y.; Li, Y.; Zhu, J.; Liu, Y.; Hu, X.; et al. Sequence and analysis of rice chromosome 4. Nature 2002, 420, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Shulaev, V.; Sargent, D.J.; Crowhurst, R.N.; Mockler, T.C.; Folkerts, O.; Delcher, A.L.; Jaiswal, P.; Mockaitis, K.; Liston, A.; Mane, S.P.; et al. The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 2010, 43, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Al-Dous, E.K.; George, B.; Al-Mahmoud, M.E.; Al-Jaber, M.Y.; Wang, H.; Salameh, Y.M.; Al-Azwani, E.K.; Chaluvadi, S.; Pontaroli, A.C.; DeBarry, J.; et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat. Biotechnol. 2011, 29, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Zhu, G.; Zhang, J.; Xu, X.; Yu, Q.; Zheng, Z.; Zhang, Z.; Lun, Y.; Li, S.; Wang, X.; et al. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 2014, 46, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhang, J.; Sun, H.; Salse, J.; Lucas, W.J.; Zhang, H.; Zheng, Y.; Mao, L.; Ren, Y.; Wang, Z.; et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 2013, 45, 51–58. [Google Scholar] [CrossRef] [PubMed]
- The 3000 rice genomes project. The 3000 rice genomes project. GigaScience 2014, 3, 7. [Google Scholar]
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Annu. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef] [PubMed]
- Phillippy, A.M. New advances in sequence assembly. Genome Res. 2017, 27, xi–xiii. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Guyot, R.; Yahiaoui, N.; Keller, B. Cacta transposons in Triticeae. A diverse family of high-copy repetitive elements. Plant Physiol. 2003, 132, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.C.; Gu, Y.Q.; Puiu, D.; Wang, H.; Twardziok, S.O.; Deal, K.R.; Huo, N.; Zhu, T.; Wang, L.; Wang, Y.; et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 2017. [Google Scholar] [CrossRef] [PubMed]
- McCoy, R.C.; Taylor, R.W.; Blauwkamp, T.A.; Kelley, J.L.; Kertesz, M.; Pushkarev, D.; Petrov, D.A.; Fiston-Lavier, A.S. Illumina truseq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PLoS ONE 2014, 9, e106689. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hsieh, C.L.; Young, A.; Zhang, Z.; Ren, X.; Zhao, Z. Illumina synthetic long read sequencing allows recovery of missing sequences even in the “finished” C. elegans genome. Sci. Rep. 2015, 5, 10814. [Google Scholar] [CrossRef] [PubMed]
- Voskoboynik, A.; Neff, N.F.; Sahoo, D.; Newman, A.M.; Pushkarev, D.; Koh, W.; Passarelli, B.; Fan, H.C.; Mantalas, G.L.; Palmeri, K.J.; et al. The genome sequence of the colonial chordate, Botryllus schlosseri. eLife 2013, 2, e00569. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Quick, J.; Simpson, J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 2015, 12, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; Gurtowski, J.; Ethe-Sayers, S.; Deshpande, P.; Schatz, M.C.; McCombie, W.R. Oxford nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 2015, 25, 1750–1756. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.J.; Liem, M.; Jong-Raadsen, S.A.; Dufour, S.; Weltzien, F.A.; Swinkels, W.; Koelewijn, A.; Palstra, A.P.; Pelster, B.; Spaink, H.P.; et al. Rapid de novo assembly of the European eel genome from nanopore sequencing reads. Sci. Rep. 2017, 7, 7213. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; Malla, S.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. bioRxiv 2017. [Google Scholar] [CrossRef]
- Schmidt, M.H.; Vogel, A.; Denton, A.K.; Istace, B.; Wormit, A.; van de Geest, H.; Bolger, M.E.; Alseekh, S.; Mass, J.; Pfaff, C.; et al. De novo assembly of a new Solanum pennellii accession using nanopore sequencing. Plant Cell 2017, 29, 2336–2348. [Google Scholar] [CrossRef] [PubMed]
- Michael, T.P.; Jupe, F.; Bemm, F.; Motley, S.T.; Sandoval, J.P.; Loudet, O.; Weigel, D.; Ecker, J.R. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. bioRxiv 2017. [Google Scholar] [CrossRef]
- Zhang, W.; Ciclitira, P.; Messing, J. PacBio sequencing of gene families—A case study with wheat gluten genes. Gene 2014, 533, 541–546. [Google Scholar] [CrossRef] [PubMed]
- VanBuren, R.; Bryant, D.; Edger, P.P.; Tang, H.; Burgess, D.; Challabathula, D.; Spittle, K.; Hall, R.; Gu, J.; Lyons, E.; et al. Single-molecule sequencing of the desiccation- tolerant grass Oropetium thomaeum. Nature 2015, 527, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, D.E.; Ho, Y.S.; Lightfoot, D.J.; Schmöckel, S.M.; Li, B.; Borm, T.J.A.; Ohyanagi, H.; Mineta, K.; Michell, C.T.; Saber, N.; et al. The genome of Chenopodium quinoa. Nature 2017, 542, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Willing, E.M.; Rawat, V.; Mandakova, T.; Maumus, F.; James, G.V.; Nordstrom, K.J.; Becker, C.; Warthmann, N.; Chica, C.; Szarzynska, B.; et al. Genome expansion of Arabis alpina linked with retrotransposition and reduced symmetric DNA methylation. Nat. Plants 2015, 1, 14023. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Laclette, E.; Lyons, E.; Hernández-Guzmán, G.; Pérez-Torres, C.A.; Carretero-Paulet, L.; Chang, T.-H.; Lan, T.; Welch, A.J.; Juárez, M.J.A.; Simpson, J.; et al. Architecture and evolution of a minute plant genome. Nature 2013, 498, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Renner, T.; Ibarra-Laclette, E.; Farr, K.M.; Chang, T.-H.; Cervantes-Pérez, S.A.; Zheng, C.; Sankoff, D.; Tang, H.; Purbojati, R.W.; et al. Long-read sequencing uncovers the adaptive topography of a carnivorous plant genome. Proc. Natl. Acad. Sci. USA 2017, 114, E4435–E4441. [Google Scholar] [CrossRef] [PubMed]
- Bombarely, A.; Moser, M.; Amrad, A.; Bao, M.; Bapaume, L.; Barry, C.S.; Bliek, M.; Boersma, M.R.; Borghi, L.; Bruggmann, R.; et al. Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat. Plants 2016, 2, 16074. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, D.; Wang, X.; Ji, C.; Cheng, F.; Liu, B.; Hu, Z.; Chen, S.; Pental, D.; Ju, Y.; et al. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 2016, 48, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- PacBiosciences. Preparing Arabidopsis genomic DNA for size-selected ~20 kb SMRTbell libraries. Available online: www.pacb.com/wp-content/uploads/2015/2009/Shared-Protocol-Preparing-Arabidopsis-DNA-for-2020-kb-SMRTbell-Libraries.pdf (accessed on 15 October 2013).
- Peterson, D.G.; Boehm, K.S.; Stack, S.M. Isolation of milligram quantities of nuclear DNA from tomato (Lycopersicon esculentum), a plant containing high levels of polyphenolic compounds. Plant Mol. Biol. Rep. 1997, 15, 148–153. [Google Scholar] [CrossRef]
- Bashir, A.; Klammer, A.; Robins, W.P.; Chin, C.-S.; Webster, D.; Paxinos, E.; Hsu, D.; Ashby, M.; Wang, S.; Peluso, P.; et al. A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotechnol. 2012, 30, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Schatz, M.C.; Walenz, B.P.; Martin, J.; Howard, J.T.; Ganapathy, G.; Wang, Z.; Rasko, D.A.; McCombie, W.R.; Jarvis, E.D.; et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012, 30, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Huddleston, J.; Chaisson, M.J.P.; Hill, C.M.; Kronenberg, Z.N.; Munson, K.M.; Malig, M.; Raja, A.; Fiddes, I.; Hillier, L.W.; et al. Long-read sequence assembly of the Gorilla genome. Science 2016, 352. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O'Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Marcais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The Masurca Genome Assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Puiu, D.; Luo, M.C.; Zhu, T.; Koren, S.; Marcais, G.; Yorke, J.A.; Dvorak, J.; Salzberg, S.L. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the Masurca mega-reads algorithm. Genome Res. 2017, 27, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Denisov, G.; Walenz, B.; Halpern, A.L.; Miller, J.; Axelrod, N.; Levy, S.; Sutton, G. Consensus generation and variant detection by Celera assembler. Bioinformatics 2008, 24, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Belton, J.M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.E.; Russell, J.; et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Jiao, W.B.; Accinelli, G.G.; Hartwig, B.; Kiefer, C.; Baker, D.; Severing, E.; Willing, E.M.; Piednoel, M.; Woetzel, S.; Madrid-Herrero, E.; et al. Improving and correcting the contiguity of long-read genome assemblies of three plant species using optical mapping and chromosome conformation capture data. Genome Res. 2017, 27, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Moll, K.M.; Zhou, P.; Ramaraj, T.; Fajardo, D.; Devitt, N.P.; Sadowsky, M.J.; Stupar, R.M.; Tiffin, P.; Miller, J.R.; Young, N.D.; et al. Strategies for optimizing Bionano and Dovetail explored through a second reference quality assembly for the legume model, Medicago truncatula. BMC Genom. 2017, 18, 578. [Google Scholar] [CrossRef] [PubMed]
- Putnam, N.H.; O’Connell, B.L.; Stites, J.C.; Rice, B.J.; Blanchette, M.; Calef, R.; Troll, C.J.; Fields, A.; Hartley, P.D.; Sugnet, C.W.; et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016, 26, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M.; et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Chain, P.S.; Grafham, D.V.; Fulton, R.S.; Fitzgerald, M.G.; Hostetler, J.; Muzny, D.; Ali, J.; Birren, B.; Bruce, D.C.; Buhay, C.; et al. Genomics. Genome project standards in a new era of sequencing. Science 2009, 326, 236–237. [Google Scholar] [CrossRef] [PubMed]
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012, 13, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Liu, H.; Zhang, J.; Yang, S.; Kong, G.; Chu, J.S.C.; Chen, N.; Wang, D. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Categories | 1st Generation | 2nd Generation | 3rd Generation | |||||
|---|---|---|---|---|---|---|---|---|
| Platform | Sanger | Illumina | PacBio | Nanopore | ||||
| HiSeq2500–High output | HiSeq2500–Rapid mode | MiSeq | Synthetic Long reads | 10× Genomics | ||||
| Read length | 800 bp | 2 × 125 bp | 2 × 250 bp | 2 × 300 bp | ~100 Kb | up to 100 Kb | 10−15 Kb | up to 200 Kb |
| Yield/Cell | 80 Kb | 450−500 Gb | 125–150 Gb | 13–15 Gb | See HiSeq2500 | See HiSeq2500 | 5–10 Gb | up to 1.5 Gb |
| Instrument Time | 3 h | 6 days | 60 h | 21–56 h | See HiSeq2500 | See HiSeq2500 | 4 h | 2 days |
| Price/Gb | $1,000,000 | $30 | $40 | $110 | $1000 | See HiSeq2500 + $500/sample | $125 | $750 |
| Features | De novo sequencing small genomes with BAC–BAC | De novo sequencing small genomes, resequencing and correcting sequence | De novo sequencing complex genomes | Order assembled contigs into scaffolds | De novo sequencing complex genomes, filling gaps and improving assembly | |||
| Species | Mean Subread Length | Number of Reads | Coverage of SMRT | Genome Size (Mb) | Contig N50 (Mb) | Assembly |
|---|---|---|---|---|---|---|
| Utricularia gibba | 10,385 | 702,640 | 88 | 82 | 3.4 | HGAP |
| Oropetium thomaeum | 12,872 | 1,400,150 | 72 | 245 | 2.4 | HGAP |
| Chenopodium quinoa | 12,444 | 6,037,280 | 100 | 1500 | 1.7 | SMRT-make |
| Zea mays | 11,700 | NA | 65 | 2300 | 1.1 | PBcR; Falcon |
| Helianthus annuus | 10,300 | 32,000,000 | 102 | 3300 | NA | PBcR |
| Assembly Parameters | Version 3 | Version 4 |
|---|---|---|
| Platform | Sanger and 454 | PacBio and Bionano |
| Contig # | 140,000 | 2958 |
| Contig N50 | 19 Kb | 1180 Kb |
| Scaffold # | 61,161 | 625 |
| Scaffold N50 | 76 Kb | 9.5 Mb |
| Centromeres | Partial | Yes |
| Telomeres | Partial | Yes |
| Gap | 10% missing | 3% missing |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Lin, F.; An, D.; Wang, W.; Huang, R. Genome Sequencing and Assembly by Long Reads in Plants. Genes 2018, 9, 6. https://doi.org/10.3390/genes9010006
Li C, Lin F, An D, Wang W, Huang R. Genome Sequencing and Assembly by Long Reads in Plants. Genes. 2018; 9(1):6. https://doi.org/10.3390/genes9010006
Chicago/Turabian StyleLi, Changsheng, Feng Lin, Dong An, Wenqin Wang, and Ruidong Huang. 2018. "Genome Sequencing and Assembly by Long Reads in Plants" Genes 9, no. 1: 6. https://doi.org/10.3390/genes9010006
APA StyleLi, C., Lin, F., An, D., Wang, W., & Huang, R. (2018). Genome Sequencing and Assembly by Long Reads in Plants. Genes, 9(1), 6. https://doi.org/10.3390/genes9010006