
Two Novel Biallelic Variants in the FARSA Gene: The First Italian Case and a Literature Review

 , , ,
, , ,  , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Genetic Studies
2.2. Clinical Exome Sequencing
2.3. Effect and Prediction
3. Results
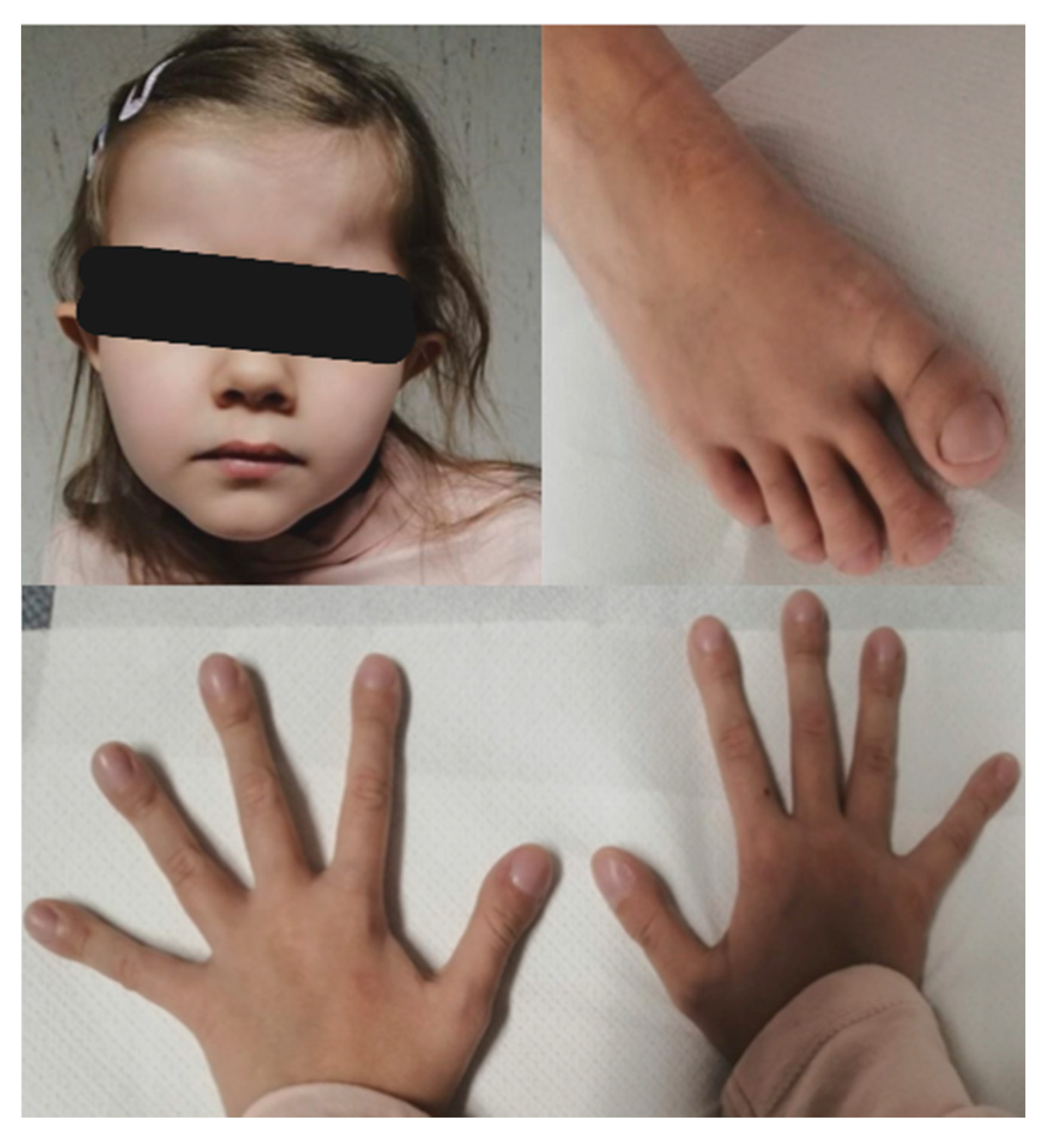
3.1. Clinical Description
3.2. Genetic and Bioinformatic Analyses
4. Discussion

{kind=link}
{kind=link}
| Index Patient (Presented Here) | 1 patient Described [6] | 4 Patients Described [17] | 1 Patient Described [18] | 3 Patients Described [1] | 1 Patient Described [5] | 1 Patient Described [7] | Σ | ||
|---|---|---|---|---|---|---|---|---|---|
| Variants | c.737T>C, c.799C>T | c.1172 T>C, c.1211G>A | P1: c.883C>T, c.883C>T; P2:c.883C>T, c.883C>T; P3:c.1066G>A, c.1066G>A; P4:c.1066G>A, c.1066G>A | c.1040C>T, c.1424G>A | P1: c.1210C>T, c.1254G>C; P2: c.883C>T, c.883C>T; P3: c.829T>G, c.829T>G | c.766T>C, c.1230C>A | c.982A>G, c.1109T>C | ||
| Respiratory system | interstitial lung disease | yes | yes | 4 | yes | 3 | yes | yes | 12/12 |
| cholesterol pneumonitis/foamy macrophages | 4 | 3 | yes | 8/12 | |||||
| pulmonary alveolar proteinosis | 1 | 1/12 | |||||||
| cystic lung disease | yes | yes | 2 | yes | yes | 6/12 | |||
| digital clubbing | yes | 4 | 2 | yes | 8/12 | ||||
| Growth and Nutrition | failure to thrive | yes | yes | 4 | yes | yes | yes | 9/12 | |
| feeding difficulty/diarrhea/vomiting | yes | yes | 2 | yes | 3 | yes | yes | 10/12 | |
| Liver | hepatomegaly/hepatosplenomegaly | yes | 3 | yes | 2 | yes | 8/12 | ||
| Chronic cytolysis and cholestasis | 4 | 4/12 | |||||||
| abnormal liver values (blood) | yes | yes | yes | 3 | yes | yes | 8/12 | ||
| liver steatosis/hyperechogenicity | yes | 2 | yes | 2 | yes | 7/12 | |||
| Nervous system | hypotonia | yes | yes | 1 | yes | 2 | yes | yes | 8/12 |
| neurodevelopmental (speech delay/ motor delay) | yes | yes | 3 | yes | 2 | yes | 9/12 | ||
| microcephaly | yes | yes | 4 | yes | 7/12 | ||||
| brain cysts | yes | 1 | yes | 3/12 | |||||
| brain calcifications | yes | yes | 2/12 | ||||||
| Cerebral vasculopathy | 3 | 1 | 4/12 | ||||||
| stroke | 2 | 2/12 | |||||||
| white matter lesions | yes | yes | yes | 1 | 4/12 | ||||
| brain aneurysm | 2 | 2/12 | |||||||
| Skeletal system | pectus carinatum/excavatum | yes | 2 | 1 | yes | 5/12 | |||
| joint hyperflexibility | yes | 2 | 2 | yes | yes | 6/12 | |||
| Scoliosis | yes | yes | 2/12 | ||||||
| Aseptic osteomyelitis | 1 | 1/12 | |||||||
| osteopenia | yes | 1 | 2/12 | ||||||
| Arachnodactyly | yes | 2 | 1 | yes | 5/12 | ||||
| Endocrine system | growth hormone resistance/deficiency | 1 | 2 | 3/12 | |||||
| Adrenal insufficiency | yes | 1/12 | |||||||
| Hypopituitarism | yes | 1/12 | |||||||
| Micropenis, cryptorchidy | 1 | 1/12 | |||||||
| hypothyroidism | yes | yes | yes | yes | 4/12 | ||||
| Dysmorphic features | face/body appearance | yes | yes | 4 | 1 | yes | 8/12 | ||
| Eye | abnormal eye movement/nystagmus | 1 | 1/12 | ||||||
| reticular opacities | 1 | 1/12 | |||||||
| myopic chorioretinitis | yes | 1/12 | |||||||
| Ear | sensorineural hearing loss | 1 | 1/12 | ||||||
| Cardiovascular system | structural heart/vessel defects | yes | yes | 2 | yes | 5/12 | |||
| Immune system | abnormal blood cell counts/leucocytosis | yes | yes | 4 | yes | yes | yes | 9/12 | |
| hypergammaglobulinemia | yes | 1/12 | |||||||
| hypoalbuminemia | yes | yes | 4 | 3 | yes | yes | 11/12 | ||
| Urinary system | vesicoureteral reflux | yes | 1/12 | ||||||
| nephrolithiasis | 1 | yes | 2/12 | ||||||
| tubulopathy | yes | 1 | 2/12 | ||||||
| proteinuria | yes | 1/12 | |||||||
| Skin/Connective Tissue | poor wound healing/frail skin | 2 | 1 | 3/12 | |||||
| Cutis laxa | yes | 2 | 3/12 | ||||||
| hernia (Abdominal/Inguinal) | 2 | yes | 3/12 | ||||||
| abnormal subcutaneous fat tissue | yes | 4 | 1 | yes | 7/12 | ||||
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schuch, L.A.; Forstner, M.; Rapp, C.K.; Li, Y.; Smith, D.E.C.; Mendes, M.I.; Delhommel, F.; Sattler, M.; Emiralioğlu, N.; Taskiran, E.Z.; et al. FARS1-related disorders caused by bi-allelic mutations in cytosolic phenylalanyl-tRNA synthetase genes: Look beyond the lungs! Clin. Genet. 2021, 99, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.C.; Han, J.M.; Kim, S. Aminoacyl-tRNA synthetases and amino acid signaling. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118889. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Luo, Q.; Han, C.; Zhou, J.; Gong, J.; Chun, L.; Xu, X.S.; Liu, J. Cytoplasmic and mitochondrial aminoacyl-tRNA synthetases differentially regulate lifespan in Caenorhabditis elegans. iScience 2022, 25, 105266. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, G.; Hu, S.; Yin, C.; Zhao, P.; Zhou, X.; Shao, S.; Liu, R.; Hu, W.; Liu, G.L.; et al. FARSB Facilitates Hepatocellular Carcinoma Progression by Activating the mTORC1 Signaling Pathway. Int. J. Mol. Sci. 2023, 24, 16709. [Google Scholar] [CrossRef] [PubMed]
- Krenke, K.; Szczałuba, K.; Bielecka, T.; Rydzanicz, M.; Lange, J.; Koppolu, A.; Płoski, R. FARSA mutations mimic phenylalanyl-tRNA synthetase deficiency caused by FARSB defects. Clin. Genet. 2019, 96, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Chen, Y.; Hu, X.; Qi, Z.; Guo, J.; Li, Y.; Hao, C. Phenylalanyl-tRNA synthetase deficiency caused by biallelic variants in FARSA gene and literature review. BMC Med. Genom. 2023, 16, 245. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sun, Y.; Chen, M.; Dong, M. A rare interstitial lung disease in children caused by novel mutations of FARSA gene. Pediatr. Pulmonol. 2024, 59, 3720–3723. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Yau, C.; Ahmed, A.A. A pan-cancer genome-wide analysis reveals tumour dependencies by induction of nonsense-mediated decay. Nat. Commun. 2017, 8, 15943. [Google Scholar] [CrossRef] [PubMed]
- American Association of Neurological Surgeons (AANS); American Society of Neuroradiology (ASNR); Cardiovascular and Interventional Radiology Society of Europe (CIRSE); Canadian Interventional Radiology Association (CIRA); Congress of Neurological Surgeons (CNS); European Society of Minimally Invasive Neurological Therapy (ESMINT); European Society of Neuroradiology (ESNR); European Stroke Organization (ESO); Society for Cardiovascular Angiography and Interventions (SCAI); Society of Interventional Radiology (SIR); et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed]
- Biagini, T.; Chillemi, G.; Mazzoccoli, G.; Grottesi, A.; Fusilli, C.; Capocefalo, D.; Castellana, S.; Vescovi, A.L.; Mazza, T. Molecular dynamics recipes for genome research. Brief Bioinform. 2018, 19, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, D.; Singh, S.; Wairkar, S. Advanced Delivery Strategies of Nintedanib for Lung Disorders and Beyond: A Comprehensive Review. AAPS PharmSciTech 2024, 25, 150. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M. ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Castellana, S.; Mazza, T. Congruency in the prediction of pathogenic missense mutations: State-of-the-art web-based tools. Brief Bioinform. 2013, 14, 448–459. [Google Scholar] [CrossRef]
- Griese, M.; Schwerk, N.; Carlens, J.; Wetzke, M.; Emiralioğlu, N.; Kiper, N.; Lange, J.; Krenke, K.; Seidl, E.; Collaborators, C. Minimal important difference in childhood interstitial lung diseases. Thorax 2023, 78, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Charbit-Henrion, F.; Goguyer-Deschaumes, R.; Borensztajn, K.; Mirande, M.; Berthelet, J.; Rodrigues-Lima, F.; Khiat, A.; Frémond, M.; Bader-Meunier, B.; Rodari, M.M.; et al. Systemic inflammatory syndrome in children with FARSA deficiency. Clin. Genet. 2022, 101, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Ko, S.; Kang, H.; Kim, M.J.; Moon, J.; Lim, B.C.; Kim, K.J.; Choi, M.; Choi, H.-J.; Chae, J.-H. Fatal systemic disorder caused by biallelic variants in FARSA. Orphanet J. Rare Dis. 2022, 17, 306. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lomuscio, S.; Cocciadiferro, D.; Petrizzelli, F.; Liorni, N.; Mazza, T.; Allegorico, A.; Ullmann, N.; Novelli, G.; Cutrera, R.; Novelli, A. Two Novel Biallelic Variants in the FARSA Gene: The First Italian Case and a Literature Review. Genes 2024, 15, 1573. https://doi.org/10.3390/genes15121573
Lomuscio S, Cocciadiferro D, Petrizzelli F, Liorni N, Mazza T, Allegorico A, Ullmann N, Novelli G, Cutrera R, Novelli A. Two Novel Biallelic Variants in the FARSA Gene: The First Italian Case and a Literature Review. Genes. 2024; 15(12):1573. https://doi.org/10.3390/genes15121573
Chicago/Turabian StyleLomuscio, Sonia, Dario Cocciadiferro, Francesco Petrizzelli, Niccolò Liorni, Tommaso Mazza, Annalisa Allegorico, Nicola Ullmann, Giuseppe Novelli, Renato Cutrera, and Antonio Novelli. 2024. "Two Novel Biallelic Variants in the FARSA Gene: The First Italian Case and a Literature Review" Genes 15, no. 12: 1573. https://doi.org/10.3390/genes15121573
APA StyleLomuscio, S., Cocciadiferro, D., Petrizzelli, F., Liorni, N., Mazza, T., Allegorico, A., Ullmann, N., Novelli, G., Cutrera, R., & Novelli, A. (2024). Two Novel Biallelic Variants in the FARSA Gene: The First Italian Case and a Literature Review. Genes, 15(12), 1573. https://doi.org/10.3390/genes15121573