
Molecular Evolution of the Bactericidal/Permeability-Increasing Protein (BPIFA1) Regulating the Innate Immune Responses in Mammals
 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval and Analysis
2.2. Selection Analysis
2.3. Recombination Analysis
2.4. Protein-Protein Interactions Analysis
2.5. Structural Analysis of BPIFA1 Protein
3. Results
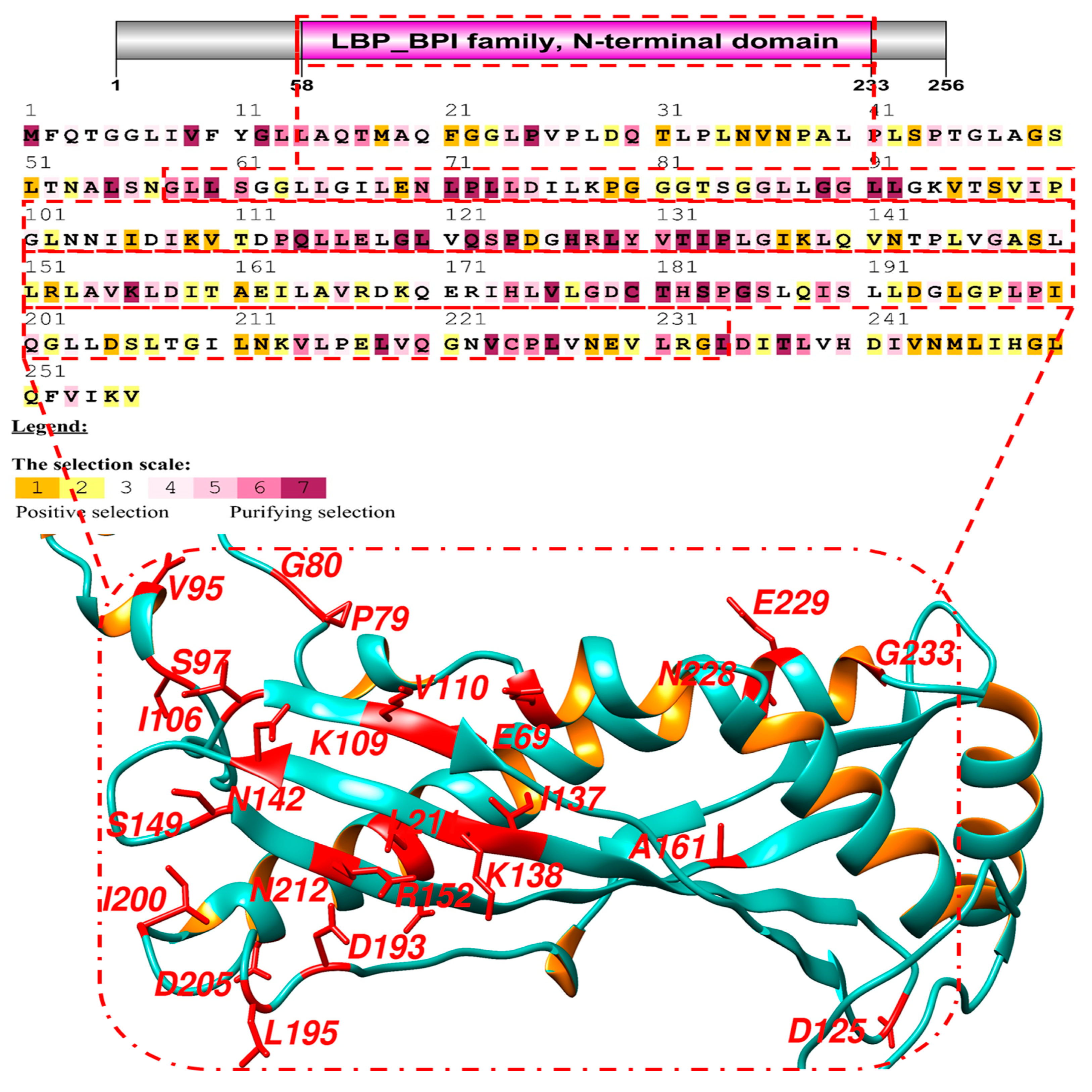
3.1. Molecular Evolution of BPIFA1 Gene
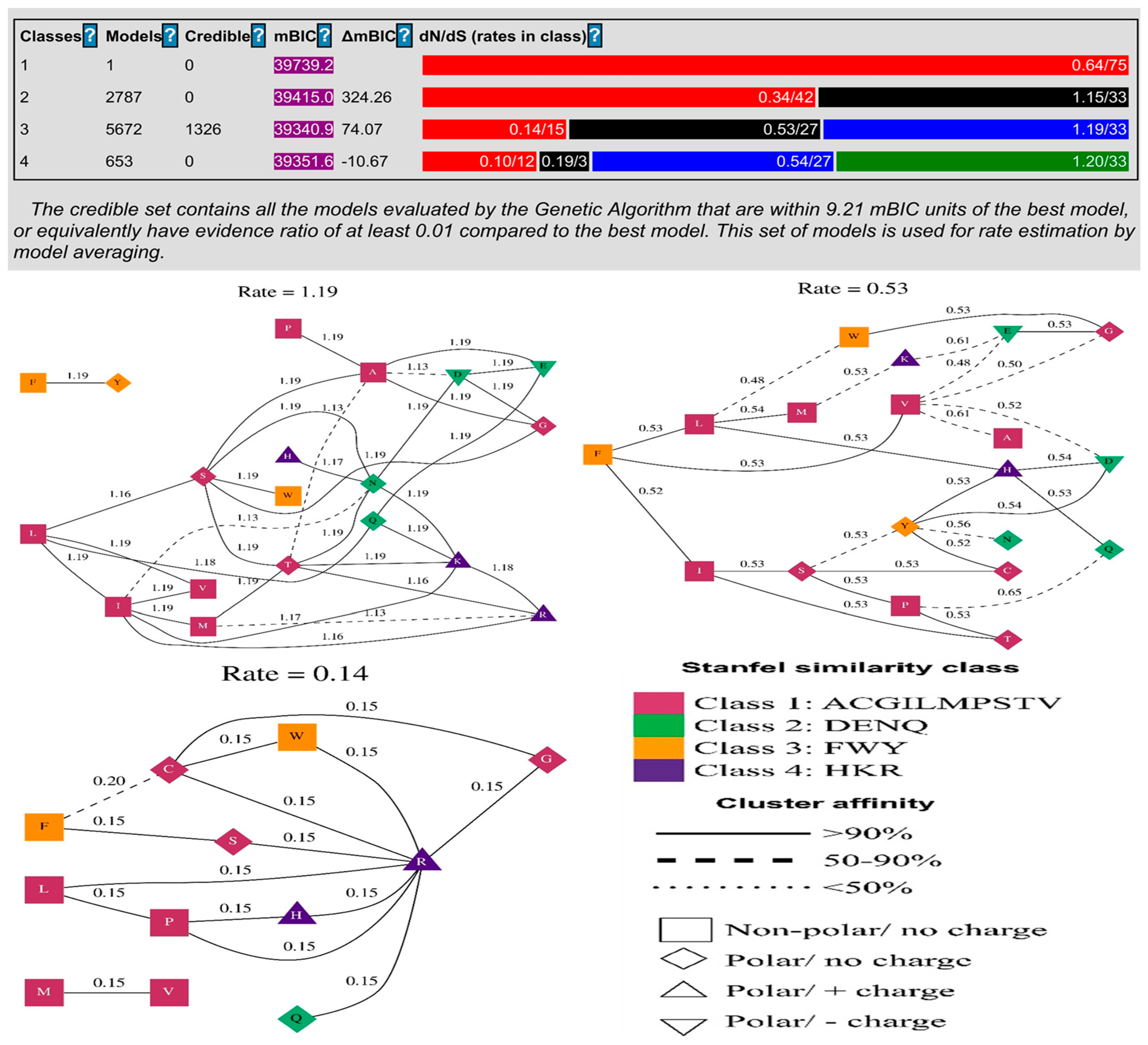
3.2. Adaptive Selection of BPIFA1 Gene
3.3. Recombination Analysis
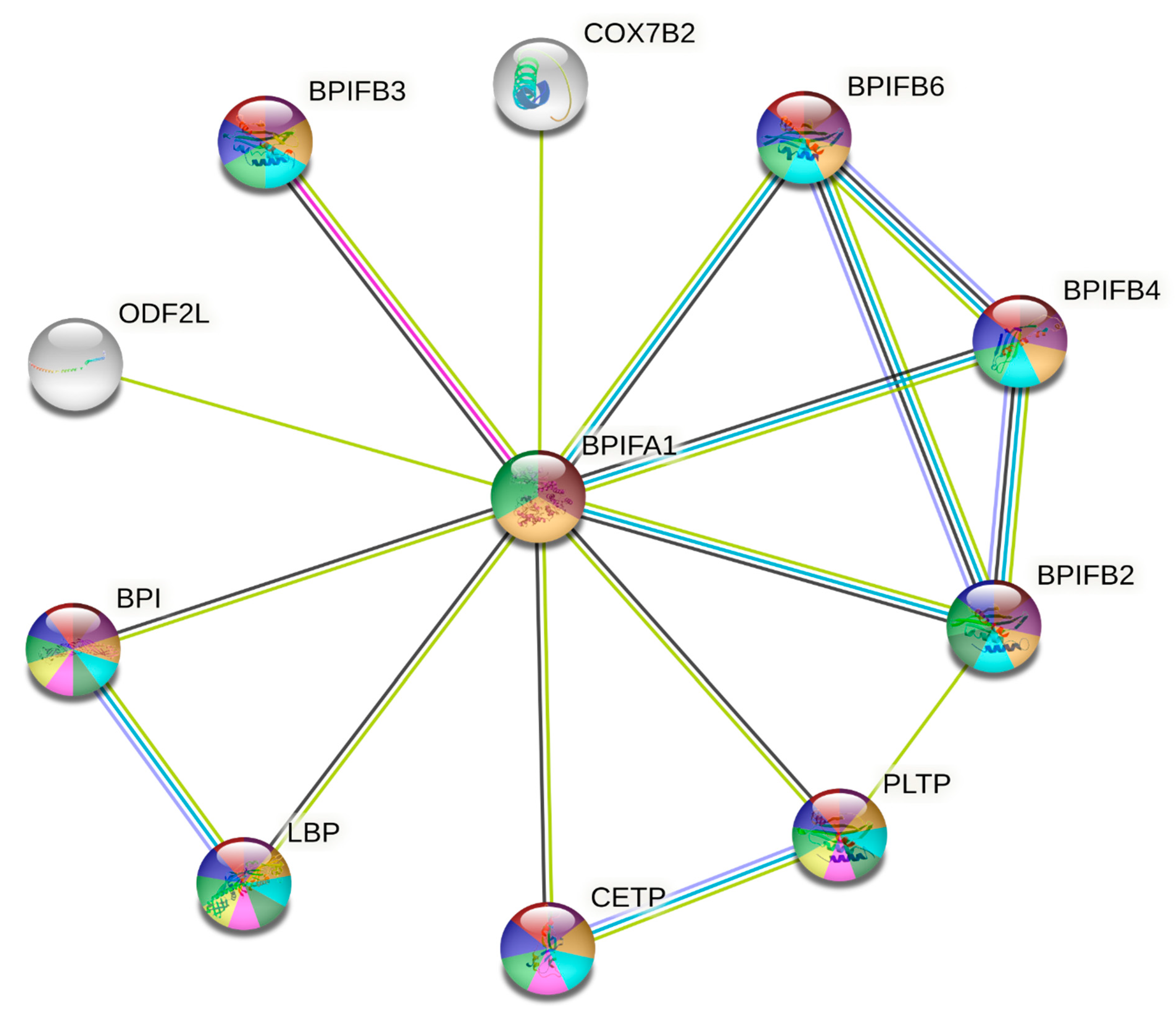
3.4. Protein-Protein Interactions and Ligand Binding Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.; Xu, P.; Wang, L.; Feng, M.; Chen, D.; Yu, X.; Lu, Y. Molecular biology of BPIFB1 and its advances in disease. Ann. Transl. Med. 2020, 8, 651. [Google Scholar] [CrossRef] [PubMed]
- Saferali, A.; Tang, A.C.; Strug, L.J.; Quon, B.S.; Zlosnik, J.; Sandford, A.J.; Turvey, S.E. Immunomodulatory function of the cystic fibrosis modifier gene BPIFA1. PLoS ONE 2020, 15, e0227067. [Google Scholar] [CrossRef] [PubMed]
- Nam, B.-H.; Moon, J.-Y.; Park, E.-H.; Kim, Y.-O.; Kim, D.-G.; Kong, H.J.; Kim, W.-J.; Jee, Y.J.; An, C.M.; Park, N.G.; et al. Antimicrobial Activity of Peptides Derived from Olive Flounder Lipopolysaccharide Binding Protein/Bactericidal Permeability-Increasing Protein (LBP/BPI). Mar. Drugs 2014, 12, 5240–5257. [Google Scholar] [CrossRef] [PubMed]
- Kirschning, C.J.; Au-Young, J.; Lamping, N.; Reuter, D.; Pfeil, D.; Seilhamer, J.J.; Schumann, R.R. Similar organization of the lipopolysaccharide-binding protein (LBP) and phospholipid transfer protein (PLTP) genes suggests a common gene family of lipid-binding proteins. Genomics 1997, 46, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, A.; Marathe, S.A.; Joglekar, M.; Chakravortty, D. Bactericidal/permeability increasing protein: A multifaceted protein with functions beyond LPS neutralization. Innate Immun. 2012, 19, 339–347. [Google Scholar] [CrossRef]
- Wright, S.D.; Ramos, R.A.; Tobias, P.S.; Ulevitch, R.J.; Mathison, J.C. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef]
- Shao, Y.; Li, C.; Che, Z.; Zhang, P.; Zhang, W.; Duan, X.; Li, Y. Cloning and characterization of two lipopolysaccharide-binding protein/bactericidal permeability–increasing protein (LBP/BPI) genes from the sea cucumber Apostichopus japonicus with diversified function in modulating ROS production. Dev. Comp. Immunol. 2015, 52, 88–97. [Google Scholar] [CrossRef]
- Schaefer, N.; Li, X.; Seibold, M.A.; Jarjour, N.N.; Denlinger, L.C.; Castro, M.; Coverstone, A.M.; Teague, W.G.; Boomer, J.; Bleecker, E.R. The effect of BPIFA1/SPLUNC1 genetic variation on its expression and function in asthmatic airway epithelium. JCI Insight 2019, 4, e127237. [Google Scholar] [CrossRef]
- Britto, C.J.; Cohn, L. Bactericidal/permeability-increasing protein fold–containing family member A1 in airway host protection and respiratory disease. Am. J. Respir. Cell Mol. Biol. 2015, 52, 525–534. [Google Scholar] [CrossRef]
- Musa, M.; Wilson, K.; Sun, L.; Mulay, A.; Bingle, L.; Marriott, H.M.; LeClair, E.E.; Bingle, C.D. Differential localisation of BPIFA1 (SPLUNC1) and BPIFB1 (LPLUNC1) in the nasal and oral cavities of mice. Cell Tissue Res. 2012, 350, 455–464. [Google Scholar] [CrossRef]
- Tsou, Y.-A.; Tung, M.-C.; Alexander, K.A.; Chang, W.-D.; Tsai, M.-H.; Chen, H.-L.; Chen, C.-M. The role of BPIFA1 in upper airway microbial infections and correlated diseases. BioMed Res. Int. 2018, 2018, 2021890. [Google Scholar] [CrossRef] [PubMed]
- Caikauskaite, R. BPIFA1 Interactions with Bacteria and Their Importance for Airway Host Defence. Ph.D. Thesis, University of Sheffield, Sheffield, UK, 2018. [Google Scholar]
- Xu, Y.; Tao, Z.; Jiang, Y.; Liu, T.; Xiang, Y. Overexpression of BPIFB1 promotes apoptosis and inhibits proliferation via the MEK/ERK signal pathway in nasopharyngeal carcinoma. Int. J. Clin. Exp. Pathol. 2019, 12, 356. [Google Scholar] [PubMed]
- Mulay, A. The Role of BPIFA1 in Otitis Media. Ph.D. Thesis, University of Sheffield, Sheffield, UK, 2016. [Google Scholar]
- Yang, D.; Han, Y.; Chen, L.; Cao, R.; Wang, Q.; Dong, Z.; Liu, H.; Zhang, X.; Zhang, Q.; Zhao, J. A bactericidal permeability-increasing protein (BPI) from manila clam Ruditapes philippinarum: Investigation on the antibacterial activities and antibacterial action mode. Fish Shellfish. Immunol. 2019, 93, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Slodkowicz, G.; Goldman, N. Integrated structural and evolutionary analysis reveals common mechanisms underlying adaptive evolution in mammals. Proc. Natl. Acad. Sci. USA 2020, 117, 5977–5986. [Google Scholar] [CrossRef] [PubMed]
- Barrier, M.; Bustamante, C.D.; Yu, J.; Purugganan, M.D. Selection on rapidly evolving proteins in the Arabidopsis genome. Genetics 2003, 163, 723–733. [Google Scholar] [CrossRef]
- Bazykin, G.A.; Kondrashov, A.S. Major role of positive selection in the evolution of conservative segments of Drosophila proteins. Proc. R. Soc. B Biol. Sci. 2012, 279, 3409–3417. [Google Scholar] [CrossRef]
- Turner, L.M.; Chuong, E.B.; Hoekstra, H.E. Comparative analysis of testis protein evolution in rodents. Genetics 2008, 179, 2075–2089. [Google Scholar] [CrossRef]
- Peng, J.; Svetec, N.; Zhao, L. Intermolecular Interactions Drive Protein Adaptive and Coadaptive Evolution at Both Species and Population Levels. Mol. Biol. Evol. 2022, 39, msab350. [Google Scholar] [CrossRef]
- Cox, S.L.; O’Siorain, J.R.; Fagan, L.E.; Curtis, A.M.; Carroll, R.G. Intertwining roles of circadian and metabolic regulation of the innate immune response. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2022; pp. 1–13. [Google Scholar]
- Hochachka, P.W.; Somero, G.N. Biochemical Adaptation: Mechanism and Process in Physiological Evolution; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Brooks, A.N.; Turkarslan, S.; Beer, K.D.; Yin Lo, F.; Baliga, N.S. Adaptation of cells to new environments. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 544–561. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Ahmad, H.I.; Liu, G.; Jiang, X.; Liu, C.; Chong, Y.; Huarong, H. Adaptive molecular evolution of MC1R gene reveals the evidence for positive diversifying selection in indigenous goat populations. Ecol. Evol. 2017, 7, 5170–5180. [Google Scholar] [CrossRef]
- Asif, A.R.; Awais, M.; Qadri, S.; Ahmad, H.I.; Du, X. Positive selection of IL-33 in adaptive immunity of domestic Chinese goats. Ecol. Evol. 2017, 7, 1954–1963. [Google Scholar] [CrossRef]
- Ahmad, H.I.; Ahmad, M.J.; Adeel, M.M.; Asif, A.R.; Du, X. Positive selection drives the evolution of endocrine regulatory bone morphogenetic protein system in mammals. Oncotarget 2018, 9, 18435. [Google Scholar] [CrossRef]
- Ahmad, M.J.; Ahmad, H.I.; Adeel, M.M.; Liang, A.; Hua, G.; Murtaza, S.; Mirza, R.H.; Elokil, A.; Ullah, F.; Yang, L. Evolutionary Analysis of Makorin Ring Finger Protein 3 Reveals Positive Selection in Mammals. Evol. Bioinform. 2019, 15, 1176934319834612. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Poon, A.F.; Frost, S.D.; Pond, S.L. Detecting signatures of selection from DNA sequences using Datamonkey. Methods Mol. Biol. Clifton N. J. 2009, 537, 163–183. [Google Scholar] [CrossRef]
- Ahmad, H.I.; Liu, G.; Jiang, X.; Edallew, S.G.; Wassie, T.; Tesema, B.; Yun, Y.; Pan, L.; Liu, C.; Chong, Y. Maximum-likelihood approaches reveal signatures of positive selection in BMP15 and GDF9 genes modulating ovarian function in mammalian female fertility. Ecol. Evol. 2017, 7, 8895–8902. [Google Scholar] [CrossRef]
- Ahmad, H.I.; Liu, G.; Jiang, X.; Liu, C.; Fangzheng, X.; Chong, Y.; Ijaz, N.; Huarong, H. Adaptive selection at agouti gene inferred breed specific selection signature within the indigenous goat populations. Asian-Australas. J. Anim. Sci. 2017. [Google Scholar] [CrossRef]
- Bielawski, J.P.; Yang, Z. Maximum likelihood methods for detecting adaptive evolution after gene duplication. J. Struct. Funct. Genom. 2003, 3, 201–212. [Google Scholar] [CrossRef]
- Kelley, L.A.; Sternberg, M.J. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 2009, 4, 363–371. [Google Scholar] [CrossRef]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.R.; Liao, B.Y.; Zhuang, S.M.; Zhang, J. Protein misinteraction avoidance causes highly expressed proteins to evolve slowly. Proc. Natl. Acad. Sci. USA 2012, 109, E831–E840. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef]
- Li, H.; Zhao, X.; Wang, J.; Zong, M.; Yang, H. Bioinformatics analysis of gene expression profile data to screen key genes involved in pulmonary sarcoidosis. Gene 2017, 596, 98–104. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. Protein structure and function prediction using I-TASSER. Curr. Protoc. Bioinform. 2015, 52, 5.8.1–5.8.15. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Nosil, P.; Funk, D.J.; Ortiz-Barrientos, D. Divergent selection and heterogeneous genomic divergence. Mol. Ecol. 2009, 18, 375–402. [Google Scholar] [CrossRef]
- Yeaman, S.; Whitlock, M.C. The genetic architecture of adaptation under migration–selection balance. Evolution 2011, 65, 1897–1911. [Google Scholar] [CrossRef]
- Feder, J.L.; Egan, S.P.; Nosil, P. The genomics of speciation-with-gene-flow. Trends Genet. 2012, 28, 342–350. [Google Scholar] [CrossRef]
- Yang, Z.; dos Reis, M. Statistical properties of the branch-site test of positive selection. Mol. Biol. Evol. 2011, 28, 1217–1228. [Google Scholar] [CrossRef]
- Murrell, D.F.; Daniel, B.S.; Joly, P.; Borradori, L.; Amagai, M.; Hashimoto, T.; Caux, F.; Marinovic, B.; Sinha, A.A.; Hertl, M.; et al. Definitions and outcome measures for bullous pemphigoid: Recommendations by an international panel of experts. J. Am. Acad. Dermatol. 2012, 66, 479–485. [Google Scholar] [CrossRef]
- Ahmad, H.I.; Afzal, G.; Iqbal, M.N.; Shokrollahi, B.; Mansoor, M.K.; Chen, J. Positive Selection Drives the Adaptive Evolution of Mitochondrial Antiviral Signaling (MAVS) Proteins-Mediating Innate Immunity in Mammals. Front. Vet. Sci. 2022, 8, 814765. [Google Scholar] [CrossRef]
- Xia, X. Nucleotide substitution models and evolutionary distances. In Bioinformatics and the Cell; Springer: Berlin/Heidelberg, Germany, 2018; pp. 269–314. [Google Scholar]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Andreani, J.; Guerois, R. Evolution of protein interactions: From interactomes to interfaces. Arch. Biochem. Biophys. 2014, 554, 65–75. [Google Scholar] [CrossRef]
- Wagner, A. Neutralism and selectionism: A network-based reconciliation. Nat. Rev. Genet. 2008, 9, 965–974. [Google Scholar] [CrossRef]
- Halperin, I.; Wolfson, H.; Nussinov, R. Protein-protein interactions: Coupling of structurally conserved residues and of hot spots across interfaces. Implications for docking. Structure 2004, 12, 1027–1038. [Google Scholar] [CrossRef]
- Pechmann, S.; Levy, E.D.; Tartaglia, G.G.; Vendruscolo, M. Physicochemical principles that regulate the competition between functional and dysfunctional association of proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 10159–10164. [Google Scholar] [CrossRef]
- Bergstrom, T.; Gyllensten, U. Evolution of Mhc class II polymorphism: The rise and fall of class II gene function in primates. Immunol. Rev. 1995, 143, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.X.; Zhao, S.M.; Cheng, M.L.; Guo, L.; Ye, R.Q.; Liu, W.Q.; Gao, S.Z. Cloning and expression levels of genes relating to the ovulation rate of the Yunling black goat. Biol. Reprod. 2009, 80, 219–226. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | n | Lc | S | Model | lnL | 2Δl M3 vs. M0 | 2Δl M2 vs. M1 | 2Δl M8 vs. M7 | Parameter Estimates | PAML Site Model (M8) ω > 1 | SLAC (p < 0.05) | FEL (p < 0.05) | FUBAR (post pr. 0.9) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BPIFA1 | 34 | 504 | 49 | M0 (one-ratio) | −18,981.403 | 422.86 | 64.5 | 93.63 | one ratio | 125, 129, 130, 178, 184, 242, 259, 270, 304, 385, 401, 402, 425, 434, 437 | 130, 270, 385, 402, 425, 455 | 130, 259, 402, 425 | 130, 270, 385, 402, 425 |
| M1 Nearly Neutral (2 categories) | −18,802.441 | p1 = 0.45328, p2 = 0.54672 | |||||||||||
| ω 1 = 0.24322, ω 2 = 1.00000 | |||||||||||||
| M2 Positive Selection (3 categories) | −18,770.141 | p1 = 0.43064, p2 = 0.45653, p3 = 0.11283 | |||||||||||
| ω 1 = 0.26160, ω 2 = 1.00000, ω 3 = 2.62492 | |||||||||||||
| M3 Discrete (3 categories) | −18,769.971 | p1 = 0.41335, p2 =0.46115, p3 =0.12550 | |||||||||||
| M7 β (10 categories) | −18,829.824 | p = 1.04797, q = 0.59014 | |||||||||||
| M8 β&Ꙍ > 1 (11 categories) | −18,783.01 | p0 = 0.78568, p = 1.86405 q = 1.74778 | |||||||||||
| p1 = 0.21432, ω = 1.93377 |
| Gene | Model | Positively Selected Sites | Amino Acids | Posterior pr. (ω > 1) | Post Mean ± SE for w |
|---|---|---|---|---|---|
| BPIFA1 | M8 β&Ꙍ > 1 (11 categories) | 71 | P | 0.555 | 1.652 ± 0.876 |
| 94 | H | 0.503 | 1.548 ± 0.897 | ||
| 106 | H | 0.618 | 1.762 ± 0.863 | ||
| 125 | G | 0.962 * | 2.356 ± 0.403 | ||
| 128 | Q | 0.938 | 2.318 ± 0.463 | ||
| 129 | D | 0.998 ** | 2.414 ± 0.283 | ||
| 130 | P | 0.615 | 1.787 ± 0.793 | ||
| 172 | L | 0.874 | 2.214 ± 0.580 | ||
| 177 | W | 0.907 | 2.268 ± 0.529 | ||
| 178 | E | 0.963 * | 2.359 ± 0.399 | ||
| 184 | A | 0.997 ** | 2.412 ± 0.288 | ||
| 190 | G | 0.764 | 2.030 ± 0.721 | ||
| 192 | L | 0.634 | 1.795 ± 0.842 | ||
| 207 | V | 0.795 | 2.084 ± 0.686 | ||
| 208 | S | 0.812 | 2.113 ± 0.661 | ||
| 213 | L | 0.652 | 1.829 ± 0.833 | ||
| 216 | H | 0.669 | 1.864 ± 0.809 | ||
| 242 | Q | 0.989 * | 2.399 ± 0.320 | ||
| 260 | G | 0.621 | 1.799 ± 0.788 | ||
| 262 | V | 0.992 ** | 2.404 ± 0.309 | ||
| 270 | N | 0.999 ** | 2.416 ± 0.278 | ||
| 304 | L | 0.975 * | 2.377 ± 0.367 | ||
| 308 | E | 0.633 | 1.816 ± 0.789 | ||
| 356 | P | 0.908 | 2.270 ± 0.527 | ||
| 385 | Q | 0.980 * | 2.385 ± 0.349 | ||
| 390 | V | 0.531 | 1.650 ± 0.798 | ||
| 401 | H | 1.000 ** | 2.416 ± 0.277 | ||
| 402 | Q | 0.991 ** | 2.403 ± 0.313 | ||
| 403 | L | 0.68 | 1.891 ± 0.777 | ||
| 417 | S | 0.937 | 2.316 ± 0.463 | ||
| 432 | E | 1.000 ** | 2.416 ± 0.278 | ||
| 434 | Q | 0.993 ** | 2.406 ± 0.304 | ||
| 436 | W | 0.964 * | 2.360 ± 0.395 | ||
| 437 | G | 0.969 * | 2.368 ± 0.383 | ||
| 452 | L | 0.808 | 2.104 ± 0.671 | ||
| 459 | C | 0.702 | 1.921 ± 0.788 | ||
| 460 | A | 0.511 | 1.584 ± 0.864 | ||
| 468 | T | 0.697 | 1.903 ± 0.814 | ||
| 469 | Q | 0.705 | 1.925 ± 0.794 |
| Codon | α | β- | Pr. [β = β−] | β+ | Pr. [β = β+] | p-Value | q-Value |
|---|---|---|---|---|---|---|---|
| 130 | 0.0000 | 0.0000 | 0.2042 | 3.4440 | 0.7957 | 0.0078 | 0.2638 |
| 167 | 0.4663 | 0.1038 | 0.7853 | 5.3438 | 0.2146 | 0.0066 | 0.2596 |
| 168 | 0.7252 | 0.2013 | 0.8497 | 28.492 | 0.1502 | 0.0008 | 0.0612 |
| 190 | 0.0000 | 0.0000 | 0.7071 | 14.030 | 0.2928 | 0.0035 | 0.1774 |
| 243 | 0.3055 | 0.0426 | 0.8016 | 3.2988 | 0.1983 | 0.0086 | 0.2561 |
| 265 | 0.6382 | 0.0000 | 0.7209 | 9.2337 | 0.2790 | 0.0086 | 0.2720 |
| 289 | 0.3685 | 0.0771 | 0.9177 | 15.304 | 0.0822 | 0.0052 | 0.2184 |
| 313 | 0.2462 | 0.2462 | 0.8605 | 21.499 | 0.1394 | 0.0071 | 0.2576 |
| 314 | 0.4211 | 0.3321 | 0.8708 | 70.669 | 0.1291 | 0.0001 | 0.0160 |
| 391 | 1.0888 | 0.3282 | 0.9122 | 105.28 | 0.0877 | 0.0041 | 0.0070 |
| 401 | 1.4302 | 0.8308 | 0.8019 | 364.04 | 0.1980 | 0.0096 | 0.2549 |
| 402 | 0.3620 | 0.3125 | 0.3515 | 25.937 | 0.6484 | 0.0040 | 0.1878 |
| 403 | 0.4970 | 0.4970 | 0.9236 | 442.56 | 0.0763 | 0.0021 | 0.1190 |
| 425 | 0.0000 | 0.0000 | 1.0000 | 1.1829 | 1.0000 | 0.0020 | 0.1319 |
| 432 | 0.8816 | 0.3808 | 0.7310 | 46.308 | 0.2680 | 0.0005 | 0.0502 |
| 437 | 0.9684 | 0.0118 | 0.7491 | 24.911 | 0.2508 | 0.0031 | 0.0090 |
| 452 | 0.3900 | 0.3654 | 0.8800 | 330.91 | 0.1199 | 0.0001 | 0.0145 |
| 455 | 0.4795 | 0.2852 | 0.8823 | 1362.1 | 0.1176 | 0.0051 | 0.0044 |
| 457 | 0.5413 | 0.0000 | 0.7556 | 10.723 | 0.2443 | 0.0088 | 0.2471 |
| Name | B | LRT | Test p-Value | Uncorrected p-Value | ω Distribution over Sites |
|---|---|---|---|---|---|
| Node43 | 0 | 30.9022 | 0.000 | 0.000 | ω1 = 1.00 (90%) |
| ω2 = 568 (9.7%) | |||||
| Node48 | 0 | 57.7808 | 0.000 | 0.000 | ω1 = 0.632 (90%) |
| ω2 = 1000 (10%) | |||||
| Node58 | 0 | 35.478 | 0.000 | 0.000 | ω1 = 0.784 (82%) |
| ω2 = 38.9 (18%) | |||||
| OTOLEMUR_GARNETTII | 0 | 35.1949 | 0.000 | 0.000 | ω1 = 0.00 (84%) |
| ω2 = 3850 (16%) | |||||
| PHYSETER_CATODON | 0 | 46.7818 | 0.000 | 0.000 | ω1 = 0.498 (85%) |
| ω2 = 37.2 (15%) | |||||
| PTEROPUS_ALECTO | 0 | 39.4366 | 0.000 | 0.000 | ω1 = 1.00 (96%) |
| ω2 = 881 (4.3%) | |||||
| Node9 | 0 | 22.9271 | 0.0002 | 0.000 | ω1 = 1.00 (85%) |
| ω2 = 9090 (15%) | |||||
| ECHINOPS_TELFAIRI | 0 | 22.2294 | 0.0003 | 0.000 | ω1 = 0.262 (87%) |
| ω2 = 1000 (13%) | |||||
| PUMA_CONCOLOR | 0 | 20.5045 | 0.0006 | 0.000 | ω1 = 1.00 (98%) |
| ω2 = 220 (2.2%) | |||||
| OCHOTONA_PRINCEPS | 0 | 16.9285 | 0.0038 | 0.0001 | ω1 = 0.486 (85%) |
| ω2 = 102 (15%) | |||||
| Node1 | 0 | 15.9753 | 0.0061 | 0.0001 | ω1 = 0.00 (96%) |
| ω2 = 543 (3.5%) | |||||
| Node42 | 0 | 15.9466 | 0.0061 | 0.0001 | ω1 = 0.00 (97%) |
| ω2 = 1000 (3.0%) | |||||
| CHINCHILLA_LANIGERA | 0 | 15.7558 | 0.0065 | 0.0001 | ω1 = 0.260 (89%) |
| ω2 = 1000 (11%) | |||||
| CASTOR_CANADENSIS | 0 | 14.0154 | 0.0154 | 0.0003 | ω1 = 0.217 (90%) |
| ω2 = 1000 (10%) |
| Codon | dS | dN | dN Leaves | dN/dS | Normalized dN-dS | p-Value |
|---|---|---|---|---|---|---|
| 85 | 9.97170 | 4.39904 | 0.85722 | 4.4116 | 0.3100 | 0.090973 |
| 105 | 0.22288 | 19.2224 | 0.00000 | 86.244 | 1.33923 | 0.050153 |
| 113 | 0.16321 | 1.21552 | 1.14983 | 7.4470 | 0.07417 | 0.076543 |
| 130 | 0.02672 | 1.38927 | 3.15186 | 51.992 | 0.09604 | 0.050535 |
| 168 | 0.97226 | 3.28401 | 0.62962 | 3.3780 | 0.16295 | 0.086927 |
| 172 | 0.87119 | 3.38718 | 1.12971 | 3.8880 | 0.17734 | 0.069473 |
| 207 | 0.71507 | 4.49186 | 0.73823 | 6.2820 | 0.26621 | 0.007236 |
| 271 | 0.45387 | 9.03290 | 0.52358 | 19.902 | 0.60471 | 0.037150 |
| 346 | 0.00000 | 1.01879 | 0.08543 | Infinite | 0.07181 | 0.027248 |
| 385 | 0.36337 | 3.73078 | 1.22177 | 10.267 | 0.23736 | 0.017551 |
| 390 | 0.52418 | 2.17195 | 1.06171 | 4.1430 | 0.11614 | 0.054228 |
| 402 | 0.53356 | 38.1391 | 1.54115 | 71.480 | 2.65073 | 0.037807 |
| 410 | 0.30457 | 2.07557 | 0.82994 | 6.8150 | 0.12483 | 0.052322 |
| 425 | 0.00000 | 0.67135 | 1.33338 | Infinite | 0.04732 | 0.015383 |
| 432 | 1.38386 | 8.15529 | 1.07013 | 5.8930 | 0.47730 | 0.037254 |
| 445 | 0.00000 | 2.59648 | 0.00000 | Infinite | 0.18302 | 0.026423 |
| 455 | 0.95951 | 3.98406 | 0.34305 | 4.1520 | 0.21319 | 0.089371 |
| GO-term | Description | Count in Gene Set | False Discovery Rate |
|---|---|---|---|
| GO:0043030 | regulation of macrophage activation | 3 of 40 | 0.00086 |
| GO:0051707 | response to other organisms | 6 of 1173 | 0.0045 |
| GO:0034375 | high-density lipoprotein particle remodeling | 2 of 17 | 0.0052 |
| GO:0043032 | positive regulation of macrophage activation | 2 of 23 | 0.0057 |
| GO:0019730 | antimicrobial humoral response | 3 of 143 | 0.0057 |
| GO:0010874 | regulation of cholesterol efflux | 2 of 21 | 0.0057 |
| GO:0006955 | immune response | 6 of 1560 | 0.0057 |
| GO:0009617 | response to bacterium | 4 of 555 | 0.0076 |
| GO:0042742 | defense response to bacterium | 3 of 250 | 0.0098 |
| GO:0042116 | macrophage activation | 2 of 41 | 0.0098 |
| GO:0032720 | negative regulation of tumor necrosis factor production | 2 of 48 | 0.0098 |
| GO:0019731 | antibacterial humoral response | 2 of 47 | 0.0098 |
| GO:0006952 | defense response | 5 of 1234 | 0.0098 |
| GO:0006869 | lipid transport | 3 of 272 | 0.0098 |
| GO:0001818 | negative regulation of cytokine production | 3 of 245 | 0.0098 |
| GO:0032496 | response to lipopolysaccharide | 3 of 298 | 0.0100 |
| GO:0032677 | regulation of interleukin-8 production | 2 of 67 | 0.0116 |
| GO:0015914 | phospholipid transport | 2 of 73 | 0.0127 |
| GO:0098542 | defense response to other organisms | 4 of 859 | 0.0148 |
| GO:0051704 | multi-organism process | 6 of 2514 | 0.0175 |
| GO:0050829 | antimicrobial humoral immune response | 2 of 95 | 0.0188 |
| GO:0061844 | antimicrobial peptide | 2 of 107 | 0.021 |
| GO:0032675 | regulation of interleukin-6 production | 2 of 112 | 0.0234 |
| GO:0071222 | cellular response to lipopolysaccharide | 2 of 146 | 0.0341 |
| GO:0002274 | myeloid leukocyte activation | 3 of 574 | 0.0386 |
| GO:0002699 | positive regulation of the immune effector process | 2 of 186 | 0.0492 |
| COACH Results | ||||
| C-Score | Cluster Size | Ligand Name | Predicted Binding Residue | |
| BPIFA1 | 0.04 | 3 | PC1 | 51, 54, 64, 118, 120, 131, 133, 157, 163, 181, 219, 223, 227, 230, 231 |
| 0.04 | 3 | BPH | 10, 14, 18 | |
| 0.04 | 3 | 1BP1A00 | 51, 54, 55, 57, 58, 168, 173, 174, 226, 227, 228, 231, 234, 237, 238 | |
| 0.03 | 2 | XE | 165, 235, 238, 239 | |
| 0.03 | 2 | 36E | 200, 204 | |
| 0.03 | 2 | DCW | 187, 216, 219 | |
| 0.03 | 2 | 2CV | 191, 204, 208, 211 | |
| 0.03 | 2 | XE | 5, 8, 9, 56, 57 | |
| 0.03 | 2 | CRT | 211, 215, 218 | |
| 0.03 | 2 | 3E8TA00 | 54, 58, 64, 84, 134, 137, 150, 155, 159, 188, 203, 211, 215, 227, 231 | |
| TM-site | ||||
| C-score | Cluster size | Ligand name | Predicted binding residues | |
| BPIFA1 | 0.19 | 3 | BPH(1),2CV(1) | 10, 14, 18 |
| 0.17 | 2 | DCW(1) | 187, 216, 219 | |
| 0.14 | 1 | CRT(1) | 211, 215, 218 | |
| 0.13 | 1 | CLA | 8, 12 | |
| 0.13 | 1 | 2CV | 191, 204, 208, 211 | |
| S-site | ||||
| C-score | Cluster size | Ligands name | Predicted binding residues | |
| BPIFA1 | 0.15 | 3 | NEH, CLA, CU | 216, 222, 223, 224, 225 |
| 0.12 | 1 | MG | 179, 180 | |
| 0.10 | 1 | FOL | 215, 218 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, H.I.; Khan, F.A.; Khan, M.A.; Imran, S.; Akhtar, R.W.; Pandupuspitasari, N.S.; Negara, W.; Chen, J. Molecular Evolution of the Bactericidal/Permeability-Increasing Protein (BPIFA1) Regulating the Innate Immune Responses in Mammals. Genes 2023, 14, 15. https://doi.org/10.3390/genes14010015
Ahmad HI, Khan FA, Khan MA, Imran S, Akhtar RW, Pandupuspitasari NS, Negara W, Chen J. Molecular Evolution of the Bactericidal/Permeability-Increasing Protein (BPIFA1) Regulating the Innate Immune Responses in Mammals. Genes. 2023; 14(1):15. https://doi.org/10.3390/genes14010015
Chicago/Turabian StyleAhmad, Hafiz Ishfaq, Faheem Ahmed Khan, Musarrat Abbas Khan, Safdar Imran, Rana Waseem Akhtar, Nuruliarizki Shinta Pandupuspitasari, Windu Negara, and Jinping Chen. 2023. "Molecular Evolution of the Bactericidal/Permeability-Increasing Protein (BPIFA1) Regulating the Innate Immune Responses in Mammals" Genes 14, no. 1: 15. https://doi.org/10.3390/genes14010015
APA StyleAhmad, H. I., Khan, F. A., Khan, M. A., Imran, S., Akhtar, R. W., Pandupuspitasari, N. S., Negara, W., & Chen, J. (2023). Molecular Evolution of the Bactericidal/Permeability-Increasing Protein (BPIFA1) Regulating the Innate Immune Responses in Mammals. Genes, 14(1), 15. https://doi.org/10.3390/genes14010015