
An Intron c.103-3T>C Variant of the AMELX Gene Causes Combined Hypomineralized and Hypoplastic Type of Amelogenesis Imperfecta: Case Series and Review of the Literature


 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. The AI Patients
2.2. Histological Analysis
2.3. Molecular Genetic Analysis
3. Results
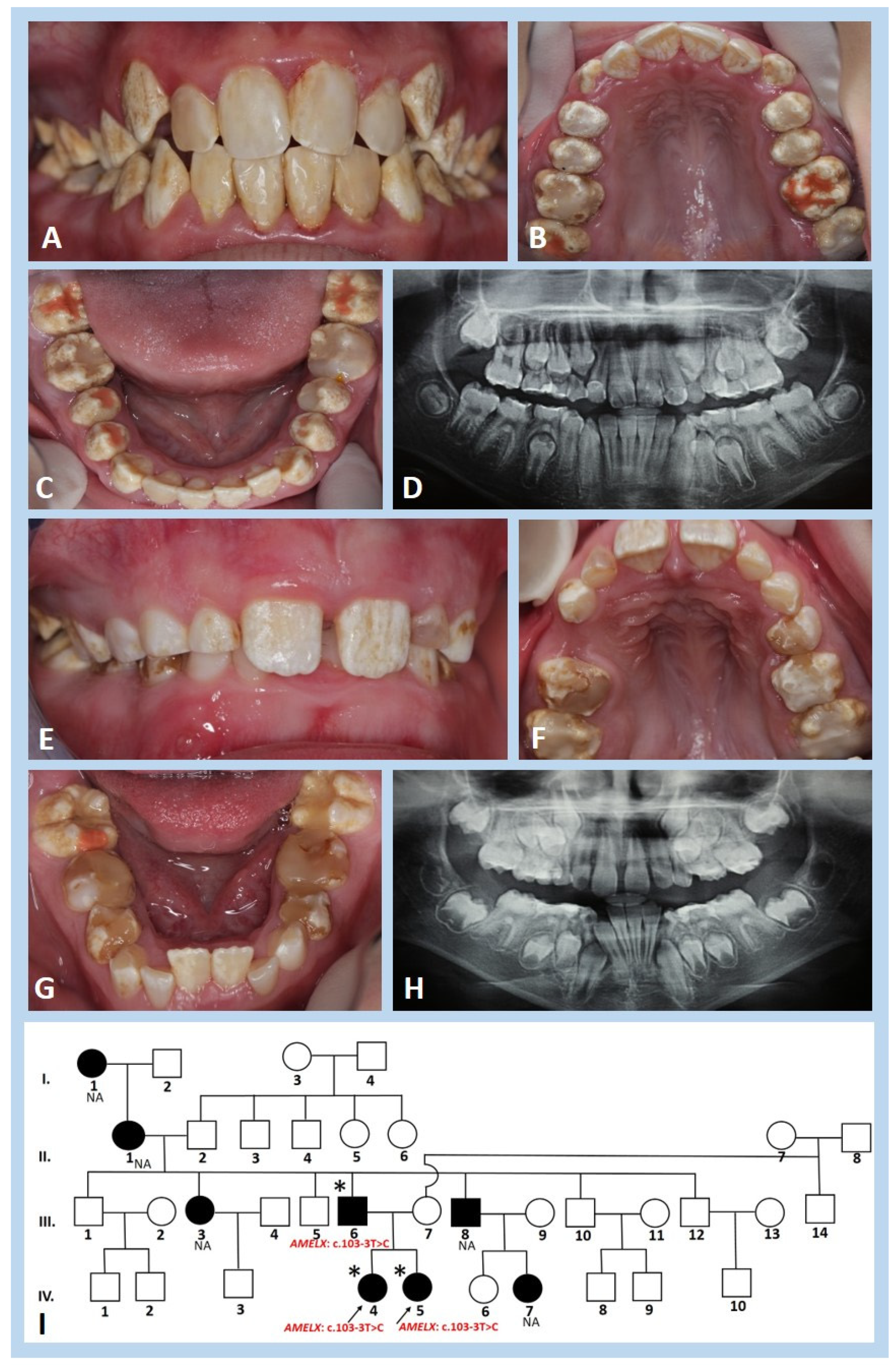
3.1. Clinical and Radiographic Findings
3.2. Histological and Ultrastructural Analysis
3.3. Molecular Genetic Analysis
3.4. Literature Review

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| c DNA | Protein | Enamel Phenotype | Reference |
|---|---|---|---|
| c.860C>A | p.S287X | Generalised hypocalcified | [38] |
| c.891T>A | p.Y297X | Generalised hypocalcified | [39] |
| c.906T>G | p.Y302X | Hypocalcified; hypocalcified, polished-looking enamel surface | [40]; [41] * |
| c.923dupT | p.V309RfsX16 | Hypocalcified | [41] |
| c.923_924delTC | p.L308RfsX16 | Generalised hypocalcified | [38] |
| c.931dupC | p.V311RfsX14 | Hypocalcified | [42] |
| c.973C>T | p.R325X | Hypocalcified | [41,42,43,44] |
| c.1024T>A | p.S342T | Hypocalcified | [45] |
| c.1130_1131delinsAA | p.S377X | Hypocalcified | [42] |
| c.1147G>T | p.E383X | Hypocalcified | [42] |
| c.1192C>T | p.Q398X | Hypocalcified; not described * | [43,46,47]; [17] * |
| c.1222A>T | p.K408X | Hypocalcified | [48] |
| c.1243G>T | p.E415X | Generalised hypocalcified | [39] |
| c.1261G>T | p.E421X | Generalised hypocalcified | [49] |
| c.1282C>T | p.Q428X | Hypomineralised | [16] |
| c.1289C>A | p.S430X | Not described *; hypomature | [17] *; [16] |
| c.1330C>T | p.Q444X | Hypocalcified | [46,47] |
| c.1354C>T | p.Q452X | Generalised hypocalcified | [18,40,41,50,51] |
| c.1366C>T | p.Q456X | Hypocalcified | [47] |
| c.1369C>T | p.Q457X | Hypocalcified | [51] |
| c.1374C>A | p.Y458X | Generalised hypocalcified, small focal areas of normal-looking enamel | [52] |
| c.1379G>A | p.W460X | Generalised hypocalcified | [38,53] |
| c.1380G>A | p.W460X | Generalised hypocalcified | [39] |
| c.1387C>T | p.Q463X | Hypocalcified | [54] |
| c.1408C>T | p.Q470X | Generalised hypocalcified | [38] |
| c.1669G>T | p.G557C | Hypocalcified, attenuated | [55] |
| c.1872_1873delCC | p.L625AfsX79 | Localised hypocalcified | [38] |
| c.1915A>T | p.K639X | Hypocalcified | [51] |
| c.1993C>T | p.Q665X | Generalised hypoplastic/hypomineralisation; less severe phenotype | [56] |
| c.2029C>T | p.Q677X | Generalised hypocalcified; not described * | [18,39,41,56]; [16,17] * |
| c.2080G>T | p.E694X | Localised hypocalcified | [38] |

| cDNA | Protein | Enamel Phenotypes | Reference |
|---|---|---|---|
| c.-11311600_X705268del | p.0? | Hypoplasia/hypomineralisation | [57] |
| c.-39356_X6166del | p.0? | Snow-capped appearance | [58] |
| c.-21552_X67556del | p.0? | Snow-capped appearance | [58] |
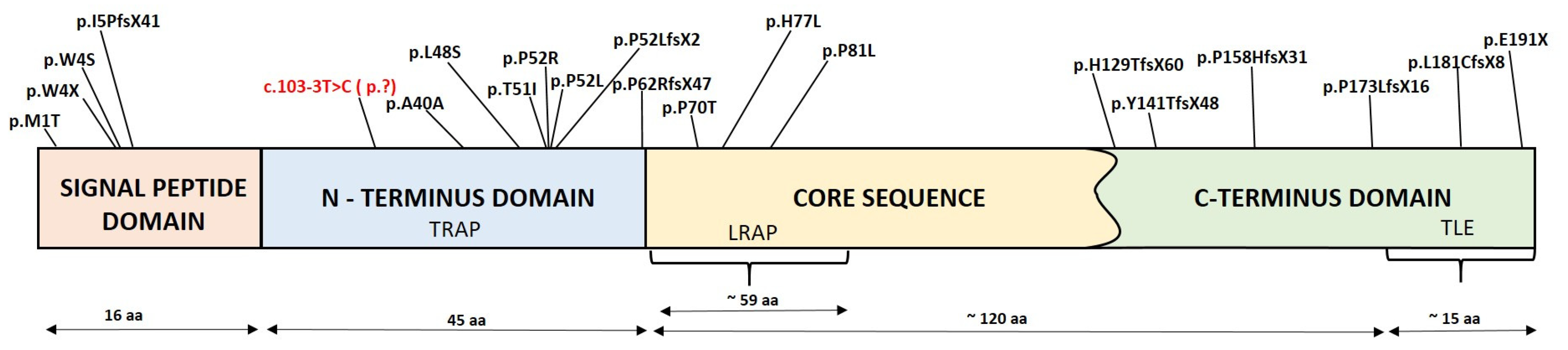
| c.2T>C | p.M1T | Hypoplasia | [59] |
| c.11G>A | p.W4X | Hypoplasia | [60] |
| c.11G>C | p.W4S | Hypoplasia | [59] |
| c.13_22delATTTTATTTG | p.I5PfsX41 | Hypoplasia | [61] |
| c.103-3T>C | Hypoplasia/hypomineralisation | In this study | |
| c.120T>C | p.A40A | Hypoplasia/hypomineralisation | [62] |
| c.143T>C | p.L48S | Hypoplasia/hypomineralisation | [15] |
| c.152C>T | p.T51I | Hypoplasia/hypomineralisation | [63] |
| c.155C>G | p.P52R | Hypoplasia | [64] |
| c.155C>T | p.P52L | Hypoplasia | [16] |
| c.155delC | p.P52LfsX2 | Hypoplasia/hypomineralisation | [17,65,66] |
| c.185delC | p.P62RfsX47 | Hypoplasia/hypomineralisation | [67] |
| c.208C>A | p.P70T | Hypomaturation | [17,18,68,69,70] |
| c.230A>T | p.H77L | Hypomaturation | [1] |
| c.242C>T | p.P81L | Hypoplasia | [29] |
| c.385delC | p.H129TfsX60 | Hypoplasia | [71] |
| c.420delC | p.Y141TfsX48 | Hypoplasia | [72] |
| c.473delC | p.P158HfsX31 | Hypoplasia; ** | [63]; [17] ** |
| c.518delC | p.P173LfsX16 | Hypoplasia | [56] |
| c.541delC | p.L181CfsX8 | Hypoplasia | [1,73] |
| c.571G>T | p.E191X | Hypoplasia | [63] |
| c.X362843_X367565del | p.? | Hypomineralisation | [74] |

4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACP4 | gene encodes acid phosphatase 4 |
| ACMG | American College of Medical Genetics |
| AI | amelogenesis imperfecta |
| AMBN | gene encodes ameloblastin |
| AMELX | gene encodes amelogenin |
| AMTN | gene encodes amelotin |
| C4orf26 | gene encodes odontogenesis-associated phosphoprotein |
| COL17A1 | gene encodes collagen type XVII α 1 chain |
| DEJ | dentin–enamel junction |
| DLX3 | gene encodes distal-less homeobox 3 |
| DNA | deoxyribonucleic acid |
| ECM | extracellular matrix |
| ENAM | gene encodes enamelin |
| FAM20A | gene encodes golgi-associated secretory pathway pseudokinase |
| FAM83H | gene encodes family with sequence similarity 83 member H |
| GPR68 | gene encodes G protein-coupled receptor 68 |
| ITGB6 | gene encodes integrin subunit β 6 |
| KLK4 | gene encodes kallikrein-related peptidase 4 |
| LAMA3 | gene encodes laminin subunit α 3 |
| LAMB3 | gene encodes laminin subunit β 3 |
| LRAP | leucin-rich amelogenin protein |
| MMP20 | gene encodes matrix metallopeptidase 20 |
| mRNA | messenger ribonucleic acid |
| PCR | polymerase chain reaction |
| SEI | secondary electrons image |
| SEM | scanning electron microscopy |
| SLC24A4 | gene encodes solute carrier family 24 member 4 |
| SP6 | gene encodes Sp6 transcription factor |
| STIM1 | gene encodes stromal interaction molecule 1 |
| TLE | teleopeptide |
| TRAP | tyrosine-rich amelogenin protein |
| VUS | variant with uncertain significance |
| WDR72 | gene encodes WD repeat domain 72 |
| WES | whole exome sequencing |
References
- Hart, P.S.; Aldred, M.J.; Crawford, P.J.M.; Wright, N.J.; Wright, J.T. Amelogenesis imperfecta phenotype-genotype correlations with two amelogenin gene mutations. Arc. Oral. Biol. 2002, 47, 261–265. [Google Scholar] [CrossRef]
- Smith, C.E.L.; Poulter, J.A.; Antanaviciute, A.; Kirkham, J.; Brookes, S.J.; Inglehearn, C.F.; Mighell, A.J. Amelogenesis imperfecta; Genes, Proteins, and Pathways. Front. Physiol. 2017, 8, 435. [Google Scholar] [CrossRef] [PubMed]
- Gadhia, K.; McDonald, S.; Arkutu, N.; Malik, K. Amelogenesis imperfecta: An introduction. Br. Dent. J. 2012, 212, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Altug-Atac, A.T.; Erdem, D. Prevalence and distribution of dental anomalies in orthodontic patients. Am. J. Orthod. Dentofac. Orthoped. 2007, 131, 510–514. [Google Scholar] [CrossRef]
- Bäckman, B.; Angmar-Månsson, B. Mineral distribution in the enamel of teeth with amelogenesis imperfecta as determined by quantitative microradiography. Scan. J. Dent. Res. 1994, 102, 193–197. [Google Scholar] [CrossRef]
- Sedano, H.O. Congenital oral anomalies in Argentinian children. Community Dent. Oral. Epidemiol. 1975, 3, 61–63. [Google Scholar] [CrossRef]
- Chosack, K.; Eidelman, E.; Wisotski, I.; Cohen, T. Amelogenesis imperfecta among Israeli Jews and the desciption of a new type of local hypoplastic autosomal recessive amelogenesis imperfecta. Oral Surg. Oral Med. Oral Pathol. 1979, 47, 148–156. [Google Scholar] [CrossRef]
- Witkop, C.J.; Sauk, J.J. Heritable defects of enamel. In Oral Facial Genetics; Stewart, R., Prescott, G.H., Eds.; CV Mosby Company: Maryland Heights, MO, USA, 1976; pp. 151–226. [Google Scholar]
- Goncalves-Filho, A.J.; Moda, L.B.; Oliveira, R.P.; Ribeiro, A.L.; Pinheiro, J.J.; Alver-Junior, S.R. Prevalence of dental anomalies on panoramic radiographs in a population of the state of Pará, Brazil. Indian J. Dent. Res. 2014, 25, 648–652. [Google Scholar] [CrossRef]
- Gupta, S.K.; Saxena, P.; Jain, S.; Jain, D. Prevalence and distribution of selected developmental dental anomalies in an Indian population. J. Oral Sci. 2011, 53, 231–238. [Google Scholar] [CrossRef]
- Herrera-Atoche, J.R.; Agüayo-de-Pau, M.D.; Escoffié-Ramírez, M.; Aguilar-Ayala, F.J.; Carrillo-Ávila, B.A.; Rejón-Peraza, M.E. Impacted Maxillary Canine Prevalence and Its Association with Other Dental Anomalies in a Mexican Population. Int. J. Dent. 2017, 2017, 7326061. [Google Scholar] [CrossRef]
- Bilge, N.H.; Yeşiltepe, S.; Ağırman, K.T.; Çağlayan, F.; Bilge, O.M. Investigation of prevalence of dental anomalies by using digital panoramic radiographs. Folia Morphol. 2018, 77, 323–328. [Google Scholar] [CrossRef]
- Hu, J.C.C.; Yamakoshi, Y.; Yamakoshi, F.; Krebsbach, P.H.; Simmer, J.P. Proteomics and genetics of dental enamel. Cells Tissue Organs 2005, 181, 219–231. [Google Scholar] [CrossRef]
- Kim, Y.J.; Lee, Y.; Zhang, H.; Song, J.S.; Hu, J.C.C.; Simmer, J.P.; Kim, J.W. Amelogenesis imperfecta among Israeli Jews and the desciption of a new type of local hypoplastic autosomal recessive amelogenesis imperfecta. Genes 2021, 12, 346. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kang, J.; Seymen, F.; Koruyucu, M.; Zhang, H.; Kasimoglu, Y.; Bayram, M.; Tuna-Ince, E.B.; Bayrak, S.; Tuloglu, N.; et al. Alteration of exon definition causes amelogenesis imperfecta. J. Dent. Res. 2020, 99, 410–418. [Google Scholar] [CrossRef]
- Prasad, M.K.; Geoffroy, V.; Vicaire, S.; Jost, B.; Dumas, M.; Le Gras, S.; Switala, M.; Gasse, B.; Laugel-Haushalter, V.; Paschaki, M.; et al. A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J. Med. Genet. 2016, 53, 98–110. [Google Scholar] [CrossRef]
- Wright, J.T.; Torain, M.; Long, K.; Seow, K.; Crawford, P.; Aldred, M.J.; Hart, P.S.; Hart, T.C. Amelogenesis imperfecta: Genotype phenotype studies in 71 families. Cells Tissue Organs 2011, 194, 279–283. [Google Scholar] [CrossRef]
- Chan, H.C.; Estrella, N.M.R.P.; Milkovich, R.N.; Kim, J.W.; Simmer, J.P.; Hu, J.C.C. Target gene analyses of 39 amelogenesis imperfecta kindreds. Eur. J. Oral Sci. 2011, 119 (Suppl. S1), 311–323. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, Y.; Yang, Y.; Qin, M. Novel ENAM and LAMB3 mutations in Chinese families with hypoplastic amelogenesis imperfecta. PLoS ONE 2015, 10, e0116514. [Google Scholar] [CrossRef]
- Poulter, J.A.; Murillo, G.; Brookes, S.J.; Smith, C.E.L.; Parry, D.A.; Silva, S.; Kirkham, J.; Inglehearn, C.F.; Mighell, A.J. Deletion of ameloblastin exon 6 is associated with amelogenesis imperfecta. Hum. Mol. Genet. 2014, 23, 5317–5324. [Google Scholar] [CrossRef]
- Prasad, M.K.; Laouina, S.; El Alloussi, M.; Dollfus, H.; Bloch-Zupan, A. Amelogenesis Imperfecta: 1 Family, 2 Phenotypes, and 2 Mutated Genes. J. Dent. Res. 2016, 95, 1457–1463. [Google Scholar] [CrossRef]
- Cho, S.H.; Seymen, F.; Lee, K.E.; Lee, S.K.; Kweon, Y.S.; Kim, K.J.; Jung, S.E.; Song, S.J.; Yildirim, M.; Bayram, M.; et al. Novel FAM20A Mutation in Hypoplastic Amelogenesis Imperfecta. Hum. Mutat. 2012, 33, 91–94. [Google Scholar] [CrossRef]
- Wang, S.K.; Choi, M.; Richardson, A.S.; Reid, B.M.; Lin, B.P.; Wang, S.J.; Kim, J.W.; Simmer, J.P.; Hu, J.C.C. ITGB6 loss-of-function mutations cause autosomal recessive amelogenesis imperfecta. Hum. Mol. Genet. 2014, 23, 2157–2163. [Google Scholar] [CrossRef]
- Seymen, F.; Park, J.C.; Lee, K.E.; Lee, H.K.; Lee, D.S.; Koruyucu, M.; Gencay, K.; Bayram, M.; Tuna, E.B.; Lee, Z.H.; et al. Novel MMP20 and KLK4 mutations in amelogenesis imperfecta. J. Dent. Res. 2015, 94, 1063–1069. [Google Scholar] [CrossRef]
- Hentschel, J.; Tatun, D.; Parkhomchuk, D.; Kurth, I.; Schimmel, B.; Heinrich-Weltzien, R.; Bertzbach, S.; Peters, H.; Beetz, C. Identification of the first multi-exonic WDR72 deletion in isolated amelogenesis imperfecta, and generation of a WDR72-specific copy number screening tool. Gene 2016, 590, 1–4. [Google Scholar] [CrossRef]
- Herzog, C.R.; Reid, B.M.; Seymen, F.; Koruyucu, M.; Tuna, E.B.; Simmer, J.P.; Hu, J.C.C. Hypomaturation amelogenesis imperfecta caused by a novel SLC24A4 mutation. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2015, 119, e77–e81. [Google Scholar] [CrossRef]
- Parry, D.A.; Brookes, S.J.; Logan, C.V.; Poulter, J.A.; El-Sayed, W.; Al-Bahlani, S.; Al Harasi, S.; Sayed, J.; Rif, E.M.; Shore, R.C.; et al. Mutations in C4orf26, encoding a peptide with in vitro hydroxyapatite crystal nucleation and growth activity, cause amelogenesis imperfecta. Am. J. Hum. Genet. 2012, 91, 565–571. [Google Scholar] [CrossRef]
- Parry, D.A.; Smith, C.E.L.; El-Sayed, W.; Poulter, J.A.; Shore, R.C.; Logan, C.V.; Mogi, C.; Sato, K.; Okajima, F.; Harada, A.; et al. Mutations in the pH-Sensing G-protein-coupled receptor GPR68 cause Amelogenesis Imperfecta. Am. J. Hum. Genet. 2016, 99, 984–990. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, Y.J.; Kang, J.; Shin, T.J.; Hyun, H.K.; Lee, S.H.; Lee, Z.H.; Kim, J.W. A novel AMELX mutation causes hypoplastic amelogenesis imperfecta. Arch. Oral Biol. 2017, 76, 61–65. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Richards, S.; Nazneen, A.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human splicing finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Marín, A. Characterization and prediction of alternative splice sites. Gene 2006, 366, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Hebsgaard, S.M.; Korning, P.G.; Tolstrup, N.; Engelbrecht, J.; Rouze, P.; Brunak, S. Splice site prediction in Arabidopsis thaliana DNA by combining local and global sequence information. Nucleic Acids Res. 1996, 24, 3439–3452. [Google Scholar] [CrossRef] [PubMed]
- Raponi, M.; Kralovicova, J.; Copson, E.; Divina, P.; Eccles, D.; Johnson, P.; Baralle, D.; Vorechovsky, I. Prediction of single-nucleotide substitutions that result in exon skipping: Identification of a splicing silencer in BRCA1 exon 6. Hum. Mutat. 2011, 32, 436–444. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, w452–w457. [Google Scholar] [CrossRef]
- Wright, J.T.; Frazier-Bowers, S.; Simmons, D.; Alexander, K.; Crawford, P.; Han, S.T.; Hart, P.S.; Hart, T.C. Phenotypic variation in the FAM83H-associated amelogenesis imperfecta. J. Dent. Res. 2009, 88, 356–360. [Google Scholar] [CrossRef]
- Lee, S.K.; Hu, J.C.C.; Bartlett, J.D.; Lee, K.E.; Lin, B.P.J.; Simmer, J.P.; Kim, J.W. Mutational spectrum of FAM83H: The C-terminal portion is required for tooth enamel calcification. Hum. Mutat. 2008, 29, E95–E99. [Google Scholar] [CrossRef]
- Haubek, D.; Gjørup, L.; Jensen, L.G.; Juncker, I.; Nyegaard, M.; Børglum, A.D.; Poulsen, S.; Hertz, J.M. Limited phenotypic variation of hypocalcified amelogenesis imperfecta in a Danish five-generation family with a novel FAM83H nonsense mutation. Int. J. Paediatr. Dent. 2011, 21, 407–412. [Google Scholar] [CrossRef]
- Song, Y.L.; Wang, C.N.; Zhang, C.Z.; Yang, K.; Bian, Z. Molecular characterization of amelogenesis imperfecta in Chinese patients. Cells Tissues Organs 2012, 196, 271–279. [Google Scholar] [CrossRef]
- Xin, W.; Wenjun, W.; Man, Q.; Yuming, Z. Novel FAM83H mutations in patients with amelogenesis imperfecta. Sci. Rep. 2017, 7, 6075. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, S.K.; Lee, Z.H.; Park, J.C.; Lee, K.E.; Lee, M.H.; Park, J.T.; Seo, B.M.; Hu, J.C.C.; Simmer, J.P. FAM83H mutation in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am. J. Hum. Genet. 2008, 82, 489–494. [Google Scholar] [CrossRef]
- Zhang, C.; Song, Y.; Bian, Z. Ultrastructural analysis of the teeth affected by amelogenesis imperfecta resulting from FAM83H mutation and review of the literature. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2015, 119, e69–e76. [Google Scholar] [CrossRef]
- Pourhashemi, S.J.; Motlagh, M.G.; Meighani, G.; Takaloo, A.E.; Mansouri, M.; Mohandes, F.; Mirzaii, M.; Khoshzaban, A.; Moshtaghi, F.; Abedkhodjasteh, H.; et al. Missense Mutation in Fam83H gene in Iranian Patients with Amelogenesis imperfecta. Iran. J. Public Health 2014, 43, 1680–1687. [Google Scholar]
- Ding, Y.; Estrella, M.R.P.; Hu, Y.Y.; Chan, H.L.; Zhang, H.D.; Kim, J.W.; Simmer, J.P.; Hu, J.C.C. FAM83H is associated with intracellular vesicles and ADHCAI. J. Dent. Res. 2009, 88, 991–996. [Google Scholar] [CrossRef]
- Hart, P.S.; Becerik, S.; Cogulu, D.; Emingil, G.; Ozdemir-Ozenen, D.; Han, S.T.; Sulima, P.P.; Firatli, E.; Hart, T.C. Novel FAM83H mutation in Turkish families with autosomal dominant hypocalcified amelogenesis imperfecta. Clin. Genet. 2009, 75, 401–404. [Google Scholar] [CrossRef]
- Yu, S.; Quan, J.; Wang, X.; Sun, X.; Zhang, X.; Liu, Y.; Zhang, C.; Zheng, S. A novel FAM83H mutation in one Chinese family with autosomal-dominant hypocalcification amelogenesis imperfecta. Mutagenesis 2018, 33, 333–340. [Google Scholar] [CrossRef]
- Nowwarote, N.; Theerapanon, T.; Osathanon, T.; Pavasant, P.; Porntaveetus, T.; Shotelersuk, V. Amelogenesis imperfecta: A novel FAM83H mutation and characteristics of periodontal ligament cells. Oral Dis. 2018, 24, 1522–1531. [Google Scholar] [CrossRef]
- Hyun, H.K.; Lee, S.K.; Lee, K.E.; Kang, H.Y.; Kim, E.J.; Choung, P.H.; Kim, J.W. Identification of a novel FAM83H mutation and microhardness of an affected molar in autosomal dominant hypocalcified amelogenesis imperfecta. Int. Endod. J. 2009, 42, 1039–1043. [Google Scholar] [CrossRef]
- Wang, S.K.; Hu, Y.; Yang, J.; Smith, C.E.L.; Richardson, A.S.; Yamakoshi, Y.; Lee, Y.L.; Seymen, F.; Kuruyucu, M.; Gencay, K.; et al. Fam83h null mice support a neomorphic mechanism for human ADHCAI. Mol. Genet. Genom. Med. 2015, 4, 46–67. [Google Scholar] [CrossRef]
- El-Sayed, W.; Shore, R.C.; Parry, D.A.; Inglehearn, C.F.; Mighell, A.J. Hypomaturation amelogenesis imperfecta due to WDR72 mutations: A novel mutation and ultrastructural analyses of deciduous teeth. Cells Tissues Organs 2010, 194, 60–66. [Google Scholar] [CrossRef]
- Wang, S.K.; Hu, Y.; Simmer, J.P.; Seymen, F.; Estrella, N.M.R.P.; Pal, S.; Reid, B.M.; Yildirim, M.; Bayram, M.; Bartlett, J.D.; et al. Novel KLK4 and MMP20 Mutations Discovered by Whole-exome Sequencing. J. Dent. Res. 2013, 93, 266–271. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Intachai, W.; Auychai, P. All enamel is not created equal: Supports from a novel FAM83H mutation. Am. J. Med. Genet. A 2016, 170A, 273–276. [Google Scholar] [CrossRef]
- Urzúa, B.; Martínez, C.; Ortega-Pinto, A.; Adorno, D.; Morales-Bozo, I.; Riadi, G.; Jara, L.; Plaza, A.; Lefimil, C.; Lozano, C.; et al. Novel missense mutation of the FAM83H gene causes retention of amelogenin and a mild clinical phenotype of hypocalcified enamel. Arch. Oral Biol. 2015, 60, 1356–1367. [Google Scholar] [CrossRef]
- Lee, S.K.; Lee, K.E.; Jeong, T.S.; Hwang, Y.H.; Kim, S.; Hu, J.C.C.; Simmer, J.P.; Kim, J.W. FAM83H mutations cause ADHCAI and alter intracellular protein localization. J. Dent. Res. 2011, 90, 377–381. [Google Scholar] [CrossRef]
- Hobson, G.M.; Gibson, C.W.; Aragon, M.; Yuan, Z.; Davis-Williams, A.; Banser, L.; Kirkham, J.; Brook, A.H. A large X-chromosomal deletion is associated with microphthalmia with Linear Skin Defects (MLS) and Amelogenesis Imperfecta (XAI). Am. J. Med. Genet. 2009, 149A, 1698–1705. [Google Scholar] [CrossRef]
- Hu, J.C.; Chan, H.-C.; Simmer, S.G.; Seymen, F.; Richardson, A.S.; Hu, Y.; Milkovich, R.N.; Estrella, N.M.; Yildirim, M.; Bayram, M.; et al. Amelogenesis imperfecta in two families with defined AMELX deletions in ARHGAP6. PLoS ONE 2011, 7, e52052. [Google Scholar] [CrossRef]
- Kim, J.W.; Simmer, J.P.; Hu, Y.Y.; Lin, B.P.L.; Boyd, C.; Wright, J.T.; Yamada, C.J.M.; Rayes, S.K.; Feigal, R.J.; Hu, J.C.C. Amelogenin p.M1t and p.W4s mutations underlying hypoplastic x-linked amelogenesis imperfecta. J. Dent. Res. 2004, 83, 378–383. [Google Scholar] [CrossRef]
- Sekiguchi, H.; Kiyoshi, M.; Yakushiji, M. DNA diagnosis of X-linked amelogenesis imperfecta using PCR detection method of the human amelogenin gene. Dent. Jpn. 2001, 37, 109–112. [Google Scholar]
- Lagerström-Fermér, M.; Nilsson, M.; Bäckman, B.; Salido, E.; Shapiro, L.; Pettersson, U.; Landegren, U. Amelogenin signal peptide mutation: Correlation between mutations in the amelogenin gene (AMGX) and manifestations of X-linked amelogenesis imperfecta. Genomics 1995, 26, 159–162. [Google Scholar] [CrossRef]
- Cho, E.S.; Kim, K.J.; Lee, K.E.; Lee, E.J.; Yun, Y.; Lee, M.J.; Shin, T.J.; Hyun, H.K.; Kim, Y.J.; Lee, S.H.; et al. Alteration of conserved alternative splicing in AMELX causes enamel defects. J. Dent. Res. 2014, 93, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Lench, N.J.; Winter, G.B. Characterisation of molecular defects in X-lined amelogenesis imperfecta (AIH1). Hum. Mutat. 1995, 5, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Kida, M.; Sakiyama, Y.; Matsuda, A.; Takabayashi, S.; Ochi, H.; Sekiguchi, H.; Minamitake, S.; Ariga, T. A novel missense mutation (p.P52r) in amelogenin gene causing X-linked amelogenesis imperfecta. J. Dent. Res. 2007, 86, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Aldred, M.J.; Crawford, P.J.; Roberts, E.; Thomas, N.S. Identification of a nonsense mutation in the amelogenin gene (AMELX) in a family with X-linked amelogenesis imperfecta (AIH1). Hum. Genet. 1992, 90, 413–416. [Google Scholar] [CrossRef]
- Lench, N.J.; Brook, A.H.; Winter, G.B. SSCP detection of a nonsense mutation in exon 5 of the amelogenin gene (AMGX) causing X-linked amelogenesis imperfecta (AIH1). Hum. Mol. Genet. 1994, 3, 827–828. [Google Scholar] [CrossRef]
- Duan, X.; Yang, S.; Zhang, H.; Wu, J.; Zhang, Y.; Ji, D.; Tie, L.; Boerkoel, C.F. A Novel AMELX mutation, its phenotypic features, and skewed X inactivation. J. Dent. Res. 2019, 98, 870–878. [Google Scholar] [CrossRef]
- Collier, P.M.; Sauk, J.J.; Rosenbloom, S.J.; Yuan, Z.A.; Gibson, C.W. An amelogenin gene defect associated with human X-linked amelogenesis imperfecta. Arch. Oral Biol. 1997, 42, 235–242. [Google Scholar] [CrossRef]
- Hart, S.; Hart, T.; Gibson, C.; Wright, J.T. Mutational analysis of X-linked amelogenesis imperfecta in multiple families. Arch. Oral Biol. 2000, 45, 79–86. [Google Scholar] [CrossRef]
- Ravassipour, D.B.; Hart, P.S.; Hart, T.C.; Ritter, A.V.; Yamauchi, M.; Gibson, C.; Wright, J.T. Unique enamel phenotype associated with amelogenin gene (AMELX) codon 41 point mutation. J. Dent. Res. 2000, 79, 1476–1481. [Google Scholar] [CrossRef]
- Sekiguchi, H.; Alaluusua, S.; Minaguchi, K.; Yakushiji, M. A new mutation in the amelogenin gene causes X-linked amelogenesis imperfecta. J. Dent. Res. 2001, 80, 617. [Google Scholar]
- Greene, S.R.; Yuan, Z.A.; Wright, J.T.; Amjad, H.; Abrams, W.R.; Buchanan, J.A.; Trachtenberg, D.I.; Gibson, C.W. A new frameshift mutation encoding a truncated amelogenin leads to X-linked amelogenesis imperfecta. Arch. Oral Biol. 2002, 47, 211–217. [Google Scholar] [CrossRef]
- Kindelan, S.A.; Brook, A.H.; Gangemi, L.; Lench, N.; Wong, F.S.; Fearne, J.; Jackson, Z.; Foster, G.; Stringer, B.M. Detection of a novel mutation in X-Iinked amelogenesis imperfecta. J. Dent. Res. 2000, 79, 1978–1982. [Google Scholar] [CrossRef]
- Lagerström, M.; Dahl, N.; Nakahori, Y.; Nakagome, Y.; Bäckman, B.; Landregren, U.; Pattersson, U. A deletion in the amelogenin gene (AMG) causes X-linked amelogenesis imperfecta (AIH1). Genomics 1991, 10, 971–975. [Google Scholar] [CrossRef]
- Salido, E.C.; Yen, P.H.; Koprivnikar, K.; Yu, L.C.; Shapiro, L.J. The human enamel protein gene amelogenin is expressed from both the X and the Y chromosomes. Am. J. Hum. Genet. 1992, 50, 303–316. [Google Scholar]
- Fincham, A.G.; Simmer, J.P. Amelogenin proteins of developing dental enamel. Ciba Found. Symp. 1997, 205, 118–130; discussion 130–134. [Google Scholar] [CrossRef]
- Migeon, B.R. Choosing the active X: The human version of X inactivation. Trends Genet. 2017, 33, 899–909. [Google Scholar] [CrossRef]
- Snead, M.L.; Zhu, D.H.; Lei, Y.; Luo, W.; Bringas, P.O., Jr.; Sucov, H.M. A simplified genetic design for mammalian enamel. Biomaterials 2011, 32, 3151–3157. [Google Scholar] [CrossRef][Green Version]
- Baralle, D.; Baralle, M. Splicing in action: Assessing disease causing sequence changes. J. Med. Genet. 2005, 42, 737–748. [Google Scholar] [CrossRef]
- Fincham, A.G.; Moradian-Oldak, J.; Simmer, J.P.; Sarte, P.; Lau, E.C.; Diekwisch, T.; Slavkin, H.C. Self-assembly of a recombinant amelogenin protein generates supramolecular structures. J. Struct. Biol. 1994, 112, 103–109. [Google Scholar] [CrossRef]
- Stahl, J.; Nakano, Y.; Horst, J.; Zhu, L.; Le, M.; Zhang, Y.; Liu, H.; Li, W.; Den Besten, P.K. Exon4 Amelogenin Transcripts in Enamel Biomineralization. J. Dent. Res. 2015, 94, 836–842. [Google Scholar] [CrossRef]



| Number | Family (F) | Gender | Predicted Mode of Inheritance | Phenotype * | |
|---|---|---|---|---|---|
| Hypomineralisation Type | Hypoplastic Type | ||||
| 1 | F1 | Female | ** | + | |
| 2 | F2 | Female | ** | + | |
| 3 | F3 | Male | AD | + | |
| 4 | Male | + | |||
| 5 | F4 | Male | AD | + | |
| 6 | Female | + | |||
| 7 | Female | + | |||
| 8 | Male | + | |||
| 9 | F5 | Female | ** | + | |
| 10 | F6 | Female | ** | + | |
| 11 | F7 | Female | AD | + | |
| 12 | Male | + | |||
| 13 | F8 | Male | AD | + | |
| 14 | Female | + | |||
| 15 | F9 | Female | AD | + | |
| 16 | F10 | Female | ** | + | |
| 17 | F11 | Female | AR | + | + |
| 18 | F12 | Male | AR | + | |
| 19 | F13 | Male | AD | + | + |
| 20 | Female | + | + | ||
| 21 | F14 | Female | AR | + | |
| 22 | F15 | Male | AD | + | + |
| 23 | F16 | Male | AD | + | + |
| 24 | Male | + | + | ||
| 25 | F17 | Female | AD | + | + |
| 26 | Male | + | + | ||
| 27 | F18 | Female | AR | + | |
| 28 | F19 | Female | ** | + | |
| 29 | F20 | Male | AD | + | |
| 30 | Male | + | |||
| 31 | F21 | Male | AD | + | |
| 32 | F22 | Female | ** | + | |
| 33 | F23 | Female | ** | + | |
| 34 | F24 | Male | AD | + | |
| 35 | Male | + | |||
| 36 | F25 | Female | X-linked | + | + |
| 37 | Female | + | + | ||
| 38 | F26 | Male | ** | + | |
| 39 | F27 | Female | X-linked | + | + |
| 40 | F28 | Female | ** | + | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leban, T.; Trebušak Podkrajšek, K.; Kovač, J.; Fidler, A.; Pavlič, A. An Intron c.103-3T>C Variant of the AMELX Gene Causes Combined Hypomineralized and Hypoplastic Type of Amelogenesis Imperfecta: Case Series and Review of the Literature. Genes 2022, 13, 1272. https://doi.org/10.3390/genes13071272
Leban T, Trebušak Podkrajšek K, Kovač J, Fidler A, Pavlič A. An Intron c.103-3T>C Variant of the AMELX Gene Causes Combined Hypomineralized and Hypoplastic Type of Amelogenesis Imperfecta: Case Series and Review of the Literature. Genes. 2022; 13(7):1272. https://doi.org/10.3390/genes13071272
Chicago/Turabian StyleLeban, Tina, Katarina Trebušak Podkrajšek, Jernej Kovač, Aleš Fidler, and Alenka Pavlič. 2022. "An Intron c.103-3T>C Variant of the AMELX Gene Causes Combined Hypomineralized and Hypoplastic Type of Amelogenesis Imperfecta: Case Series and Review of the Literature" Genes 13, no. 7: 1272. https://doi.org/10.3390/genes13071272
APA StyleLeban, T., Trebušak Podkrajšek, K., Kovač, J., Fidler, A., & Pavlič, A. (2022). An Intron c.103-3T>C Variant of the AMELX Gene Causes Combined Hypomineralized and Hypoplastic Type of Amelogenesis Imperfecta: Case Series and Review of the Literature. Genes, 13(7), 1272. https://doi.org/10.3390/genes13071272