
In-Silico Investigation of Effects of Single-Nucleotide Polymorphisms in PCOS-Associated CYP11A1 Gene on Mutated Proteins

 , ,
, ,  ,
,  , ,
, ,  and
and
Abstract
1. Introduction
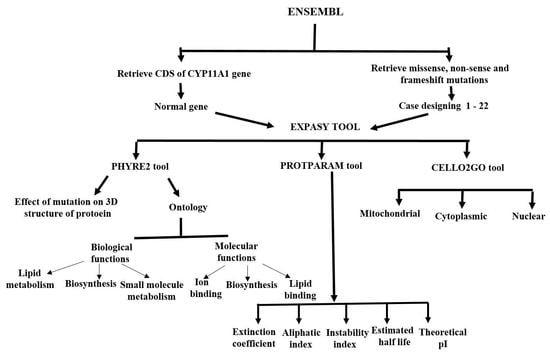
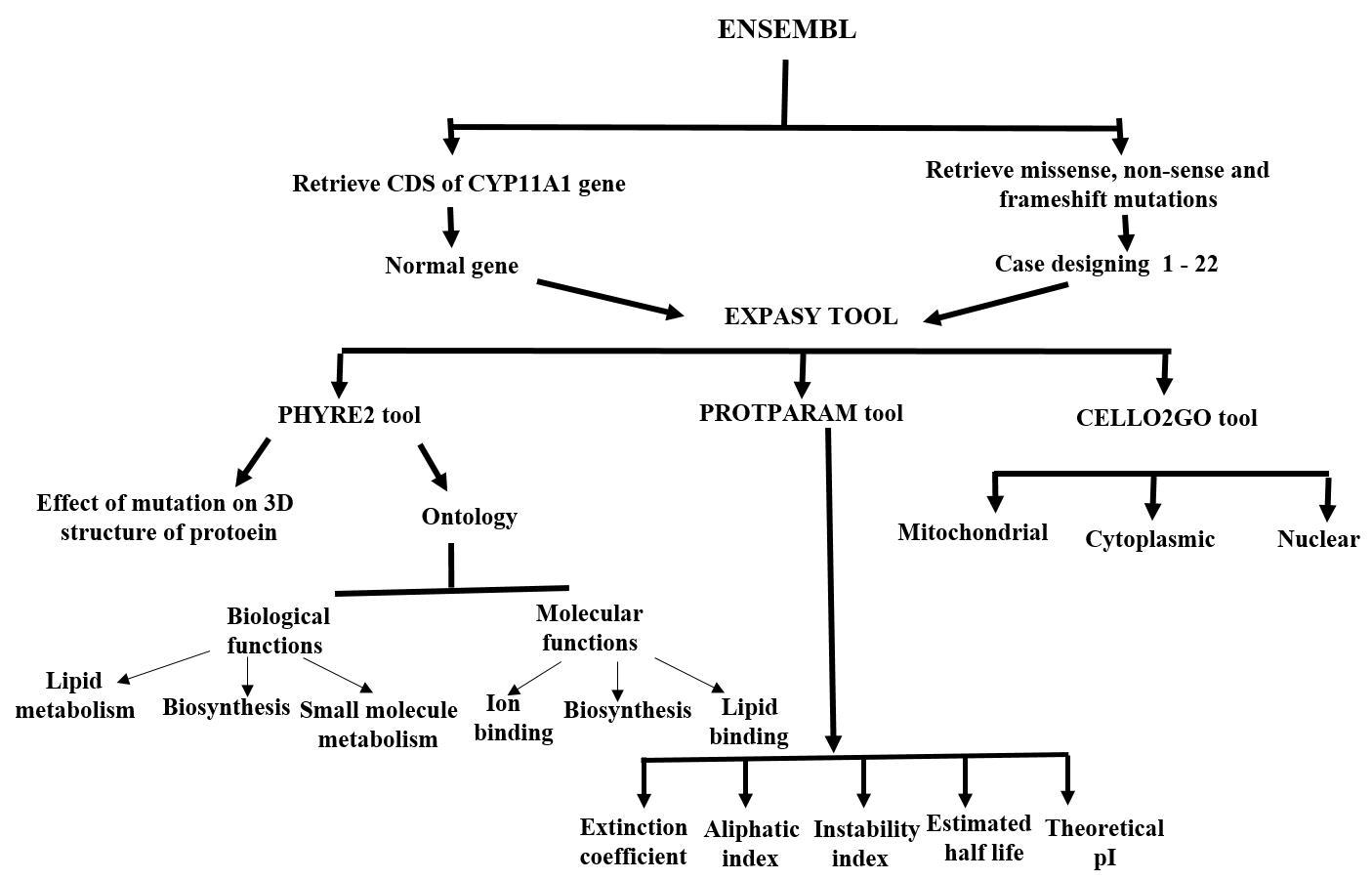
2. Materials and Methods
2.1. Retrieving CDS of CYP11A1
2.2. Retrieving SNPs and Construction of Mutated CDS
2.3. Translation of CDS into Protein Sequence
2.4. Effect of SNPs on Stability of Mutated Proteins
2.5. Sub-Cellular Localization and Ontology of Wild Type and Mutated Proteins
2.6. Physicochemical Features
2.7. 3-D and 2-D Structures and Trans-Membrane Topology
3. Results
3.1. Effect of SNPs on Mutated Protein Stability
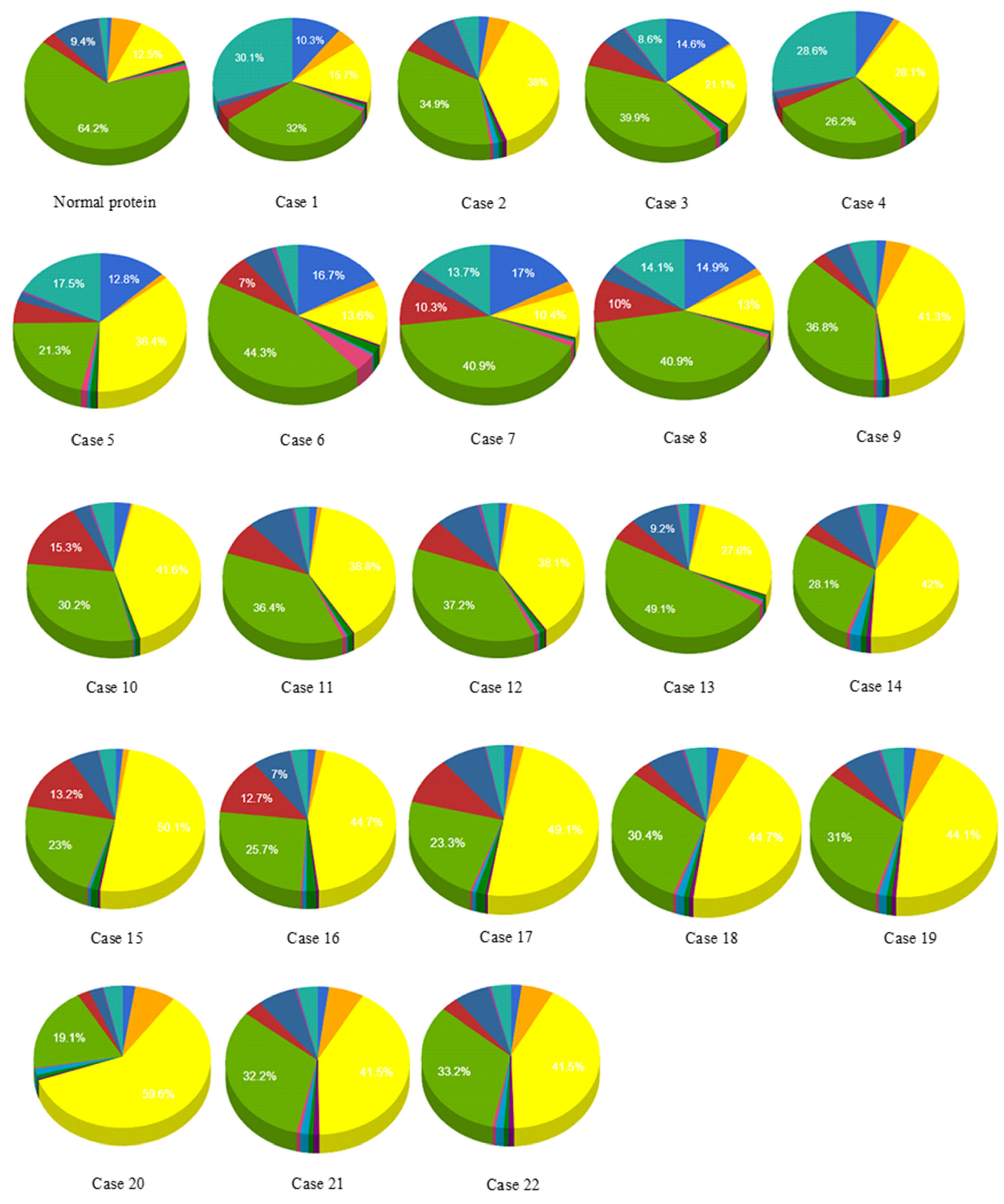
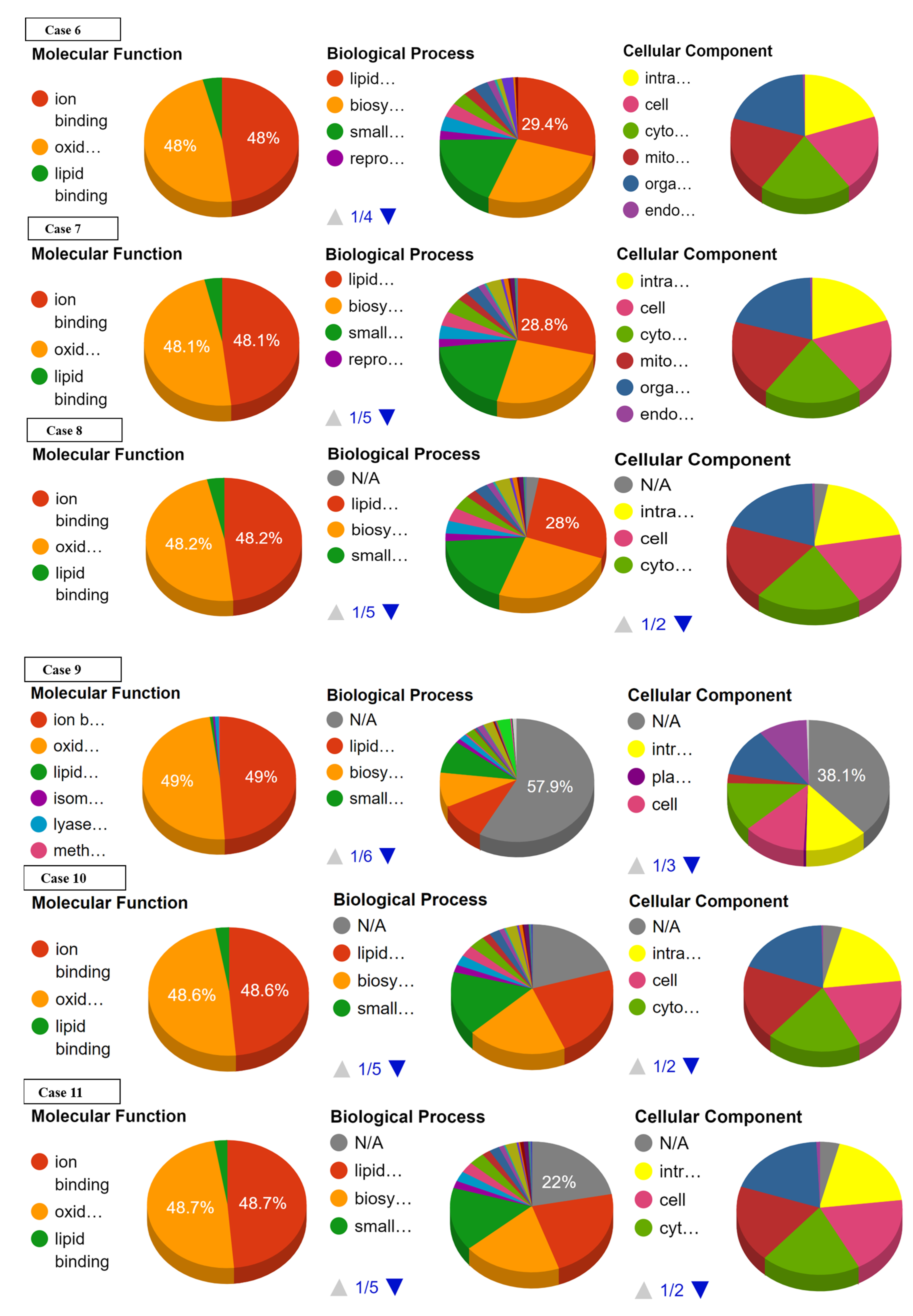
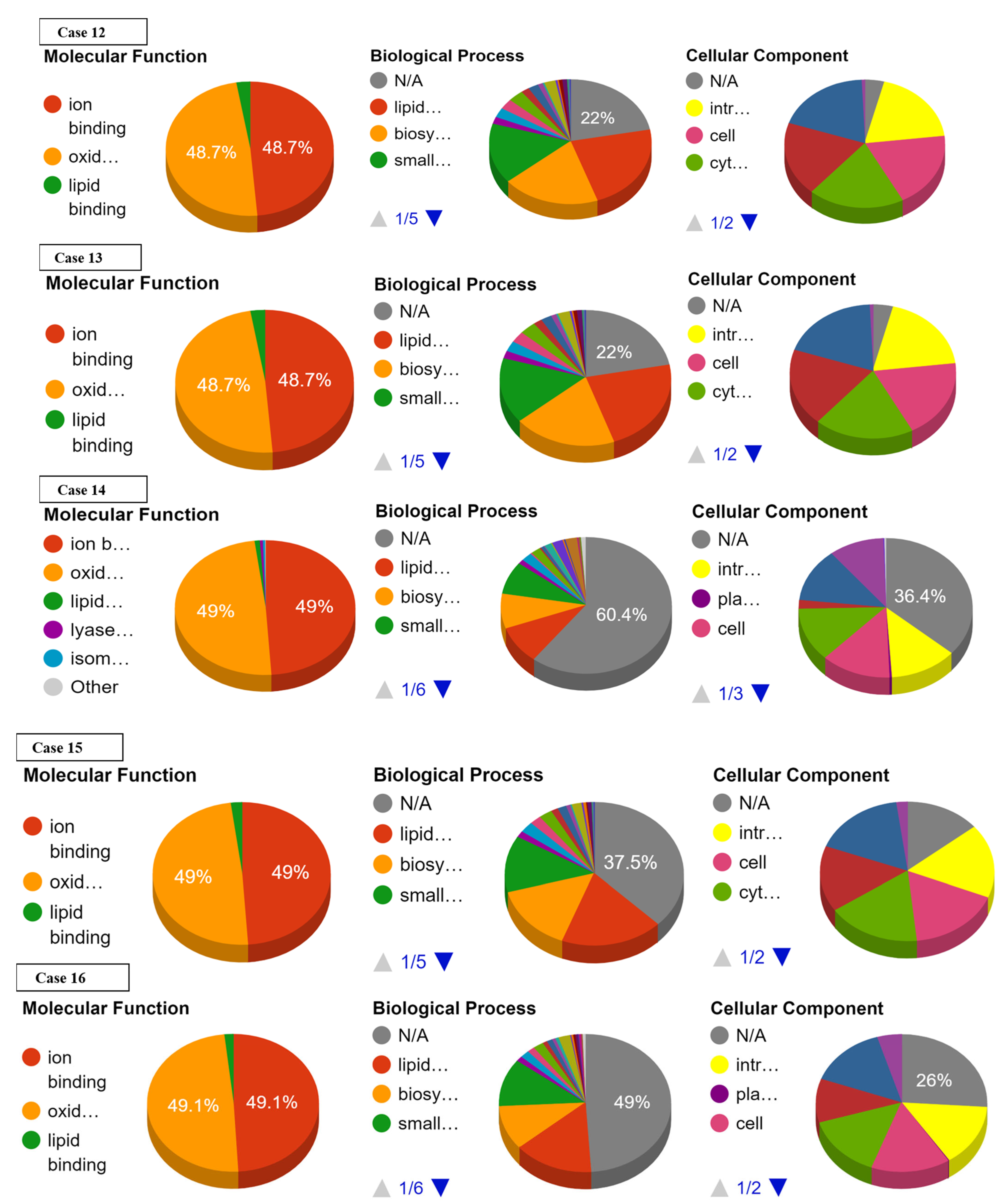
3.2. Effect of SNPs on Sub-Cellular Localization of Proteins
3.3. Effect of SNPs on Ontology of Mutated Proteins
3.4. Effect of SNPs on Physicochemical Parameters of Mutated Proteins
3.5. Effect of SNPs on 3D Structures of Mutated Proteins
3.6. Effect of SNPs on Secondary Structure of Mutated Proteins
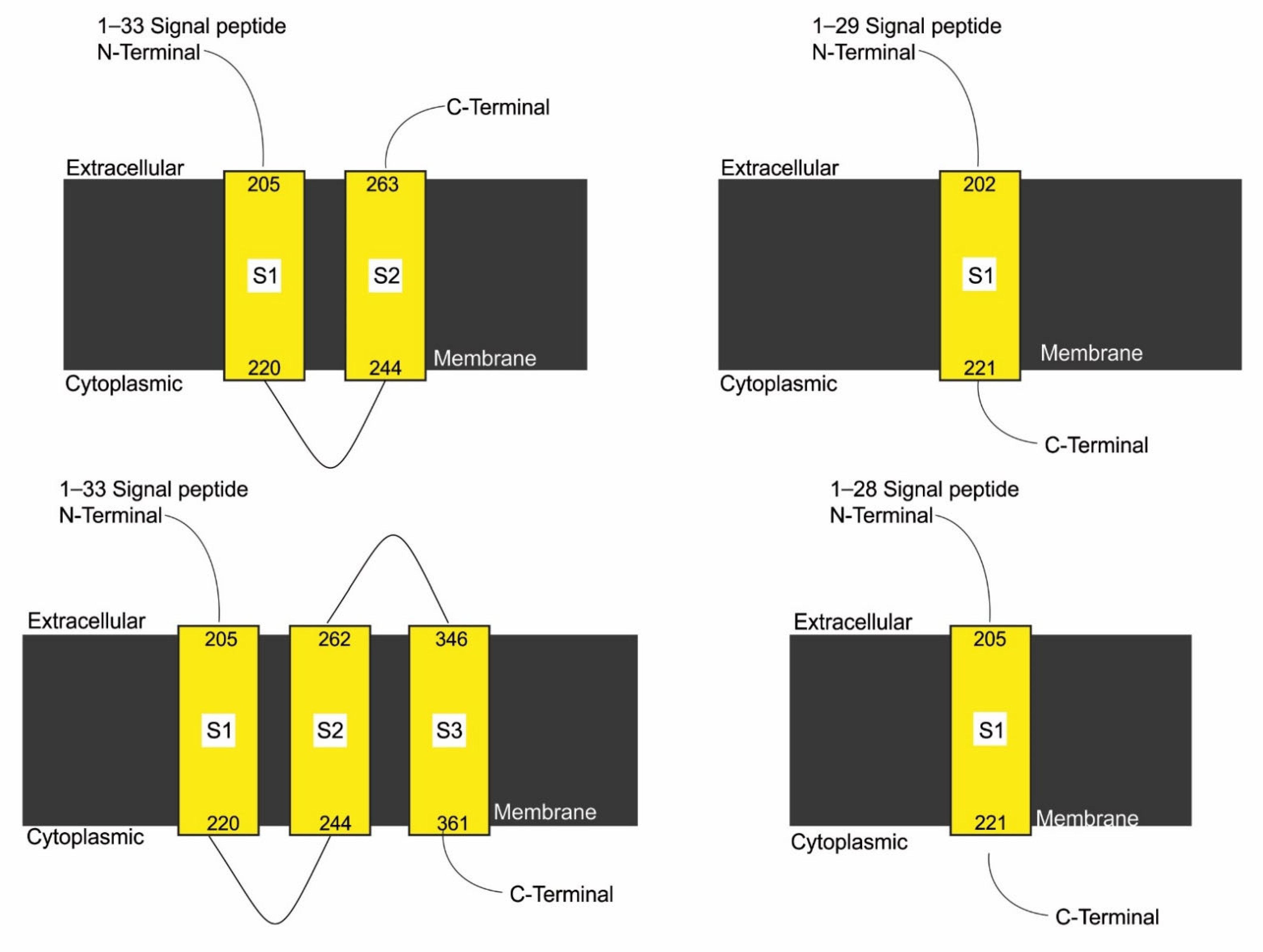
3.7. Effect of SNPs on Trans-Membrane Topology of Mutated Proteins
3.8. Nature of SNPs Effect
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Bulsara, J.; Patel, P.; Soni, A.; Acharya, S. A review: Brief insight into Polycystic Ovarian syndrome. Endocr. Metab. Sci. 2021, 3, 100085. [Google Scholar] [CrossRef]
- Balen, A.H.; Morley, L.C.; Misso, M.; Franks, S.; Legro, R.S.; Wijeyaratne, C.N.; Stener-Victorin, E.; Fauser, B.C.J.M.; Norman, R.J.; Teede, H. The management of anovulatory infertility in women with polycystic ovary syndrome: An analysis of the evidence to support the development of global WHO guidance. Hum. Reprod. Update 2016, 22, 687–708. [Google Scholar] [CrossRef] [PubMed]
- Deswal, R.; Narwal, V.; Dang, A.; Pundir, C.S. The prevalence of polycystic ovary syndrome: A brief systematic review. J. Hum. Reprod. Sci. 2020, 13, 261. [Google Scholar]
- Franks, S. Diagnosis of polycystic ovarian syndrome: In defense of the Rotterdam criteria. J. Clin. Endocrinol. Metab. 2006, 91, 786–789. [Google Scholar] [CrossRef] [PubMed]
- Azziz, R.; Carmina, E.; Dewailly, D.; Diamanti-Kandarakis, E.; Escobar-Morreale, H.F.; Futterweit, W.; Janssen, O.E.; Legro, R.S.; Norman, R.J.; Taylor, A.E. Criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: An androgen excess society guideline. J. Clin. Endocrinol. Metab. 2006, 91, 4237–4245. [Google Scholar] [CrossRef]
- Farhadi-Azar, M.; Behboudi-Gandevani, S.; Rahmati, M.; Mahboobifard, F.; Pouya, E.K.; Tehrani, F.R.; Azizi, F. The Prevalence of Polycystic Ovary Syndrome, Its Phenotypes and Cardio-Metabolic Features in a Community Sample of Iranian Population: Tehran Lipid and Glucose Study. Front. Endocrinol. 2022, 13, 825528. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, Q.; Hao, Y.; Jiao, M.; Wang, X.; Jiang, S.; Han, L. Measuring the global disease burden of polycystic ovary syndrome in 194 countries: Global Burden of Disease Study 2017. Hum. Reprod. 2021, 36, 1108–1119. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Kouli, C.R.; Bergiele, A.T.; Filandra, F.A.; Tsianateli, T.C.; Spina, G.G.; Zapanti, E.D.; Bartzis, M.I. A survey of the polycystic ovary syndrome in the Greek island of Lesbos: Hormonal and metabolic profile. J. Clin. Endocrinol. Metab. 1999, 84, 4006–4011. [Google Scholar] [CrossRef]
- Deeks, A.A.; Gibson-Helm, M.E.; Paul, E.; Teede, H.J. Is having polycystic ovary syndrome a predictor of poor psychological function including anxiety and depression? Hum. Reprod. 2011, 26, 1399–1407. [Google Scholar] [CrossRef]
- Louwers, Y.V.; Laven, J.S.E. Characteristics of polycystic ovary syndrome throughout life. Ther. Adv. Reprod. Health 2020, 14, 2633494120911038. [Google Scholar] [CrossRef]
- Liu, A.L.; Xie, H.J.; Xie, H.Y.; Liu, J.; Yin, J.; Hu, J.S.; Peng, C.Y. Association between fat mass and obesity associated (FTO) gene rs9939609 A/T polymorphism and polycystic ovary syndrome: A systematic review and meta-analysis. BMC Med. Genet. 2017, 18, 89. [Google Scholar] [CrossRef]
- Rojas, J.; Chávez, M.; Olivar, L.; Rojas, M.; Morillo, J.; Mejías, J.; Calvo, M.; Bermúdez, V. Polycystic ovary syndrome, insulin resistance, and obesity: Navigating the pathophysiologic labyrinth. Int. J. Reprod. Med. 2014, 2014, 719050. [Google Scholar] [CrossRef] [PubMed]
- Puttabyatappa, M.; Padmanabhan, V. Ovarian and extra-ovarian mediators in the development of polycystic ovary syndrome. J. Mol. Endocrinol. 2018, 61, R161–R184. [Google Scholar] [CrossRef]
- Ashraf, S.; Nabi, M.; Rashid, F.; Amin, S. Hyperandrogenism in polycystic ovarian syndrome and role of CYP gene variants: A review. Egypt. J. Med. Hum. Genet. 2019, 20, 25. [Google Scholar] [CrossRef]
- Walters, K.A.; Gilchrist, R.B.; Ledger, W.L.; Teede, H.J.; Handelsman, D.J.; Campbell, R.E. New perspectives on the pathogenesis of PCOS: Neuroendocrine origins. Trends Endocrinol. Metab. 2018, 29, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Broekmans, F.J.; Visser, J.A.; Laven, J.S.E.; Broer, S.L.; Themmen, A.P.N.; Fauser, B.C. Anti-Müllerian hormone and ovarian dysfunction. Trends Endocrinol. Metab. 2008, 19, 340–347. [Google Scholar] [CrossRef]
- Kumar, S. Is environmental exposure associated with reproductive health impairments? J. Turk. Ger. Gynecol. Assoc. 2008, 9, 60–69. [Google Scholar]
- Kumar, S. Tobacco and areca nut chewing—Reproductive impairments: An overview. Reprod. Toxicol. 2013, 36, 12–17. [Google Scholar] [CrossRef]
- Sedha, S.; Gautam, A.K.; Verma, Y.; Ahmad, R.; Kumar, S. Determination of in vivo estrogenic potential of Di-isobutyl phthalate (DIBP) and Di-isononyl phthalate (DINP) in rats. Environ. Sci. Pollut. Res. 2015, 22, 18197–18202. [Google Scholar] [CrossRef]
- Papalou, O.; Diamanti-Kandarakis, E. The role of stress in PCOS. Expert Rev. Endocrinol. Metab. 2017, 12, 87–95. [Google Scholar] [CrossRef]
- Zhang, B.; Zhou, W.; Shi, Y.; Zhang, J.; Cui, L.; Chen, Z.J. Lifestyle and environmental contributions to ovulatory dysfunction in women of polycystic ovary syndrome. BMC Endocr. Disord. 2020, 20, 19. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, A.; Ramirez, S.; Yong, E.L. Androgen receptor gene CAG trinucleotide repeats in anovulatory infertility and polycystic ovaries. J. Clin. Endocrinol. Metab. 2000, 85, 3484–3488. [Google Scholar] [CrossRef] [PubMed]
- Xita, N.; Tsatsoulis, A.; Chatzikyriakidou, A.; Georgiou, I. Association of the (TAAAA) n repeat polymorphism in the sex hormone-binding globulin (SHBG) gene with polycystic ovary syndrome and relation to SHBG serum levels. J. Clin. Endocrinol. Metab. 2003, 88, 5976–5980. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Haijian, F.A.N.; Yong, W.; Xiaoke, W.U. Genetic polymorphism of CYP11α (tttta) n allele in Chinese women. Chin. J. Public Health 2006, 22, 221–222. [Google Scholar]
- Escobar-Morreale, H.F.; San Millán, J.L.; Smith, R.R.; Sancho, J.; Witchel, S.F. The presence of the 21-hydroxylase deficiency carrier status in hirsute women: Phenotype-genotype correlations. Fertil. Steril. 1999, 72, 629–638. [Google Scholar] [CrossRef]
- Marszalek, B.; Laciński, M.; Babych, N.; Capla, E.; Biernacka-Lukanty, J.; Warenik-Szymankiewicz, A.; Trzeciak, W.H. Investigations on the genetic polymorphism in the region of CYP17 gene encoding 5′-UTR in patients with polycystic ovarian syndrome. Gynecol. Endocrinol. 2001, 15, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Petry, C.J.; Ong, K.K.; Michelmore, K.F.; Artigas, S.; Wingate, D.L.; Balen, A.H.; de Zegher, F.; Ibanez, L.; Dunger, D.B. Association of aromatase (CYP 19) gene variation with features of hyperandrogenism in two populations of young women. Hum. Reprod. 2005, 20, 1837–1843. [Google Scholar] [CrossRef]
- Urbanek, M.; Wu, X.; Vickery, K.R.; Kao, L.C.; Christenson, L.K.; Schneyer, A.; Legro, R.S.; Driscoll, D.A.; Strauss III, J.F.; Dunaif, A. Allelic variants of the follistatin gene in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2000, 85, 4455–4461. [Google Scholar] [CrossRef]
- Rajkhowa, M.; Taibot, J.A.; Jones, P.W.; Pettersson, K.; Haavisto, A.M.; Huhtaniemi, I.; Clayton, R.N. Prevalence of an immunological LH β-subunit variant in a UK population of healthy women and women with polycystic ovary syndrome. Clin. Endocrinol. 1995, 43, 297–303. [Google Scholar] [CrossRef]
- Esinler, I.; Aktas, D.; Otegen, U.; Alikasifoglu, M.; Yarali, H.; Tuncbileke, E. CYP1A1 gene polymorphism and polycystic ovary syndrome. Reprod. Biomed. Online 2008, 16, 356–360. [Google Scholar] [CrossRef]
- Settas, N.; Dracopoulou-Vabouli, M.; Dastamani, A.; Katsikis, I.; Chrousos, G.; Panidis, D.; Dacou-Voutetakis, C. CYP21A2 mutations in women with polycystic ovary syndrome (PCOS). Horm. Metab. Res. 2013, 45, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, M.O.; Xu, N.; Azziz, R. Association of CYP3A7* 1C and serum dehydroepiandrosterone sulfate levels in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 2909–2912. [Google Scholar] [CrossRef] [PubMed]
- San Millán, J.L.; Cortón, M.; Villuendas, G.; Sancho, J.; Peral, B.; Escobar-Morreale, H.F. Association of the polycystic ovary syndrome with genomic variants related to insulin resistance, type 2 diabetes mellitus, and obesity. J. Clin. Endocrinol. Metab. 2004, 89, 2640–2646. [Google Scholar] [CrossRef][Green Version]
- Michelmore, K.; Ong, K.; Mason, S.; Bennett, S.; Perry, L.; Vessey, M.; Balen, A.; Dunger, D.B. Clinical features in women with polycystic ovaries: Relationships to insulin sensitivity, insulin gene VNTR and birth weight. Clin. Endocrinol. 2001, 55, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Villuendas, G.; Tosi, F.; Sancho, J.; Moghetti, P.; San Millán, J.L. Association between the D19S884 marker at the insulin receptor gene locus and polycystic ovary syndrome. Fertil. Steril. 2003, 79, 219–220. [Google Scholar] [CrossRef]
- Escobar-Morreale, H.F.; Peral, B.; Villuendas, G.; Calvo, R.M.; Sancho, J.; San Millán, J.L. Common single nucleotide polymorphisms in intron 3 of the calpain-10 gene influence hirsutism. Fertil. Steril. 2002, 77, 581–587. [Google Scholar] [CrossRef]
- Mohlig, M.; Spranger, J.; Osterhoff, M.; Ristow, M.; Pfeiffer, A.F.H.; Schill, T.; Schlosser, H.W.; Brabant, G.; Schofl, C. The polycystic ovary syndrome per se is not associated with increased chronic inflammation. Eur. J. Endocrinol. 2004, 150, 525–532. [Google Scholar] [CrossRef]
- Unluturk, U.; Harmanci, A.; Kocaefe, C.; Yildiz, B.O. The genetic basis of the polycystic ovary syndrome: A literature review including discussion of PPAR-γ. PPAR Res. 2007, 2007, 049109. [Google Scholar] [CrossRef]
- Korhonen, S.; Romppanen, E.L.; Hiltunen, M.; Mannermaa, A.; Punnonen, K.; Hippeläinen, M.; Heinonen, S. Lack of association between C-850T polymorphism of the gene encoding tumor necrosis factor-α and polycystic ovary syndrome. Gynecol. Endocrinol. 2002, 16, 271–274. [Google Scholar] [CrossRef]
- Hong, L.; Zhang, Y.; Wang, Q.; Han, Y.; Teng, X. Effects of interleukin 6 and tumor necrosis factor-α on the proliferation of porcine theca interna cells: Possible role of these cytokines in the pathogenesis of polycystic ovary syndrome. Taiwan. J. Obstet. Gynecol. 2016, 55, 183–187. [Google Scholar] [CrossRef]
- Oksanen, L.; Tiitinen, A.; Kaprio, J.; Koistinen, H.A.; Karonen, S.L.; Kontula, K. No evidence for mutations of the leptin or leptin receptor genes in women with polycystic ovary syndrome. MHR Basic Sci. Reprod. Med. 2000, 6, 873–876. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Escobar-Morreale, H.F.; Villuendas, G.; Botella-Carretero, J.I.; Alvarez-Blasco, F.; Sanchon, R.; Luque-Ramirez, M.; San Millan, J.L. Adiponectin and resistin in PCOS: A clinical, biochemical and molecular genetic study. Hum. Reprod. 2006, 21, 2257–2265. [Google Scholar] [CrossRef] [PubMed]
- Orio, F., Jr.; Matarese, G.; Di Biase, S.; Palomba, S.; Labella, D.; Sanna, V.; Savastano, S.; Zullo, F.; Colao, A.; Lombardi, G. Exon 6 and 2 peroxisome proliferator-activated receptor-γ polymorphisms in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2003, 88, 5887–5892. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Y.; Wu, X.; Cao, Y.; Yi, L.; Fan, H.; Chen, J. Polymorphisms of the peroxisome proliferator–activated receptor-γ and its coactivator-1α genes in Chinese women with polycystic ovary syndrome. Fertil. Steril. 2006, 85, 1536–1540. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, B.O.; Haznedaroglu, I.C.; Kirazli, S.; Bayraktar, M. Global fibrinolytic capacity is decreased in polycystic ovary syndrome, suggesting a prothrombotic state. J. Clin. Endocrinol. Metab. 2002, 87, 3871–3875. [Google Scholar] [CrossRef][Green Version]
- Heinonen, S.; Korhonen, S.; Hippeläinen, M.; Hiltunen, M.; Mannermaa, A.; Saarikoski, S. Apolipoprotein E alleles in women with polycystic ovary syndrome. Fertil. Steril. 2001, 75, 878–880. [Google Scholar] [CrossRef]
- Urbanek, M.; Du, Y.; Silander, K.; Collins, F.S.; Steppan, C.M.; Strauss, J.F., III; Dunaif, A.; Spielman, R.S.; Legro, R.S. Variation in resistin gene promoter not associated with polycystic ovary syndrome. Diabetes 2003, 52, 214–217. [Google Scholar] [CrossRef]
- Rajkhowa, M.; Talbot, J.A.; Jones, P.W.; Clayton, R.N. Polymorphism of glycogen synthetase gene in polycystic ovary syndrome. Clin. Endocrinol. 1996, 44, 85–90. [Google Scholar] [CrossRef]
- Zhao, S.P.; Tang, X.M.; Shao, D.H.; Dai, H.Y.; Dai, S.Z. Association study between a polymorphism of aldosterone synthetase gene and the pathogenesis of polycystic ovary syndrome. Zhonghua Fu Chan Ke Za Zhi 2003, 38, 94–97. [Google Scholar]
- Kahsar-Miller, M.; Boots, L.R.; Azziz, R. Dopamine D3 receptor polymorphism is not associated with the polycystic ovary syndrome. Fertil. Steril. 1999, 71, 436–438. [Google Scholar] [CrossRef]
- Yazawa, T.; Imamichi, Y.; Uwada, J.; Sekiguchi, T.; Mikami, D.; Kitano, T.; Ida, T.; Sato, T.; Nemoto, T.; Nagata, S. Evaluation of 17β-hydroxysteroid dehydrogenase activity using androgen receptor-mediated transactivation. J. Steroid Biochem. Mol. Biol. 2020, 196, 105493. [Google Scholar] [CrossRef]
- Goodarzi, M.O.; Dumesic, D.A.; Chazenbalk, G.; Azziz, R. Polycystic ovary syndrome: Etiology, pathogenesis and diagnosis. Nat. Rev. Endocrinol. 2011, 7, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, X.L.; Xia, Y.; Cao, Y.; Wang, W.; Xu, P.; Che, Y.; Wu, X.; Yi, L.; Gao, Q. Association between polymorphisms of the CYP11A1 gene and polycystic ovary syndrome in Chinese women. Mol. Biol. Rep. 2012, 39, 8379–8385. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.R.; Deepika, M.L.N.; Supriya, K.; Latha, K.P.; Rao, S.S.; Rani, V.U.; Jahan, P. CYP11A1 microsatellite (tttta) n polymorphism in PCOS women from South India. J. Assist. Reprod. Genet. 2014, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Hu, K.; Shi, H.; Yu, Y.; Xu, J.; Sun, Y. The single-nucleotide polymorphism rs743572 of CYP17A1 shows significant association with polycystic ovary syndrome: A meta-analysis. Reprod. Biomed. Online 2021, 43, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Beglova, A.Y.; Elgina, S.I.; Gordeeva, L.A. Polymorphism of the CYP11A1, CYP17A1, and CYP19A1 genes in reproductive-aged women with polycystic ovary syndrome. Obstet. Gynegology 2019, 12, 148–153. [Google Scholar] [CrossRef]
- Shan, B.; Zhou, L.; Yang, S.; Yan, M.; Wang, Z.; Ouyang, Y.; Li, Z. Association between polycystic ovary syndrome (PCOS) and CYP11A1 polymorphism in Hainan, China: A case-control study. Int. J. Clin. Exp. Pathol. 2016, 9, 230–236. [Google Scholar]
- Tirosh, I.; Barkai, N.; Verstrepen, K.J. Promoter architecture and the evolvability of gene expression. J. Biol. 2009, 8, 95. [Google Scholar] [CrossRef]
- Yu, C.S.; Cheng, C.W.; Su, W.C.; Chang, K.C.; Huang, S.W.; Hwang, J.K.; Lu, C.H. CELLO2GO: A web server for protein subCELlular LOcalization prediction with functional gene ontology annotation. PLoS ONE 2014, 9, e99368. [Google Scholar] [CrossRef]
- Mohammed, A.; Hasan, J.; Joseph, D.N. Association between polycystic ovary syndrome and polymorphisms of CYP11A gene among sample of Iraqi women Introduction: Molecular Analysis: Gene Selection Polymerase Chain Reaction [PCR]. J. Univ. Shanghai Sci. Technol. 2020, 22, 712–725. [Google Scholar]
- Jiao, X.; Chen, W.; Zhang, J.; Wang, W.; Song, J.; Chen, D.; Zhu, W.; Shi, Y.; Yu, X. Variant alleles of the ESR1, PPARG, HMGA2 and MTHFR genes are associated with PCOS risk in Chinese population: A case-control study. Front. Endocrinol. 2018, 9, 504. [Google Scholar] [CrossRef] [PubMed]
- Asuncioon, M.; Calvo, R.M.; Millaan, J.L.S.; Sancho, J.; Avila, S.; Escobar-Morreale, H.F. A prospective study of the prevalence of PCOS in unselected Caucasian women from Spain. J. Endocrinol. Metab. 2000, 85, 2434–2438. [Google Scholar]
- Pusalkar, M.; Meherji, P.; Gokral, J.; Chinnaraj, S.; Maitra, A. CYP11A1 and CYP17 promoter polymorphisms associated with hyperandrogenemia in PCOS. Fertil. Steril. 2009, 92, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L. Androgen biosynthesis from cholesterol to DHEA. Mol. Cell. Endocrinol. 2002, 198, 7–14. [Google Scholar] [CrossRef]
- Gao, G.H.; Cao, Y.X.; Yi, L.; Wei, Z.L.; Xu, Y.P.; Yang, C. Polymorphism of CYP11A1 gene in Chinese patients with polycystic ovarian syndrome. Zhonghua Fu Chan Ke Za Zhi 2010, 45, 191–196. [Google Scholar]
- Diamanti-Kandarakis, E.; Bartzis, M.; Bergiele, A.T.; Tsianateli, T.C.; Kouli, C.R. Microsatellite polymorphism (tttta)n at -528 base pairs of gene CYP11α influences hyperandrogenemia in patients with PCOS. Fertil. Steril. 2000, 73, 735–745. [Google Scholar] [CrossRef]
- Gharani, N.; Waterworth, D.M.; Batty, S.; White, D.; Gilling-Smit, C.; Conway, G.S.; McCarthy, M.; Franks, S.; Williamson, R. Association of steroid synthesis gene CYP11A with polycystic ovary syndrome and hyperandrogenism. Hum. Mol. Genet. 1997, 6, 397–402. [Google Scholar] [CrossRef]
- Abdel-Mageed, W.S.; Dabous, E.; Gerguis, A. Association between polymorphisms of the CYP11A1 gene and polycystic ovary syndrome in Egyptian Female. Res. J. Appl. Biotechnol. 2016, 2, 19–28. [Google Scholar] [CrossRef]
- Shen, W.; Li, T.; Hu, Y.; Liu, H.; Song, M. Common polymorphisms in the CYP1A1 and CYP11A1 genes and polycystic ovary syndrome risk: A meta-analysis and meta-regression. Arch. Gynecol. Obstet. 2014, 289, 107–118. [Google Scholar] [CrossRef]
- Saddick, S.Y. Identifying genes associated with the development of human polycystic ovary syndrome. Saudi J. Biol. Sci. 2020, 27, 1271–1279. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, T.; Sudhir, N.; Kaur, A. Association Analysis of CYP11A1 Variants with Polycystic Ovary Syndrome: A Case-Control Study from North India. Reprod. Sci. 2021, 28, 2951–2960. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNPs # | Region of Gene | SNP ID | Nucleotide Change | Case | Consequence Type | Amino Acid Change | Amino Acid Position | SIFT | Polyphen |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Exon 1 | rs750026801 | tAt > tt | 1 | Frame shift mutation | Y > X | 85 | --- | --- |
| 2 | rs999606408 | Ccc > Tcc | 2 | Missense mutation | P > S | 7 | 0.45 | 0 | |
| 3 | rs1444841908 | Ctg > Atg | L > M | 20 | 0.01 | 0.621 | |||
| 4 | rs533078157 | aTc > aCc | I > T | 40 | 0.39 | 0.003 | |||
| 5 | rs1402720332 | AaC > aaA | N > K | 61 | 0.04 | 0.999 | |||
| 6 | rs746842413 | ttC > ttA | F > L | 82 | 0 | 0.986 | |||
| 7 | rs143655263 | Gtc > Atc | V > I | 79 | 0.44 | 0 | |||
| 8 | rs1316467116 | Cta > Gta | L > V | 60 | 0.24 | 0.018 | |||
| 9 | rs1471344818 | tCc > tTc | S > F | 41 | 0.04 | 0.838 | |||
| 10 | Exon 2 | rs546976108 | Gag > Aag | E > K | 91 | 0 | 1 | ||
| 11 | rs190764523 | Ggc > Agc | G > S | 94 | 0 | 0.998 | |||
| 12 | rs866217710 | Gat > Aat | D > N | 106 | 0.02 | 0.981 | |||
| 13 | Exon 3 | rs1258660118 | Tcg > Acg | S > T | 144 | 0.01 | 0.096 | ||
| 14 | rs1393077247 | aAg > aGg | K > R | 148 | 1 | 0.075 | |||
| 15 | rs1440123283 | gAg > gGg | E > G | 162 | 0.2 | 0.006 | |||
| 16 | rs762299364 | cTg > cCg | L > P | 182 | 0 | 0.999 | |||
| 17 | rs1400474918 | Gag > Aag | E > K | 208 | 0 | 0.952 | |||
| 18 | Exon 2 | rs776056840 | Cga > Tga | 3 | Stop gained mutation | R > * | 120 | --- | --- |
| 19 | rs779154292 | atC > at | 4 | Frame shift mutation | I > X | 102 | --- | --- | |
| 20 | rs1217014229 | Cac > CCac | 5 | H > PX | 130 | --- | --- | ||
| 21 | Exon 3 | rs549043326 | tCg > tAg | 6 | Stop gained mutation | S > * | 144 | --- | --- |
| 22 | rs1421587886 | GcA > gc | 7 | Frame shift mutation | A > X | 173 | --- | --- | |
| 23 | rs1178589612 | cTg > cg | 8 | L > X | 170 | --- | --- | ||
| 24 | Exon 4 | rs1567052760 | gCc > gTc | 9 | Missense mutation | A > V | 230 | 0.03 | 0.872 |
| 25 | rs1207802955 | Atc > Gtc | I > V | 237 | 1 | 0.007 | |||
| 26 | rs1461423064 | cTc > cCc | L > P | 248 | 0 | 0.998 | |||
| 27 | rs1416104210 | ttC > ttG | F > L | 258 | 0.9 | 0.001 | |||
| 28 | rs1596159786 | Acc > Gcc | T > A | 262 | 0.24 | 0.006 | |||
| 29 | rs1228084259 | Gct > Tct | A > S | 268 | 0.62 | 0.506 | |||
| 30 | rs1021942880 | gCa > gTa | A > V | 269 | 0 | 0.989 | |||
| 31 | Exon 5 | rs746124429 | tTc > tCc | F > S | 284 | 0.21 | 0.509 | ||
| 32 | rs1459305798 | Tgg > Cgg | W > R | 286 | 1 | 0.001 | |||
| 33 | rs1596159365 | tAc > tCc | Y > S | 298 | 0 | 0.995 | |||
| 34 | rs747101738 | Ctg > Gtg | L > V | 324 | 0.01 | 0.286 | |||
| 35 | rs1402190131 | gCa > gAa | A > E | 325 | 0 | 0.991 | |||
| 36 | rs748120824 | gAc > gGc | D > G | 329 | 0 | 0.98 | |||
| 37 | rs1424340465 | aCg > aTg | T > M | 330 | 0 | 0.997 | |||
| 38 | Exon 6 | rs139449608 | aCg > aTg | T > M | 331 | 0 | 1 | ||
| 39 | rs1046646548 | aTg > aCg | M > T | 333 | 0.11 | 0.481 | |||
| 40 | rs1567051424 | tAt > tTt | Y > F | 340 | 0.53 | 0.991 | |||
| 41 | rs531531464 | Gag > Aag | E > K | 341 | 0 | 1 | |||
| 42 | rs745719036 | cAg > cGg | Q > R | 372 | 0.05 | 0.051 | |||
| 43 | Exon 4 | rs755186597 | Cga > Tga | 10 | Stop gained mutation | R > * | 232 | --- | --- |
| 44 | Exon 5 | rs1224774813 | Cag > Tag | 11 | Q > * | 282 | --- | --- | |
| 45 | rs757299093 | Ata > ta | 12 | Frame shift mutation | I > Y | 279 | --- | --- | |
| 46 | rs1555425667 | Gag > ag | 13 | E > X | 314 | --- | --- | ||
| 47 | Exon 7 | rs867506250 | Cac > Tac | 14 | Missense mutation | H > Y | 388 | 0.43 | 0.938 |
| 48 | rs1208632679 | Tcc > Ccc | S > P | 391 | 0.01 | 0.437 | |||
| 49 | rs752776256 | Cag > Aag | Q > K | 395 | 0 | 0.967 | |||
| 50 | rs1220602604 | Ctt > Gtt | L > V | 398 | 1 | 0.037 | |||
| 51 | rs200029503 | Gta > Tta | V > L | 399 | 0.12 | 0.012 | |||
| 52 | rs1398357296 | Gtt > Ttt | V > F | 403 | 0 | 0.869 | |||
| 53 | rs1321165216 | Ctt > Att | L > I | 404 | 0.29 | 0.239 | |||
| 54 | rs944327325 | Gat > Cat | D > H | 406 | 0 | 0.748 | |||
| 55 | rs771663597 | aTt > aCt | I > T | 409 | 0 | 0.656 | |||
| 56 | rs1448075161 | Gcc > Acc | A > T | 411 | 0.03 | 0.409 | |||
| 57 | Exon 8 | rs1344040376 | Caa > Gaa | Q > E | 416 | 0.04 | 0.986 | ||
| 58 | rs1416463682 | tAt > tGt | Y > C | 420 | 0 | 0.998 | |||
| 59 | rs780138488 | Cca > Tca | P > S | 437 | 0 | 1 | |||
| 60 | rs774799229 | cTg > cCg | L > P | 463 | 0 | 0.992 | |||
| 61 | rs757055824 | Gct > Act | A > T | 468 | 0 | 1 | |||
| 62 | rs758061011 | Atg > Gtg | M > V | 472 | 0 | 0.977 | |||
| 63 | rs1326008763 | Atc > Gtc | I > V | 474 | 0.01 | 0.135 | |||
| 64 | Exon 9 | rs566280511 | Atg > Ctg | M > L | 479 | 0.18 | 0.011 | ||
| 65 | rs754698583 | Gtt > Att | V > I | 485 | 1 | 0.007 | |||
| 66 | rs775664050 | gTg > gAg | V > E | 493 | 0 | 0.997 | |||
| 67 | rs551306530 | aTg > aCg | M > T | 502 | 0.17 | 0.085 | |||
| 68 | rs148124218 | Ccc > Tcc | P > S | 513 | 0 | 0.999 | |||
| 69 | rs777925426 | Ttt > Ctt | F > L | 514 | 1 | 0 | |||
| 70 | rs200726137 | aAc > aTc | N > I | 515 | 0.06 | 0.127 | |||
| 71 | Exon 7 | rs1454328072 | Cga > Tga | 15 | Stop gained mutation | R > * | 405 | --- | --- |
| 72 | Exon 8 | rs762412759 | Cga > Tga | 16 | R > * | 424 | --- | --- | |
| 73 | rs755975808 | Cga > Tga | 17 | R > * | 439 | --- | --- | ||
| 74 | Exon 9 | rs754610565 | Cag > Tag | 18 | Q > * | 521 | --- | --- | |
| 75 | rs779413653 | Tga > Aga | 19 | R > * | 522 | --- | --- | ||
| 76 | rs765916701 | Caa > Taa | 20 | Q > * | 488 | --- | --- | ||
| 77 | rs1368450780 | Cag > Tag | 21 | Q > * | 516 | --- | --- | ||
| 78 | rs747901197 | Gaa > Taa | 22 | E > * | 517 | --- | --- |
| # | SNP ID | DDG (kcal/mole) | Stability | RI |
|---|---|---|---|---|
| 1 | rs999606408 | −0.47 | Decrease | 5 |
| 2 | rs1444841908 | −1.33 | Decrease | 8 |
| 3 | rs533078157 | −2.24 | Decrease | 9 |
| 4 | rs1402720332 | −0.31 | Decrease | 5 |
| 5 | rs746842413 | −1.20 | Decrease | 8 |
| 6 | rs143655263 | −0.21 | Decrease | 4 |
| 7 | rs1316467116 | −1.62 | Decrease | 8 |
| 8 | rs1471344818 | 0.03 | Increase | 3 |
| 9 | rs546976108 | −0.89 | Decrease | 8 |
| 10 | rs190764523 | −1.53 | Decrease | 9 |
| 11 | rs866217710 | −1.16 | Decrease | 5 |
| 12 | rs1258660118 | 0.09 | Increase | 5 |
| 13 | rs1393077247 | 0.07 | Increase | 3 |
| 14 | rs1440123283 | −1.04 | Decrease | 8 |
| 15 | rs762299364 | −1.80 | Decrease | 7 |
| 16 | rs1400474918 | −1.08 | Decrease | 8 |
| 17 | rs1567052760 | −0.35 | Decrease | 4 |
| 18 | rs1207802955 | −1.00 | Decrease | 9 |
| 19 | rs1461423064 | −1.65 | Decrease | 3 |
| 20 | rs1416104210 | −1.63 | Decrease | 8 |
| 21 | rs1596159786 | −1.31 | Decrease | 9 |
| 22 | rs1228084259 | −0.74 | Decrease | 9 |
| 23 | rs1021942880 | −0.19 | Decrease | 2 |
| 24 | rs746124429 | −1.93 | Decrease | 9 |
| 25 | rs1459305798 | −1.16 | Decrease | 9 |
| 26 | rs1596159365 | −1.41 | Decrease | 6 |
| 27 | rs747101738 | −1.47 | Decrease | 8 |
| 28 | rs1402190131 | −0.42 | Decrease | 3 |
| 29 | rs748120824 | −0.40 | Decrease | 2 |
| 30 | rs1424340465 | 0.24 | Increase | 2 |
| 31 | rs139449608 | 0.13 | Increase | 1 |
| 32 | rs1046646548 | −0.67 | Decrease | 5 |
| 33 | rs1567051424 | 0.08 | Increase | 3 |
| 34 | rs531531464 | −0.69 | Decrease | 7 |
| 35 | rs745719036 | −0.05 | Decrease | 1 |
| 36 | rs867506250 | 0.22 | Increase | 5 |
| 37 | rs1208632679 | −0.12 | Increase | 4 |
| 38 | rs752776256 | −0.56 | Increase | 1 |
| 39 | rs1220602604 | −1.75 | Decrease | 7 |
| 40 | rs200029503 | −1.63 | Decrease | 8 |
| 41 | rs1398357296 | −1.61 | Decrease | 8 |
| 42 | rs1321165216 | −1.52 | Decrease | 9 |
| 43 | rs944327325 | −0.96 | Decrease | 9 |
| 44 | rs771663597 | −2.38 | Decrease | 9 |
| 45 | rs1448075161 | −0.67 | Decrease | 7 |
| 46 | rs1344040376 | −0.22 | Decrease | 1 |
| 47 | rs1416463682 | −1.03 | Increase | 1 |
| 48 | rs780138488 | −1.58 | Decrease | 8 |
| 49 | rs774799229 | −1.62 | Decrease | 6 |
| 50 | rs757055824 | −0.69 | Decrease | 6 |
| 51 | rs758061011 | −0.69 | Decrease | 8 |
| 52 | rs1326008763 | −0.64 | Decrease | 6 |
| 53 | rs566280511 | −0.94 | Decrease | 7 |
| 54 | rs754698583 | −0.73 | Decrease | 8 |
| 55 | rs775664050 | −1.59 | Decrease | 7 |
| 56 | rs551306530 | −1.06 | Decrease | 6 |
| 57 | rs148124218 | −1.15 | Decrease | 6 |
| 58 | rs777925426 | −0.48 | Decrease | 0 |
| 59 | rs200726137 | 1.23 | Increase | 7 |
| Case ID | M | C | N | P | E | PM | GA | ER | CT | L | V |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal | 3.208 | 0.624 | 0.085 | 0.469 | 0.047 | 0.297 | 0.013 | 0.025 | 0.016 | 0.057 | 0.011 |
| Case 1 | 1.602 | 0.784 | 1.506 | 0.057 | 0.515 | 0.218 | 0.018 | 0.036 | 0.030 | 0.038 | 0.017 |
| Case 2 | 1.747 | 1.899 | 0.250 | 0.424 | 0.108 | 0.214 | 0.060 | 0.036 | 0.041 | 0.028 | 0.021 |
| Case 3 | 1.995 | 1.057 | 0.429 | 0.246 | 0.728 | 0.023 | 0.017 | 0.078 | 0.016 | 0.057 | 0.026 |
| Case 4 | 1.311 | 1.407 | 1.429 | 0.072 | 0.386 | 0.069 | 0.020 | 0.099 | 0.012 | 0.048 | 0.019 |
| Case 5 | 1.063 | 1.820 | 0.877 | 0.099 | 0.638 | 0.060 | 0.026 | 0.060 | 0.009 | 0.061 | 0.022 |
| Case 6 | 2.213 | 0.678 | 0.203 | 0.277 | 0.834 | 0.072 | 0.018 | 0.095 | 0.017 | 0.206 | 0.038 |
| Case 7 | 2.046 | 0.521 | 0.684 | 0.133 | 0.848 | 0.124 | 0.011 | 0.031 | 0.009 | 0.057 | 0.021 |
| Case 8 | 2.047 | 0.650 | 0.707 | 0.168 | 0.746 | 0.085 | 0.009 | 0.028 | 0.007 | 0.039 | 0.015 |
| Case 9 | 1.839 | 2.064 | 0.252 | 0.232 | 0.093 | 0.230 | 0.050 | 0.028 | 0.032 | 0.021 | 0.019 |
| Case 10 | 1.510 | 2.082 | 0.216 | 0.154 | 0.159 | 0.019 | 0.011 | 0.042 | 0.007 | 0.010 | 0.024 |
| Case 11 | 1.822 | 1.939 | 0.141 | 0.431 | 0.079 | 0.046 | 0.012 | 0.058 | 0.010 | 0.040 | 0.025 |
| Case 12 | 1.859 | 1.907 | 0.170 | 0.419 | 0.079 | 0.050 | 0.014 | 0.055 | 0.014 | 0.044 | 0.032 |
| Case 13 | 2.457 | 1.380 | 0.112 | 0.461 | 0.114 | 0.055 | 0.017 | 0.051 | 0.010 | 0.058 | 0.018 |
| Case 14 | 1.404 | 2.009 | 0.184 | 0.426 | 0.124 | 0.320 | 0.109 | 0.053 | 0.044 | 0.034 | 0.028 |
| Case 15 | 1.150 | 2.509 | 0.154 | 0.270 | 0.071 | 0.056 | 0.024 | 0.067 | 0.018 | 0.011 | 0.011 |
| Case 16 | 1.283 | 2.236 | 0.162 | 0.349 | 0.080 | 0.086 | 0.034 | 0.082 | 0.023 | 0.017 | 0.012 |
| Case 17 | 1.165 | 2.455 | 0.155 | 0.381 | 0.088 | 0.087 | 0.033 | 0.079 | 0.022 | 0.019 | 0.014 |
| Case 18 | 1.522 | 2.235 | 0.189 | 0.321 | 0.108 | 0.264 | 0.069 | 0.044 | 0.036 | 0.027 | 0.022 |
| Case 19 | 1.552 | 2.205 | 0.200 | 0.329 | 0.109 | 0.248 | 0.068 | 0.043 | 0.038 | 0.027 | 0.022 |
| Case 20 | 0.954 | 2.980 | 0.176 | 0.124 | 0.122 | 0.374 | 0.073 | 0.047 | 0.015 | 0.009 | 0.013 |
| Case 21 | 1.609 | 2.076 | 0.184 | 0.341 | 0.104 | 0.307 | 0.072 | 0.043 | 0.045 | 0.033 | 0.022 |
| Case 22 | 1.659 | 2.077 | 0.182 | 0.323 | 0.103 | 0.293 | 0.071 | 0.043 | 0.042 | 0.030 | 0.022 |
| Case ID | Molecular Functions | Biological Process | ||||
|---|---|---|---|---|---|---|
| Ion Binding | Oxidoreductase Activity | Lipid Binding | Lipid Metabolism | Biosynthesis | Small Molecule Metabolism | |
| Normal gene | 48.9 | 48.9 | 0.9 | 9.1 | 8.8 | 8.1 |
| 1 | 46.2 | 46.2 | 7.7 | 29.2 | 29.2 | 25.3 |
| 2 | 49 | 49 | 0.8 | 8.9 | 8.7 | 7.9 |
| 3 | 47.8 | 47.8 | 4.3 | 29.6 | 28.3 | 18.3 |
| 4 | 47.4 | 47.4 | 5.1 | 31.1 | 31.1 | 19.5 |
| 5 | 48 | 48 | 4 | 29.4 | 26.6 | 18.8 |
| 6 | 48 | 48 | 4 | 9.8 | 26.6 | 18.8 |
| 7 | 48.1 | 48.1 | 3.7 | 28.8 | 25.3 | 19.1 |
| 8 | 48.2 | 48.2 | 3.6 | 28 | 24.6 | 18.6 |
| 9 | 49 | 49 | 0.6 | 9.8 | 9.1 | 8.6 |
| 10 | 48.6 | 48.6 | 2.7 | 23.1 | 19.6 | 16 |
| 11 | 48.7 | 48.7 | 2.7 | 22.7 | 19.2 | 15.7 |
| 12 | 48.7 | 48.7 | 2.7 | 22.7 | 19.2 | 15.7 |
| 13 | 48.7 | 48.7 | 2.7 | 22.7 | 19.2 | 15.7 |
| 14 | 49 | 49 | 0.9 | 9 | 8.3 | 7.8 |
| 15 | 49 | 49 | 2.1 | 18.2 | 15 | 12.8 |
| 16 | 49.1 | 49.1 | 1.7 | 14.8 | 10.4 | 11.2 |
| 17 | 49.5 | 49.5 | 0.9 | 9.6 | 7.6 | 8 |
| 18 | 48.9 | 48.9 | 0.9 | 9.1 | 8.8 | 8.1 |
| 19 | 48.9 | 48.9 | 0.9 | 9.1 | 8.8 | 8.1 |
| 20 | 48.9 | 48.9 | 0.9 | 9 | 8.7 | 8 |
| 21 | 48.9 | 48.9 | 0.9 | 9.1 | 8.8 | 8.1 |
| 22 | 48.9 | 48.9 | 0.9 | 9.1 | 8.8 | 8.1 |
| Case ID | No. of Amino Acids | Molecular Weight | Theoretical pI | Estimated Half Life | Instability Index | Aliphatic Index | Extinction Coefficient (M−1 cm−1) |
|---|---|---|---|---|---|---|---|
| Normal | 553 | 63,785.85 | 5.83 | 30 h | 31.19 | 83.91 | 87,445 |
| Case 1 | 144 | 16,063.12 | 10.52 | 30 h | 51.45 | 54.86 | 23,490 |
| Case 2 | 555 | 64,033.25 | 6.35 | 30 h | 31.57 | 82.20 | 87,445 |
| Case 3 | 121 | 13,533.36 | 7.93 | 30 h | 34.30 | 76.45 | 16,960 |
| Case 4 | 202 | 22,095.05 | 9.33 | 30 h | 52.01 | 70.89 | 22,585 |
| Case 5 | 201 | 22,029.91 | 6.76 | 30 h | 50.14 | 74.13 | 24,075 |
| Case 6 | 143 | 16,271.72 | 9.59 | 30 h | 32.86 | 83.08 | 26,930 |
| Case 7 | 230 | 25,559.33 | 9.46 | 30 h | 43.24 | 78.00 | 38,180 |
| Case 8 | 232 | 25,978.79 | 9.56 | 30 h | 43.58 | 73.53 | 43,680 |
| Case 9 | 555 | 63,855.02 | 6.07 | 30 h | 32.76 | 83.75 | 78,965 |
| Case 10 | 237 | 26,787.50 | 7.86 | 30 h | 30.55 | 81.81 | 33,920 |
| Case 11 | 291 | 33,246.95 | 7.14 | 30 h | 27.27 | 81.72 | 47,900 |
| Case 12 | 297 | 33,847.53 | 7.85 | 30 h | 26.10 | 78.75 | 47,900 |
| Case 13 | 351 | 40,289.79 | 7.82 | 30 h | 29.51 | 78.29 | 63,495 |
| Case 14 | 547 | 63,085.11 | 6.06 | 30 h | 33.03 | 85.36 | 87,445 |
| Case 15 | 428 | 49,040.00 | 6.19 | 30 h | 26.97 | 85.63 | 66,350 |
| Case 16 | 449 | 51,374.76 | 6.19 | 30 h | 28.30 | 87.06 | 69,330 |
| Case 17 | 464 | 53,215.72 | 5.91 | 30 h | 28.98 | 84.25 | 69,330 |
| Case 18 | 552 | 63,657.72 | 5.83 | 30 h | 30.88 | 84.06 | 87,445 |
| Case 19 | 554 | 63,942.03 | 5.90 | 30 h | 31.15 | 83.75 | 87,445 |
| Case 20 | 517 | 59,607.16 | 6.02 | 30 h | 31.13 | 85.22 | 81,945 |
| Case 21 | 547 | 63,100.16 | 5.90 | 30 h | 30.99 | 84.64 | 87,445 |
| Case 22 | 548 | 63,288.29 | 5.90 | 30 h | 30.82 | 84.49 | 87,445 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muccee, F.; Bijou, O.; Harakeh, S.; Adawiyah, R.; Sayyed, R.Z.; Haghshenas, L.; Alshehri, D.; Ansari, M.J.; Ghazanfar, S. In-Silico Investigation of Effects of Single-Nucleotide Polymorphisms in PCOS-Associated CYP11A1 Gene on Mutated Proteins. Genes 2022, 13, 1231. https://doi.org/10.3390/genes13071231
Muccee F, Bijou O, Harakeh S, Adawiyah R, Sayyed RZ, Haghshenas L, Alshehri D, Ansari MJ, Ghazanfar S. In-Silico Investigation of Effects of Single-Nucleotide Polymorphisms in PCOS-Associated CYP11A1 Gene on Mutated Proteins. Genes. 2022; 13(7):1231. https://doi.org/10.3390/genes13071231
Chicago/Turabian StyleMuccee, Fatima, Osama Bijou, Steve Harakeh, Rabi’atul Adawiyah, R. Z. Sayyed, Leila Haghshenas, Dikhnah Alshehri, Mohammad Javed Ansari, and Shakira Ghazanfar. 2022. "In-Silico Investigation of Effects of Single-Nucleotide Polymorphisms in PCOS-Associated CYP11A1 Gene on Mutated Proteins" Genes 13, no. 7: 1231. https://doi.org/10.3390/genes13071231
APA StyleMuccee, F., Bijou, O., Harakeh, S., Adawiyah, R., Sayyed, R. Z., Haghshenas, L., Alshehri, D., Ansari, M. J., & Ghazanfar, S. (2022). In-Silico Investigation of Effects of Single-Nucleotide Polymorphisms in PCOS-Associated CYP11A1 Gene on Mutated Proteins. Genes, 13(7), 1231. https://doi.org/10.3390/genes13071231