
Identification of Runs of Homozygosity Islands and Genomic Estimated Inbreeding Values in Caqueteño Creole Cattle (Colombia)


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Genotypes
2.2. Genetic Structure of the Population
2.3. Runs of Homozygosity Detection
2.4. ROH Islands
3. Results
3.1. The Genetic Structure of CAQ Exhibits Low Population Stratifications
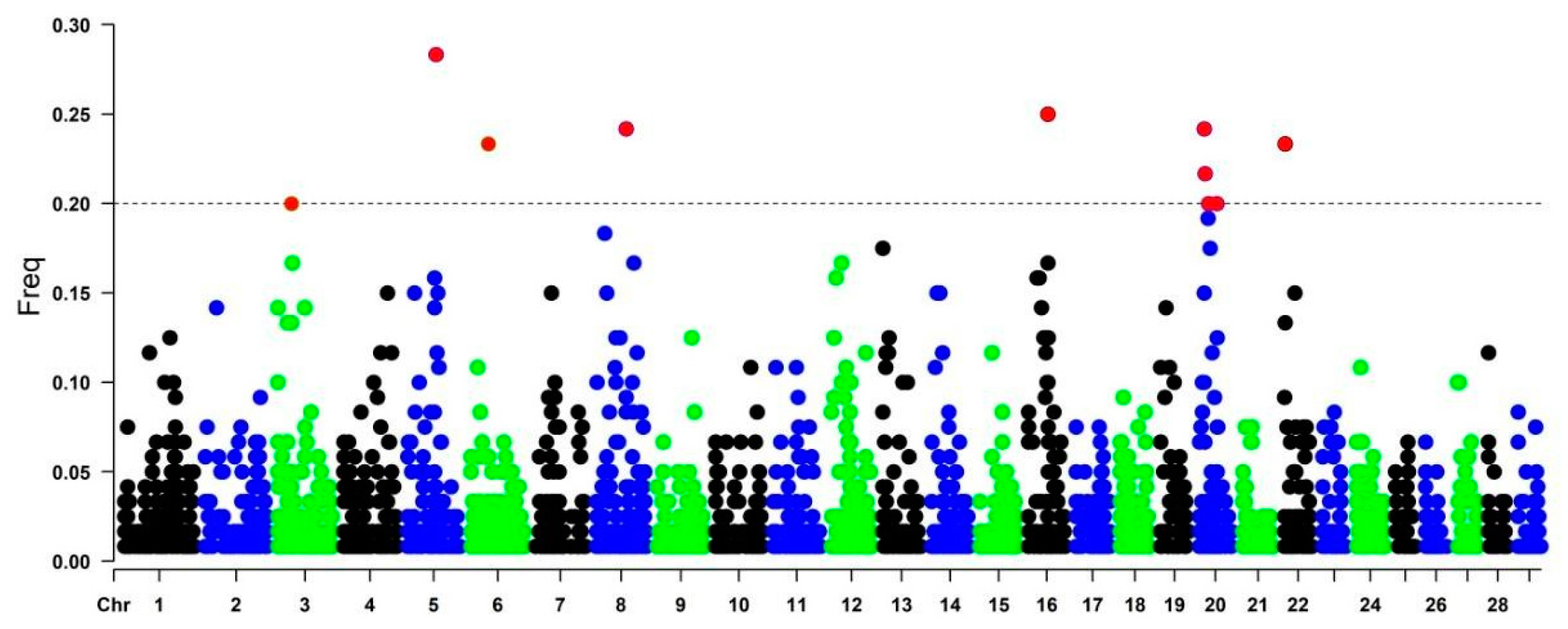
3.2. Genomic Distribution of Runs of Homozygosity
3.3. Genomic Inbreeding Coefficient (FROH)
3.4. Gene Characterization in ROH Islands
4. Discussion
4.1. Population Structure of the CAQ Population Identified Genetic Variability
4.2. Runs of Homozygosity (ROH) Characteristics
4.3. Identification of Candidate Genes within Runs of Homozygosity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Xu, L.; Bickhart, D.M.; Cole, J.B.; Schroeder, S.G.; Song, J.; Van Tassell, C.P.; Sonstegard, T.S.; Liu, G.E. Genomic Signatures Reveal New Evidences for Selection of Important Traits in Domestic Cattle. Mol. Biol. Evol. 2015, 32, 711–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejarano, D.; Pedraza, A.; Martinez Rocha, J.F.; Martínez, R. Variabilidad Genética En Subpoblaciones Comerciales de La Raza Criolla Colombiana Romosinuano. Cienc. Tecnol. Agropecu. 2012, 13, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, Y.T.; Floriano, P.L.E. Características Morfométricas y Fanerópticas de La Raza. In Genética Pura: Ganado Criollo Caqueteño; Torrijos, R.R., Ed.; Impresos Panamericanos: Florencia, Colombia, 2013; pp. 27–48. ISBN 978-9-58579-450-4. [Google Scholar]
- Barrera, G.P.; Martínez, R.; Torrijos, R.; Ramón, F. Caracterización Molecular de Una Población de Ganado Caqueteño y Su Relación Filogenética Con Razas Bovinas Criollas Colombianas. Cienc. Tecnol. Agropecu. 2006, 7, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Delgado, J.V.; Martnez, A.M.; Acosta, A.; Alvarez, L.A.; Armstrong, E.; Camacho, E.; Cañón, J.; Cortés, O.; Dunner, S.; Landi, V.; et al. Genetic characterization of Latin-American Creole cattle using microsatellite markers. Anim. Genet. 2012, 43, 2–10. [Google Scholar] [CrossRef]
- McKay, S.D.; Schnabel, R.D.; Murdoch, B.M.; Matukumalli, L.K.; Aerts, J.; Coppieters, W.; Crews, D.; Neto, E.D.; Gill, C.A.; Gao, C.; et al. An Assessment of Population Structure in Eight Breeds of Cattle Using a Whole Genome SNP Panel. BMC Genet. 2008, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Gibson, J.; Morton, N.E.; Collins, A. Extended Tracts of Homozygosity in Outbred Human Populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef] [Green Version]
- Toro Ospina, A.M.; Silva Faria, R.A.; Vercesi Filho, A.E.; dos Santos Goncalves Cyrillo, J.N.; Zerlotti Mercadante, M.E.; Curi, R.A.; Vasconcelos Silva, J.A. Genome-wide Identification of Runs of Homozygosity Islands in the Gyr Breed (Bos Indicus). Reprod. Domest. Anim. 2020, 55, 333–342. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, G.; Yang, L.; Zhu, B.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Liu, G.E.; Li, J. Genomic Patterns of Homozygosity in Chinese Local Cattle. Sci. Rep. 2019, 9, 16977. [Google Scholar] [CrossRef] [Green Version]
- Caivio-Nasner, S.; López-Herrera, A.; González-Herrera, L.G.; Rincón, J.C. Diversity Analysis, Runs of Homozygosity and Genomic Inbreeding Reveal Recent Selection in Blanco Orejinegro Cattle. J. Anim. Breed. Genet. 2021, 138, 613–627. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Sardina, M.T.; Tolone, M.; Di Gerlando, R.; Sutera, A.M.; Fontanesi, L.; Portolano, B. Genome-Wide Identification of Runs of Homozygosity Islands and Associated Genes in Local Dairy Cattle Breeds. Animal 2018, 12, 2480–2488. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Tolone, M.; Di Gerlando, R.; Fontanesi, L.; Sardina, M.T.; Portolano, B. Genomic Inbreeding Estimation in Small Populations: Evaluation of Runs of Homozygosity in Three Local Dairy Cattle Breeds. Animal 2016, 10, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Grossman, S.R.; Shylakhter, I.; Karlsson, E.K.; Byrne, E.H.; Morales, S.; Frieden, G.; Hostetter, E.; Angelino, E.; Garber, M.; Zuk, O.; et al. A Composite of Multiple Signals Distinguishes Causal Variants in Regions of Positive Selection. Science 2010, 327, 883–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic Patterns of Homozygosity in Worldwide Human Populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; MacCiotta, N.P.P. Analysis of Runs of Homozygosity and Their Relationship with Inbreeding in Five Cattle Breeds Farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chen, Z.; Zhao, W.; Guo, L.; Sun, H.; Zhu, K.; Liu, G.; Shen, X.; Zhao, X.; Wang, Q.; et al. Genome-Wide Selection Signatures Detection in Shanghai Holstein Cattle Population Identified Genes Related to Adaption, Health and Reproduction Traits. BMC Genom. 2021, 22, 747. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Howrigan, D.P.; Simonson, M.A.; Keller, M.C. Detecting Autozygosity through Runs of Homozygosity: A Comparison of Three Autozygosity Detection Algorithms. BMC Genom. 2011, 12, 460. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.C.; Visscher, P.M.; Goddard, M.E. Quantification of Inbreeding Due to Distant Ancestors and Its Detection Using Dense Single Nucleotide Polymorphism Data. Genetics 2011, 189, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Bjelland, D.W.; Weigel, K.A.; Vukasinovic, N.; Nkrumah, J.D. Evaluation of Inbreeding Depression in Holstein Cattle Using Whole-Genome SNP Markers and Alternative Measures of Genomic Inbreeding. J. Dairy Sci. 2013, 96, 4697–4706. [Google Scholar] [CrossRef]
- Zimin, A.V.; Delcher, A.L.; Florea, L.; Kelley, D.R.; Schatz, M.C.; Puiu, D.; Hanrahan, F.; Pertea, G.; Van Tassell, C.P.; Sonstegard, T.S.; et al. A Whole-Genome Assembly of the Domestic Cow, Bos Taurus. Genome Biol. 2009, 10, R42. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal Components Analysis Corrects for Stratification in Genome-Wide Association Studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Martínez, R.A.; Pérez, J.E. Características De Crecimiento En El Ganado Criollo. Rev. Corpoica-Cienc. Tecnol. Agropecu. 2006, 7, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Zambrano, M.F.B.; Flórez, J.C.R.; Rios, A.C.H.; Portilla, C.E.S.; de Jesús Bedoya Berrio, G. Evaluation of Runs of Homozygosity and Genomic Inbreeding in Holstein Cattle from Colombia. Semin. Ciências Agrárias 2020, 41, 3397–3418. [Google Scholar] [CrossRef]
- Peripolli, E.; Stafuzza, N.B.; Amorim, S.T.; Lemos, M.V.A.; Grigoletto, L.; Kluska, S.; Ferraz, J.B.S.; Eler, J.P.; Mattos, E.C.; Baldi, F. Genome-wide Scan for Runs of Homozygosity in the Composite Montana Tropical® Beef Cattle. J. Anim. Breed. Genet. 2020, 137, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Sölkner, J.; Ferenčaković, M.; Karimi, Z.; O’Brien, A.M.P.; Mészáros, G.; Eaglen, S.; Boison, S.A.; Curik, I. Extremely Non-Uniform: Patterns of Runs of Homozygosity in Bovine Populations. In Proceedings of the 10th World Congress on Genetics Applied to Livestock Production, Vancouver, BC, Canada, 17–22 August 2014; p. 3. [Google Scholar]
- Mesbah-Uddin, M. Identification of Causal Factors for Recessive Lethals in Dairy Cattle with Special Focus on Large Chromosomal Deletions. Ph.D. Thesis, Aarhus University, Aarhus, Denmark, 2019. [Google Scholar]
- Dejenie, M.Y. Genome-Wide Signature of Positive Selection, Breed-Specific SNPs and Linkage Disequilibrium in Ethiopian Indigenous and European Beef Cattle Breeds; Addis Ababa University: Addis Ababa, Ethiopia, 2021. [Google Scholar]
- Mastrangelo, S.; Ben Jemaa, S.; Ciani, E.; Sottile, G.; Moscarelli, A.; Boussaha, M.; Montedoro, M.; Pilla, F.; Cassandro, M. Genome-wide Detection of Signatures of Selection in Three Valdostana Cattle Populations. J. Anim. Breed. Genet. 2020, 137, 609–621. [Google Scholar] [CrossRef]
- Nolte, W.; Weikard, R.; Brunner, R.M.; Albrecht, E.; Hammon, H.M.; Reverter, A.; Kühn, C. Biological Network Approach for the Identification of Regulatory Long Non-Coding RNAs Associated with Metabolic Efficiency in Cattle. Front. Genet. 2019, 10, 1130. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.; Miglior, F.; Fonseca, P.; Seymour, D.; Asselstine, V.; Brito, L.; Schenkel, F. Identification of Variants Associated with Divergent Feed Efficiency Groups Using Multiple RNA-Sequencing Datasets from Dairy and Beef Cattle. In Proceedings of the World Congress on Genetics Applied to Livestock Production, Auckland, New Zealand, 11–16 February 2018; p. 773. [Google Scholar]
- Silva, D.O. Estudo de Associação Genômica Para Habilidade de Permanência No Rebanho Na Raça Nelore, Considerando Diferentes Idades. Master’s Thesis, Sao Paulo State University, Sao Paulo, Brazil, 2018. [Google Scholar]
- Wu, X.; Fang, M.; Liu, L.; Wang, S.; Liu, J.; Ding, X.; Zhang, S.; Zhang, Q.; Zhang, Y.; Qiao, L.; et al. Genome Wide Association Studies for Body Conformation Traits in the Chinese Holstein Cattle Population. BMC Genom. 2013, 14, 897. [Google Scholar] [CrossRef] [Green Version]
- Adamson, B.; Norman, T.M.; Jost, M.; Cho, M.Y.; Nuñez, J.K.; Chen, Y.; Villalta, J.E.; Gilbert, L.A.; Horlbeck, M.A.; Hein, M.Y.; et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell. 2016, 167, 1867–1882.e21. [Google Scholar] [CrossRef] [Green Version]
- Hardie, L.C.; VandeHaar, M.J.; Tempelman, R.J.; Weigel, K.A.; Armentano, L.E.; Wiggans, G.R.; Veerkamp, R.F.; de Haas, Y.; Coffey, M.P.; Connor, E.E.; et al. The Genetic and Biological Basis of Feed Efficiency in Mid-Lactation Holstein Dairy Cows. J. Dairy Sci. 2017, 100, 9061–9075. [Google Scholar] [CrossRef] [Green Version]
- Celis Parra, G.A.; Chinguad Taramuel, L.G.; Garcia Bustos, J.J. Evaluation of the Productive Characteristics of the Caqueteño Creole Cattle Breed. Trop. Anim. Health Prod. 2020, 52, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Yuan, Y.; Shi, L.; Li, Y.; Liu, L.; Sun, D. Identification of Single Nucleotide Polymorphisms of PIK3R1 and DUSP1 Genes and Their Genetic Associations with Milk Production Traits in Dairy Cows. J. Anim. Sci. Biotechnol. 2019, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Sudrajad, P.; Sharma, A.; Dang, C.G.; Kim, J.J.; Kim, K.S.; Lee, J.H.; Kim, S.; Lee, S.H. Validation of Single Nucleotide Polymorphisms Associated with Carcass Traits in a Commercial Hanwoo Population. Asian-Australas. J. Anim. Sci. 2016, 29, 1541–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas, E.; Garcia, M.D.; Wells, J.E.; Smith, T.P.L. Association of Single Nucleotide Polymorphisms in the ANKRA2 and CD180 Genes with Bovine Respiratory Disease and Presence of Mycobacterium Avium Subsp. Paratuberculosis1. Anim. Genet. 2011, 42, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Huson, H.J.; Kim, E.-S.; Godfrey, R.W.; Olson, T.A.; McClure, M.C.; Chase, C.C.; Rizzi, R.; O’Brien, A.M.P.; Van Tassell, C.P.; Garcia, J.F.; et al. Genome-Wide Association Study and Ancestral Origins of the Slick-Hair Coat in Tropically Adapted Cattle. Front. Genet. 2014, 5, 101. [Google Scholar] [CrossRef]
- Littlejohn, M.D.; Henty, K.M.; Tiplady, K.; Johnson, T.; Harland, C.; Lopdell, T.; Sherlock, R.G.; Li, W.; Lukefahr, S.D.; Shanks, B.C.; et al. Functionally Reciprocal Mutations of the Prolactin Signalling Pathway Define Hairy and Slick Cattle. Nat. Commun. 2014, 5, 5861. [Google Scholar] [CrossRef] [Green Version]
- Porto-Neto, L.R.; Bickhart, D.M.; Landaeta-Hernandez, A.J.; Utsunomiya, Y.T.; Pagan, M.; Jimenez, E.; Hansen, P.J.; Dikmen, S.; Schroeder, S.G.; Kim, E.-S.; et al. Convergent Evolution of Slick Coat in Cattle through Truncation Mutations in the Prolactin Receptor. Front. Genet. 2018, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Upadhyay, R.C.; Chandra, G.; Kumar, S.; Malakar, D.; Singh, S.V.; Singh, M.K. Genome wide Expression Analysis of the Heat Stress Response in Dermal Fibroblasts of Tharparkar (Zebu) and Karan-Fries (Zebu × Taurine) Cattle. Cell Stress Chaperones 2020, 25, 327–344. [Google Scholar] [CrossRef]
- Shen, J.; Hanif, Q.; Cao, Y.; Yu, Y.; Lei, C.; Zhang, G.; Zhao, Y. Whole Genome Scan and Selection Signatures for Climate Adaption in Yanbian Cattle. Front. Genet. 2020, 11, 94. [Google Scholar] [CrossRef]
- Ocampo, R.J.; Martínez, J.F.; Martínez, R. Assessment of Genetic Diversity and Population Structure of Colombian Creole Cattle Using Microsatellites. Trop. Anim. Health Prod. 2021, 53, 122. [Google Scholar] [CrossRef]
- Sarlo Davila, K.M.; Howell, A.; Nunez, A.; Orelien, A.; Roe, V.; Rodriguez, E.; Dikmen, S.; Mateescu, R.G. Genome-wide Association Study Identifies Variants Associated with Hair Length in Brangus Cattle. Anim. Genet. 2020, 51, 811–814. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Class | ROH (N) | ROH (%) | Mean | Min | Max | Standard Deviation |
|---|---|---|---|---|---|---|
| (ROH < 2 Mb) | 4067 | 62.82 | 1.386 | 1 | 1.99 | 0.28 |
| (ROH2–4 Mb) | 1752 | 27.06 | 2.750 | 2 | 3.99 | 0.55 |
| (ROH4–8 Mb) | 589 | 9.09 | 5.306 | 4 | 7.89 | 1.00 |
| (ROH8–16 Mb) | 65 | 1.01 | 9.848 | 8 | 14.23 | 1.80 |
| (ROH > 16 Mb) | 1 | 0.015 | 17.26 | 17.26 | 17.26 | 17.26 |
| Total ROH | 6474 | 100 | 2.20 | 1 | 17.26 | 1.50 |
| Variable | N | Mean | Standard Deviation | Sum | Minimum | Maximum |
|---|---|---|---|---|---|---|
| FROH < 2 | 15 | 0.16348 | 0.04702 | 2.45226 | 0.10206 | 0.24563 |
| FROH 2–4 | 15 | 0.12847 | 0.04268 | 1.92711 | 0.06728 | 0.21083 |
| FROH 4–8 | 15 | 0.09731 | 0.05168 | 1.45964 | 0.04909 | 0.24458 |
| FROH 8–16 | 14 | 0.02650 | 0.02937 | 0.37098 | 0.00328 | 0.09405 |
| FROH > 16 | 2 | 0.00683 | 0.0000974 | 0.01367 | 0.00676 | 0.00690 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toro-Ospina, A.M.; Herrera Rios, A.C.; Pimenta Schettini, G.; Vallejo Aristizabal, V.H.; Bizarria dos Santos, W.; Zapata, C.A.; Ortiz Morea, E.G. Identification of Runs of Homozygosity Islands and Genomic Estimated Inbreeding Values in Caqueteño Creole Cattle (Colombia). Genes 2022, 13, 1232. https://doi.org/10.3390/genes13071232
Toro-Ospina AM, Herrera Rios AC, Pimenta Schettini G, Vallejo Aristizabal VH, Bizarria dos Santos W, Zapata CA, Ortiz Morea EG. Identification of Runs of Homozygosity Islands and Genomic Estimated Inbreeding Values in Caqueteño Creole Cattle (Colombia). Genes. 2022; 13(7):1232. https://doi.org/10.3390/genes13071232
Chicago/Turabian StyleToro-Ospina, Alejandra M., Ana C. Herrera Rios, Gustavo Pimenta Schettini, Viviana H. Vallejo Aristizabal, Wellington Bizarria dos Santos, Cesar A. Zapata, and Edna Gicela Ortiz Morea. 2022. "Identification of Runs of Homozygosity Islands and Genomic Estimated Inbreeding Values in Caqueteño Creole Cattle (Colombia)" Genes 13, no. 7: 1232. https://doi.org/10.3390/genes13071232
APA StyleToro-Ospina, A. M., Herrera Rios, A. C., Pimenta Schettini, G., Vallejo Aristizabal, V. H., Bizarria dos Santos, W., Zapata, C. A., & Ortiz Morea, E. G. (2022). Identification of Runs of Homozygosity Islands and Genomic Estimated Inbreeding Values in Caqueteño Creole Cattle (Colombia). Genes, 13(7), 1232. https://doi.org/10.3390/genes13071232