Abstract
The aim of this study was to determine the suitability of the comparative genomic hybridization to microarray (aCGH) technique for prenatal diagnosis, but also to assess the frequency of chromosomal aberrations that may lead to fetal malformations but are not included in the diagnostic report. We present the results of the aCGH in a cohort of 7400 prenatal cases, indicated for invasive testing due to ultrasound abnormalities, high-risk for serum screening, thickened nuchal translucency, family history of genetic abnormalities or congenital abnormalities, and advanced maternal age (AMA). The overall chromosomal aberration detection rate was 27.2% (2010/7400), including 71.2% (1431/2010) of numerical aberrations and 28.8% (579/2010) of structural aberrations. Additionally, the detection rate of clinically significant copy number variants (CNVs) was 6.8% (505/7400) and 0.7% (57/7400) for variants of unknown clinical significance. The detection rate of clinically significant submicroscopic CNVs was 7.9% (334/4204) for fetuses with structural anomalies, 5.4% (18/336) in AMA, 3.1% (22/713) in the group of abnormal serum screening and 6.1% (131/2147) in other indications. Using the aCGH method, it was possible to assess the frequency of pathogenic chromosomal aberrations, of likely pathogenic and of uncertain clinical significance, in the groups of cases with different indications for an invasive test.
1. Introduction
Prenatal diagnosis is one of the most important achievements of modern perinatology. Intrauterine imaging of the fetus enables the early detection of a significant number of malformations and creates a chance for intrauterine treatment or surgical intervention immediately after birth. Therefore, according to the recommendations of the Polish Society of Gynecologists and Obstetricians regarding the management of prenatal diagnosis, all pregnant women should be offered prenatal screening tests that determine the risk of chromosomal abnormalities in the fetus. Invasive genetic tests should be performed when screening tests show a higher risk of having a child with a genetic defect than the normal population.
The main indications for an invasive prenatal diagnosis have mainly included ultrasound anomalies, high-risk for serum screening, family history of genetic disorders or birth defects, and advanced maternal age (AMA) (over 35 years old). Currently, the first-choice method in prenatal diagnosis is the method of comparative genomic hybridization to microarrays (aCGH). The variability of the etiology and clinical heterogeneity of these disorders require high-sensitivity and the whole genome method.
Similar indications can be found in the available literature. Luo X. et al., used chromosome microarray analysis (CMA) to evaluate 1256 prenatal cases (between 15–33 weeks of gestation) with clinical indications: non-structural ultrasound anomalies (n = 132), structural ultrasound anomalies (n = 283), high-risk for maternal serum markers (n = 44), abnormal non-invasive prenatal test results (n = 143), family history of genetic disorders (n = 288), AMA (n = 328), and other indications (n = 38) [1]. In another group led by Xiang J. et al., the research was carried out on a group of 5000 pregnancies with the following indications: anomaly on ultrasonography (n = 1055), advanced maternal age (n = 1784), abnormal result on maternal serum screening (n = 1199), abnormal NIPT results (n = 515) and other indications (n = 709). The mean age of the pregnant women qualified for the study was 32.04 years (range: 18–49 years), and the mean gestational age was 21.28 weeks (range: 9–34 weeks) [2].
The introduction of the CMA method in the late 90s, revolutionized cytogenetic diagnostics, enabling detailed results to be obtained in a short time. As a result, in recent years it has been widely used in prenatal diagnostics [3,4,5].
Congenital malformations of the fetus concern ~70% of spontaneous miscarriages, ~2–3% of live births and ~20% of stillborn. They are also one of the leading causes of infant mortality. Currently, it is estimated that about 35% of congenital malformations are genetically determined. Among the genetic causes of congenital malformations, chromosome aberrations account for approximately 24% of cases in live births and approximately 50% of spontaneous abortions [3].
Congenital malformations divide into isolated malformations or multiple malformations, which are accounted for 2/3 and 1/3 of all malformations, respectively.
It is estimated that about 6–7% of congenital malformations are caused by submicroscopic genome imbalance, which can only be detected using the microarray technique. Additionally, in ~1% of fetuses with a congenital malformation, alterations in the number of copies of DNA sequences of unknown clinical significance are found [2].
In the available literature, the detection rate of pathogenic imbalances between the different clinical groups is variable. For example, Luo X. et al., showed overall prenatal diagnostic yield was (7.8%) of 1256 pregnancies. Clinically significant genomic aberrations were identified in 1.5% of analyzed patients with non-structural ultrasound anomalies, 12.7% in fetus with structural ultrasound anomalies, 4.5% at high-risk for biochemic test, 26.6% at high-risk for NIPT, 3.8% with a burdened family history, and 2.1% with AMA [2].
The use of the aCGH microarray method to diagnose fetuses with abnormal ultrasound results is recommended by European scientists. According to the American Congress of Obstetricians and Gynecologists (ACOG) and the Society for Maternal-Fetal Medicine (SMFM), CMA should be used as a first-tier diagnostic test for all fetuses, both with the presence of fetal structural abnormalities and in patients who wish to continue prenatal diagnosis in the case of normal ultrasound of the fetus. Additionally, this test can be considered in all women undergoing prenatal screening, regardless of their age [6,7]. While the recommendations of the Canadian College of Medical Geneticists (CCMG) recommend the use of CMA when multiple fetal malformations or nuchal translucency (NT) ≥3.5 mm is detected after normal rapid screening for the most common aneuploidies [8].
Based on the above recommendations, copy number variations detected in the microarray CGH study, can be classified as pathogenic, likely pathogenic, likely benign, benign and of unknown significance (VOUS). To establish the origin of aberrations, a routine karyotype analysis by the GTG technique (G-bands after trypsin and Giemsa), FISH (Fluorescence In Situ Hybridization), aCGH, or MLPA method (Multiplex Ligation-dependent Probe Amplification) should be performed on the fetus’s parental material, depending on the type and size of the variant, and the methods available in the laboratory. Interpretation of the clinical significance of unknown CNVs is often very complicated and there is no single general rule or algorithm for the interpretation of test results.
The greatest diagnostic challenge in prenatal testing are the aberrations of unknown clinical significance. Therefore, following the recommendations issued by the European Cytogenetics Association, these abnormalities should not be reported in prenatal results [9]. As opposed to European recommendations, the CCMG indicates that variants of unknown importance, greater than a 500 kb in the case of deletions and 1 MB for duplications, should be shown in the prenatal results [8].
The VOUS, unlike likely pathogenic aberrations, do not involve genes of known pathogenicity and are absent in a few cases in the general population as likely benign. The numbers of VOUS will decrease as their importance is published in the medical literature and publicly available databases. It is assumed that the VOUS frequency remains quite variable. For example: Song T. et al., reported variants of unknown importance (VOUS) in 14/190 (7.37%) cases, Lee M.Y. et al., in 4/32 (12.5%), Yan Y. et al., in 4/76 (5.3%), and Wu X.L. et al., in 2.9% (3/104) of cases [10,11,12,13]. This difference can be explained by considering the different resolution of the microarray platform used by the authors and the differences in the interpretation of the meaning of the same VOUS [14]. In addition, a variant of unknown significance may in fact be pathogenic but has been classified as a VOUS due to insufficient data to determine its actual pathogenic significance. Therefore, a better understanding of VOUS are necessary to ensure appropriate prenatal genetic counseling [10].
To date, many studies evaluating the performance of CMA in prenatal diagnosis have been published. A study by the National Institute of Child Health and Human Development (NICHD) showed clinically significant CNV in 6% of fetuses with normal karyotype and anatomical defects [15]. In the study by Srebniak et al., a group of 1033 fetuses with abnormalities found in ultrasound was analyzed and pathogenic CNV found in 5.5% of cases [16]. In a larger study of 5000 fetuses, the prevalence of CNV was found to be 6.6% in 2462 cases with ultrasound abnormalities [17].
We present the results of a chromosomal microarray analysis in a larger cohort of 7400 cases diagnosed in the single center, the Institute of Mother and Child, from January 2014 to September 2021. We identified pathogenic aberrations and likely pathogenic abnormalities in 505 fetuses, 57 CNVs of unknown clinical significance and 17 CNVs that were classified as likely benign. Indications for the study included the following groups: prenatally diagnosed ultrasound abnormalities, high risk for screening, thickened nuchal translucency, family history of genetic abnormalities or congenital abnormalities, and advanced maternal age (AMA).
2. Materials and Methods
In the aCGH study, the DNA was isolated from amniotic fluid (n = 6620), trophoblast (n = 510), amniocyte cultures (n = 130), fixed cell sediment (n = 21), umbilical cord blood (n = 53), fetal skin fibroblast (n = 3) or fetal urine (n = 4) using the Sherlock kit (A&A Biotechnology) (Figure 1). The tests were performed on patients between 11 and 35 weeks of pregnancy. The amniocentesis was performed between 13–23 weeks of pregnancy, trophoblast biopsy between the 11th and 13th, and cordocentesis between 23–35 weeks of pregnancy.

Figure 1.
Types of materials used for extraction DNA.
2.1. Sample Types and DNA Isolation
The research was approved by the Bioethics Committee of the Institute of Mother and Child in Warsaw. All pregnant women decided to undergo aCGH testing after receiving genetic counselling and signing informed consent prior to invasive prenatal testing. The study group included a total of 7400 samples (Figure 1). The mean age of the pregnant women was 35 years (range: 18–49 years). Amniotic fluid (n = 6545), trophoblast (n = 495), amniocyte cultures (n = 114), fixed cell sediment (n = 16), umbilical cord blood (n = 41), fetal skin fibroblast (n = 3) or fetal urine (n = 4) and sample DNA (n = 56) were collected and successfully analyzed at the Department of Medical Genetics of the Institute of Mother and Child in the period from January 2014 to September 2021. Due to the poor quality of the material provided to us for DNA isolation, too early amniocentesis, incorrectly performed chorionic villus sampling and failures in cell culture, in 126 cases we obtained very poor data, which did not allow us to issue an informative result. Table 1 shows the number of tests performed in a particular year along with the number of correct, incorrect, and non-informative results. In cases where it was required, parents blood samples were taken to determine the origin of the identified aberrations.

Table 1.
Number of tests performed in a particular year along with the number of correct, incorrect, and non-informative results.
In all samples of amniotic fluid, trophoblast and fetal urine except umbilical cord blood, cell cultures were established for DNA preservation and conventional karyotype determination. Genomic DNA was immediately isolated from the fresh material. When amniotic fluid (AF) and chorionic villus sampling (CVS) was visibly contaminated, we used a cell culture that eliminates maternal cells and stimulates the growth of fetal cells and then DNA was isolated from cultured cells. The CVS villi were separated from maternal tissue under a microscope to minimize maternal cell contamination (MCC). Two to four villi were used for DNA extraction. Sample DNA was isolated from 2 to 5 mL of amniotic fluid. The AF samples were centrifuged. For CVS, incubation at 56 °C with 20 µL of proteinase K, water, and tissue lysis buffer (L1.4 buffer) was performed for at least 1 h for efficient digestion and lysis of the entire sample. For AF samples, incubation at 56 °C with 20 µL of proteinase K, water, and lysis buffer (L1.4 buffer) was performed for 45 min. Genomic DNA was extracted using a DNA isolation kit (Sherlock A&A Biotechnology, Poland) according to the manufacturer’s instructions.
2.2. Genomic Array Platform (Array Comparative Genomic Hybridization (Array CGH) Analysis and Interpretation)
The aCGH was performed using an 8x60K microarray from Oxford Gene Technology (CytoSure ISCA, v2 and v3, Oxford, UK). The array used in this study contains 60-mer oligonucleotide probes covering the entire genome with an average spatial resolution of 120 kb. The CGH methodology description is available in the manufacturer’s website. All genomic coordinates are based on a reference genome (NCBI37/hg19). The data analysis was performed using CytoSure Interpret Software (Oxford Gene Technology, Oxford, UK) and a circular binary segmentation algorithm. The calling thresholds were the deviation of the binary cyclic segmentation (CBS) segment from a zero-log ratio of +0.30 for duplication and −0.5 for deletion. Our results were classified using the CytoSure Interpret Software (Oxford Gene Technology, Oxford, UK). Quality control measures were monitored using CytoSure Interpret Software (Oxford Gene Technology). The microarray used in this analysis does not contain SNP probes, does not detect polyploidy, inversion, balanced translocation regions of absence of heterozygosity.
In this study, we report the detection rate of chromosomal microarray analysis in the detection of fetal chromosomal aberrations among 7400 fetuses with congenital malformations, high risk for screening, thickened nuchal translucency, family history of genetic abnormalities or congenital abnormalities, and advanced maternal age (AMA) in Poland and evaluate the additional diagnostic yields produced by CMA for different referral indications (Table 2).
Table 2.
Microarray results of 7400 samples according to reason for prenatal testing.
2.3. CNV Classification
The clinical relevance of each copy number variant should be considered individually using the general CNV classification. In our study, as recommended, we used five categories to classify the detected aberrations: pathogenic, likely pathogenic, variants of unknown significance (VOUS), possibly benign and benign:
- Pathogenic aberrations: CNV was classified as pathogenic if it was a large aberration of several MB, or was associated with known microdeletion/microduplication syndromes or contained known genes responsible for a specific pathology and had been previously described in specific clinical disorders.
- Likely pathogenic aberrations: CNVs that have not yet been described or have been rarely described and contain some gene/genes whose function is known and most likely may be responsible for the clinical features of the patient.
- Variants of unknown significance (VOUS): This category includes CNVs that did not have a clearly defined clinical significance at the time of publication of the study results. These changes were not included in the prenatal outcomes because the function of the genes in this region was unknown or the cause of the abnormal ultrasound examination was difficult to determine. In our study, parental inheritance of VOUS was not specified.
- Likely benign aberrations: CNVs that occur in healthy people and have only been described in a few cases in the general population, but do not represent a common polymorphism. CNV interpreted as possibly mild was not reported in a result of the study.
- Benign aberrations: do not affect the phenotype (polymorphisms occurring in the general population), which include: aberrations in the region of segmental duplication, aberrations that do not contain genes, aberrations in regions containing dose-insensitive genes frequently repeated in the Polish population and known aberrations as copy number variants described in the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home (accessed on 3 January 2022)) (path: DGV Gold Standard Variants).
All detected copy number variants (CNVs) were systematically assessed for clinical significance against those in the scientific literature and available databases: OMIM (http://www.ncbi.nlm.nih.gov/omim, (accessed on 3 January 2022)), Clinical Structural Variants (https://www.ncbi.nlm.nih.gov/dbvar/, (accessed on 3 January 2022)), Database of Genomic Variants (http://projects.tcag.ca/variation/ (accessed on 3 January 2022)), Ensembl (https://www.ensembl.org/index.html, (accessed on 3 January 2022)), and DECIPHER (http://decipher.sanger.ac.uk/, (accessed on 3 January 2022)).
3. Results
A total of 7400 samples were analyzed by aCGH from January 2014 to September 2021, of which 2010 samples yielded abnormal results (2010/7400, 27.2%), including 1431 cases with numerical chromosome anomalies (1431/7400, 19.3%) and 579 cases with CNV (579/7400, 7.8%) including pathogenic, likely pathogenic, VOUS and likely benign CNVs (Table 2). Benign, likely benign, and VOUS CNVs, were not reported in our results.
Among 1160 cases with numerical chromosome anomalies, trisomy 21 (578/7400, 7.8%) was the most common type, trisomy 18 (295/7400, 4%), trisomy 13 (130/7400, 1.7%), monosomy chromosome X (119/7400, 1.6%) and polyploidy (38/7400, 0.5%). A total of 135 CNVs (135/579, 23.3%) were inherited from a parent (97 maternal and 38 paternal) and 119 CNVs (119/579, 20.6%) were de novo in origin. A total of 446 CNVs (446/579, 77%) were classified as pathogenic (P), 59 as likely pathogenic (LP) (59/579, 10.2%), 57 CNVs (57/579, 9.8%) were classified as variants of uncertain significance (VOUS), and 17 CNVs (17/579, 2.9%) were classified as likely benign (LB) (Table 2).
3.1. Anomaly on Ultrasonography
In fetuses with multiple ultrasound defects, detection rates of aneuplidy and triploidy was 21.8% (489/2245) and CNV 9.4% (219/2245). In the subgroup with a structural anomaly in one system, detection rates of aneuploidy and triploidy was 13.7% (269/1959) and CNV 9% (176/1959). The highest percentage of abnormal results was in hydrops fetalis (59/123, 48%), followed by congenital heart disease (285/1188; 24%), or by fetal hypotrophy (19/79, 24.1%) (Table 2).
3.2. Advanced Maternal Age (AMA)
A microarray CGH analysis was performed for a total of 336 pregnant women with AMA (over 35 years old), and the average age of this group was 37.65 years (range: 35–49 years). Chromosome abnormalities were detected in 70 samples (70/336, 20.8%), including 46 cases with aneuploidy (46/336, 13.7%), 6 cases with chromosomal triploidy (6/336, 1.8%), and 18 cases with CNV (18/336, 5.4%) (Table 3). Aneuploidies included: 1 case with trisomy 13, 1 case with mosaic trisomy 14, 1 case with trisomy 15, 6 cases with trisomy 18, 35 cases with trisomy 21, 2 cases with monosomy X.
Table 3.
The aCGH results of 336 samples with indication of advanced maternal age.
3.3. Abnormal Serum Screening Results: PAPP-A
Serum screening results: PAPP-A (first-trimester test), was conducted by measuring pregnancy-associated plasma protein A concentration and the free β-hCG (human chronic gonadotropin) along with the measurement of NT in the USG.
A total of 713 samples with abnormal result in the PAPP-A test were analyzed by microarray CGH, and abnormal results were observed in 136 samples (136/713, 19%), including 110 cases with aneuploidy (110/713, 15.4%) and 26 cases with CNV (26/713, 3.6%) (Table 4).
Table 4.
The aCGH results of 713 samples with abnormal result in PAPP-A test and normal USG.
The 713 samples with abnormal results in the PAPP-A test were divided into four subgroups according to risk type: 615 cases (615/713, 86.3%) with high risk of Down syndrome, 36 cases (36/713, 5%) with high risk of Edwards syndrome, 20 cases (20/715, 2.8%) with high risk of Patau syndrome and 42 cases (42/713, 5.9%) with high risk of trisomy 21, 18 and 13. Among these, microarray confirmed 78 cases in the group of high risk of trisomy 21 syndrome, 13 cases in the group of high risk of trisomy 18, and 5 cases in the group of high risk of trisomy 13. Additionally, in the group of high risk of trisomy 21, we identified 6 cases with chromosome X monosomy. In the group of high risk of trisomy 21, 18 and 13, CGH showed 3 cases with trisomy 21, 3 cases with trisomy 18, and 2 cases with trisomy 13 (Table 4).
3.4. Other Indications
In the subgroup with thickened nuchal translucency (NT), the detection rate of abnormal results was 36.9% (212/573). In the subgroup with parental anxiety, we got only correct results in all cases (13/13, 100%) (Table 2).
The last 1533 samples had indications for prenatal testing, such as genetic disorders in family history and verification of the others prenatal tests. The microarray analysis revealed that 60 cases with genetic disorders with family history (60/197, 30.5%) and 418 cases with verification of the others prenatal tests (418/1336, 31.3%) (Table 2) had chromosomal abnormalities.
In 207 cases, indications were based on the family history: mothers who had previous pregnancies with fetal abnormalities or a child with genetic disorders, structural malformations, chromosomal abnormalities or parents who were carriers of a chromosome aberration were also assessed by CMA. In this group, clinically significant CNV was detected in 25 fetuses and numerical aberrations were found in 31 fetuses.
3.5. Discrepancy between Karyotyping and CGH Array
Cell culture performed to exclude aberrations not detectable by aCGH method analysis was made simultaneously on 5164 samples (5164/7400, 69.8%) of our cohort. Among 5164 cases with normal aCGH results, routine karyotype analysis showed 31 abnormal results with chromosomal triploidy in the female fetus (31/5264, 0.6%). Chromosomal triploidy was associated with ultrasound disorders: lymphoid holoprosencephaly, collapse of the frontal bones, VSD, abnormal departure of large vessels, omphalocele with displacement, hypotrophy, and feet and hands defects. Additionally, in 34 cases with normal CGH results, karyotype analysis showed balanced translocation or inversion.
Furthermore, CGH analysis showed the presence of microduplication/microdeletion in 265 out of 3474 cases with normal karyotype results.
We identified 57 variants of unknown clinical significance (VOUS). This group covers genetic aberrations are not generally seen in the population and, therefore, have few or no clinical evidence available to evaluate their pathogenic character. The VOUS may include benign familial variants that make no clinical function, or may be rare harmful changes that appear subsequent in a clinical phenotype.
4. Discussion
In our study, we focused on the assessment of the usefulness of the aCGH in prenatal diagnosis in the cohort of 7400 pregnancies in a single diagnostic center in Poland. Our research included a large study group mostly thanks to public funds, and strict uniform criteria for qualifying patients for invasive tests. Samples of DNA from both parents were also available in many cases, which significantly helped to classify many CNVs that were difficult to interpret. Genetic counselling was performed before sampling and after obtaining the test results.
The percentage of identified abnormalities was 27.2% (2010/7400), of which 71.2% (1431/2010) were numerical chromosomal aberrations and 28.8% (579/2010) structural aberrations (Table 2). Unfortunately, due to the poor quality of the DNA, it was not possible to obtain reliable results in 126 cases. In our study, the diagnostic success rate was 98.29% (7274/7400). For example, Wang Y. et al., using BACs-on-Beads (BoBs) assay, of the 1520 prenatal cases collected, 1428 cases were successfully analyzed, and 92 cases did not have informative results. The observation rate was 93.95% (1428/1520) [18].
The microarray method not only provides information about the presence of a CNV, but also enables the identification of the chromosomal mosaic. In our study, we identified 28 mosaic chromosomal trisomies and 19 mosaics of X monosomy. Mosaic trisomies of chromosome 8 were identified in 3 cases, chromosome 14 in 3 cases, chromosome 15 in 2 cases, chromosome 16 in 4 cases, chromosome 18 in 3 cases and chromosome 21 in 3 cases. In addition to recurring aneuploidy, we also found a mosaic of chromosomal trisomy in individual cases of chromosomes: 2, 3, 4, 5, 7, 9, 12, 13, 17 and 22. The clinical consequences of chromosomal mosaicism identified during invasive prenatal diagnosis may be difficult to assess, ranging from no apparent clinical phenotype to early fetal death. The chromosomal mosaic detection values are a log2 ratio from 0 to +0.3 or from 0 to −0.5. An example of the lowest mosaic trisomy found in prenatal diagnosis is the 6% mosaic trisomy of chromosome 4 (in 6 out of 100 analyzed cells) confirmed by FISH (from uncultured material). However, during the CGH analysis of this case, the degree of mosaicism was estimated at 25–30% (mean log ration 0.1174). This difference may result from the test procedure itself (for aCGH we used whole genomic DNA, while during the FISH procedure, the entire obtained cell sediment was not used to prepare the preparation, or some cells may not show an informative signal for the probe). Cell culture in a conventional karyotype can promote in vivo selection of euploid cells versus aneuploid cells, which increases with longer culture duration.
Clinically significant structural aberrations (pathogenic and likely pathogenic) accounted for 87.2% (505/579) of all diagnosed CNV and 6.8% (505/7400) of all performed tests. Our detection rate was higher than those presented in other works, for example in the study of Breman A. et al., in which the percentage of clinically significant CNV was 2.4%, or the publication of Chai H. and co-workers, where the detection rate was described as 2.59% [19,20]. However, the results were similar in the group of over 5000 pregnancies described by Shaffer L.G. et al., where the percentage of detected clinically significant copy number changes was 6.5%, in cases referred with abnormal ultrasound examination, while the percentage of results of uncertain clinical significance was 4.2% [21]. In another group of 4282 fetal samples reported by Wapner R.J. et al., the diagnostic efficiency was 6.0% in samples with normal conventional karyotype results, when the indication was a fetal structural defect found on ultrasound and 1.7% when the indication was advanced maternal age or abnormal screening results [15]. Breman A. et al., demonstrated a diagnostic efficiency of 4.2% in a group of 1075 prenatal samples with no known chromosomal abnormalities or familial genomic imbalance [19].
In the study by Lovrecic L. et al., the detection rate of pathogenic CNV in the group of fetuses with defects in ultrasound was 10.0%. The diagnostic efficiency was the highest in the group of cases with multiple congenital malformations (16.7%), and the lowest in the group of isolated IUGR (intrauterine growth restriction) (6.3%) [22]. The study by de Witt M.C. et al., showed that a significant submicroscopic CNV could be identified in 3.1–7.9% of fetuses with a defect limited to one system and in 9.1% of fetuses with multiple abnormalities [23].
All samples received for our study were classified into four groups, according to the indications for invasive prenatal diagnosis: abnormal ultrasound examination result, advanced maternal age, abnormal results of screening test (pregnancy-associated plasma protein A) and parental anxiety, genetic diseases present in the family and the verification of other genetic tests. As expected, the rate of chromosomal abnormalities detected by the CGH microarray was the highest in the group with abnormalities in the ultrasound examination and amounted to 27.3%, and then in the group with advanced maternal age, elevated NT, abnormal PAPP-A test result and anxiety, it amounted to 23.6% (Figure 2).
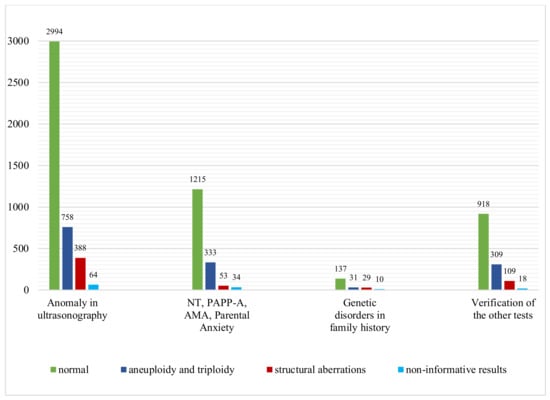
Figure 2.
Microarray CGH results of 7400 samples: number of cases with normal results, aneuploidy and triploidy and structural aberrations in four subgroups of different indications for prenatal testing.
Microdeletions were found significantly more often than microduplications: 375 and 204 respectively. In our study, clinically significant CNV (pathogenic and likely pathogenic) was detected in 7.6% (149/1959) of fetuses with isolated abnormalities or 8.2% (185/2245) in fetuses with multiple malformations identified during ultrasound examination. Among isolated defects, the highest percentage of CNV was observed in congenital heart defects—10.4%, followed by central nervous system defects with 9.1% or in fetal hypotrophy—8.4%. Studies in other centers have shown that the diagnostic efficiency of CMA in cases of multiple structural abnormalities in the fetus and isolated heart defects ranged from 3.1–10% for multiple abnormalities, and from 7.9–13.1% for isolated defects [21,24,25]. The differences in the degree of detection may be related to the appropriate selection of patients, bias, availability of reimbursed tests or the use of the previously classic karyotype method to exclude microscopically visible chromosomal aberrations in the fetus.
In the group of malformations of one system, the CGH detection rate of all aberration was the lowest for a urinary tract defect and amounted to 4.5% (6/134) including 2.2% (3/134) CNV. In the available literature, the most common pathogenic CNV in fetuses with congenital kidney and urinary tract defects is the 17q12 microdeletion [24,26,27], which causes polycystic kidney disease and the diabetic syndrome. In the study group, three fetuses had urinary tract defects and had a 17q12 microdeletion. In the case of fetal gastroschisis and achondroplasia, we did not find any abnormal results. The genetic causes of these anomalies are heterogeneous and poorly understood. Some of these abnormalities may be due to multifactorial inheritance or a mutation in the single gene, and therefore the use of CGH for diagnosis may be an inappropriate strategy.
The most common submicroscopic aberration in our study was the deletion of the 22q11.21 region (80/7400, 1.1%) (Figure 3). Ultrasound abnormalities in these fetuses included isolated and coexisting heart defects, cleft lip and palate, and congenital anomalies. The second most frequently observed structural aberration was the microdeletion in the 16p11.2 region (16/7400, 0.22%). Abnormalities occurring in these fetuses were increased neck translucency, heart defect, megacystis, ventriculomegaly, cleft lip and cleft palate, and fetal hypotrophy. Postnatal phenotype of 16p11.2 carriers is characterized by motor and speech disorder, language disorder, motor coordination difficulties, psychiatric conditions, and autistic features. If the 16p11.2 deletion is found in the fetus, it is difficult to determine the exact phenotype at birth, most people with a 16p11.2 deletion have language retardation and cognitive impairment. Aberrations with low penetrance are only reported if the USG shows fetal defects corresponding to this deletion.

Figure 3.
Number and type of clinically relevant CNVs.
Duplication of 16p13.11 occurred in 15 fetuses (15/7400, 0.21%) with, inter alia, abnormal results of biochemical test PAPP-A and polysectomy including fetal edema, ascites, and hydrocephalus (Figure 3). The 16p13.11 microduplication syndrome is associated with variable clinical features including behavioral abnormalities, pervasive developmental delay, congenital heart disease, and skeletal defects. As with the 16p11.2 deletion, the prenatally established 16p13.11 microduplication is difficult to interpret and determine the child’s phenotype at birth. Due to low penetrance, 16p13.11 duplication should not be taken into account in the prenatal result, and we did not report it in our results.
The 18p11.32p11.21 deletion was found in 15 fetuses (15/7400, 0.21%) with increased nuchal translucency, heart defect, polydactyly of the hand and multiple lethal malformations: holoprosencephaly, large cleft upper lip, no typical nose, small eyeballs, severe hypertelorism, low nape of the neck, massive edema of the head and thorax (Figure 3). The available literature describes cases of fetuses with increased NT and craniofacial defects.
The 1q21.1 deletion occurred in fourteen fetuses (14/7400, 0.19%), which always had ultrasound findings: hydrocephalus, aqueductal stenosis, pyelectasias, congenital heart defects, skeletal defects, and increased NT (Figure 3). Postnatal carriers of 1q21.1 deletion may present phenotypic variability, e.g., mental retardation, microcephaly, heart defects, short stature, hypotonia, cataract [28,29].
Pathogenic structural CNVs were not detected in the cohort of 573 fetuses with isolated increased nuchal translucency, and in the cohort of 13 fetuses in which the indication was only parental anxiety. Egloff M. et al., described a large group of fetuses with isolated increased neck translucency. In this study, pathogenic CNV was found in only 2.7% of fetuses with a neck translucency ≥3.5 mm, and nearly half of these were CNV associated with neurodevelopmental disorders: autism spectrum disorders, schizophrenia, attention deficit with hyperactivity disorder and intellectual disability [30].
In the group of referrals due to advanced maternal age, clinically significant submicroscopic CNVs were detected in 5.3% (18/336) of fetuses (Table 3), which is higher than the results obtained by Wapner R.J. et al., who found CNV in 1.7% of fetuses tested for AMA [15]. The results of our research showed that the frequency of numerical aberrations increased with age (Table 3), which is in line with previous studies [2]. However, the prevalence of structural aberrations in our cohort with AMA was the highest in the group of patients aged 35–39.
In the group of abnormal serum screening results (PAPP-A), we identified clinically significant submicroscopic CNV in 3.1% (22/713) of the fetuses (Table 4 and Table 5). Our results were higher than results showed by Xiang J et al., in which 2.5% (30/1199) of fetuses had clinically significant CNV [2]. In this group, we identified 22 unexpected, pathogenic structural aberrations. One of the more interesting cases was a female fetus (number 7408), with high risk of trisomy 21 where we identified deletion 5p14.3p14.1, of approximately 5.72 Mb in size. The aberration includes four gene encoding proteins: CDH18 (OMIM 603019), CDH12 (OMIM 600562), PRDM9 (OMIM 6097601 and CDH10 (OMIM 604555). Deletion of this region has been described in a fetus with congenital heart disease and was inherited from a mother without clinical symptoms [31].
Table 5.
22 structural aberrations detected in group of 713 samples with abnormal result on PAPP-A test and normal USG.
In male fetus (number 7121), with high risk of trisomy 21, we identified a duplication of the 20p12.3p11.1 region (approximately size 20.09 Mb). The test result was confirmed by the FISH method. Analysis of the interphase nuclei by FISH using the RP11-430K20 probe specific for the 20p12.2p12.3 region revealed the presence of two cell lines. In the first line (41.4%), three signals were found for the studied region, and in the second line (58.6%), two signals were found. The duplication included 93 protein-coding genes and was located in the region of the 20p trisomy syndrome (ORPHA: 261318), described in patients with: intellectual disability, speech development delay, impaired motor coordination and facial dysmorphic features. The literature describes a patient with excessive growth, psychomotor retardation, hypotension, features of facial dysmorphia and convulsions, who was diagnosed with mosaic trisomy 20p11.2-p12.1 [32].
In addition to aneuploidies and known recurrent pathogenic CNVs explaining the clinical abnormalities in the fetus, we highlighted 20 interesting structural aberrations that are pathogenic (n = 16) (Table 6) or likely pathogenic (n = 4) (Table 7) and were associated with the fetus phenotype.
Table 6.
Examples of interesting pathogenic CNVs which were correlated with the fetal defects.
Table 7.
Examples of interesting likely pathogenic CNVs which can be correlated with the fetal defects.
In the group of interesting pathogenic aberrations, we listed two structural aberrations in female fetuses with defects of the central nervous system (case 5653); duplication of 17q12 (approximate size 1.76 Mb) and duplication of Xq28 (approximate size 607 kb). The duplication in 17q12 includes 21 protein coding genes therein: CCL3L1 (OMIM 601395), ZNHIT3 (OMIM 604500), PIGW (OMIM 610275) and dose sensitive genes: LHX1 (OMIM 601999), AATF (OMIM 608463), ACACA (OMIM 200350), TADA2A (OMIM 602276), HNF1B (OMIM 189907). Additionally, this aberration covers a region of known 17q12 duplication syndrome (OMIM 614526) described in patients among others with: developmental delay, structural brain abnormalities, heart, and kidney defects [33,34]. Duplication of this region found in the fetus with ventriculomegaly and corpus callosum agenesis has been described in the literature, as well as in a patient with Th9 vertebrae of the thoracic spine [34]. Aberration in this region was characterized by variable expression and incomplete penetrance (21.1%) and occurred de novo. Duplication in Xq28 includes 13 protein coding genes for example: GAB3 (OMIM 300482), dose-sensitive gene DKC1 (OMIM 300126), MPP1 (OMIM 305360), SMIM96, F8 (OMIM 300841), H2AFB1 (OMIM 301037), F8A1 (OMIM 305423), FUNDC2 (OMIM 301042), CMC46, MTCP1 (OMIM 300116), BRCC3 (OMIM 300617), VBP1 (OMIM 300133) and exon 2 of the RAB39B gene (OMIM 300774) and partially encompasses the distal Xq28 microduplication syndrome region (ORPHA: 293939) with candidate genes RAB39B1 and CLIC2 (OMIM 300138). Our fetus was the only carrier of the aberration. This syndrome is reported in male patients with intellectual disability, behavioral disorders, facial dysmorphic features, and recurrent infections. Parental aCGH showed that identical aberrations were detected in a normal, asymptomatic father, which was a big surprise for us.
In the male fetus (number 7271), directed to the diagnostics due to advanced maternal age, high risk of chromosomal aberration (high risk of T21 1:29), intrauterine growth restriction, tetralogy of Fallot and syndactyly, we detected deletion 16q24.1 (approximately 653 kb in size). This deletion includes four protein coding genes: dose sensitive FOXF1 gene (OMIM 601089), MTHFSD gene (OMIM 616820), dose sensitive FOXC2 gene (OMIM 602402), FOXL1 gene (OMIM 603252) and is located in the region of microdeletion 16q24.1 (ORPHA: 352629). Deletions in the 16q24.1 region involving the FOX gene cluster, which were described in patients, involved among others: vascular dysplasia of the pulmonary alveoli with displacement of the pulmonary vessels, heart defects, gastrointestinal malformations and urinary system defects, including hydronephrosis [35,36,37]. The aCGH tests in parents showed that the identified aberration occurred de novo.
The second group of clinically significant aberrations consists of four interesting likely pathogenic CNVs (Table 7). Deletions and duplications were classified as likely pathogenic CNV if they contained candidate genes that may contribute to the abnormal phenotype. The inheritance of these aberrations was established in three cases and they were inherited from a normal mother. One of the examples was a female fetus with the referral reasons: cerebral hydrocephalus, the width of the lateral ventricle 13 mm, concave outline of the frontal bones (symptom of lemon and banana), hernia in the sacro-lumbar section and maternal age 37 (case number 2643). We have identified duplications of 10q24.31q24.32 (approximal size 658 kb) and Xq27.1 (approximal size 565 kb) regions. Duplication of the 10q24.31q24.32 region includes genes: BTRC (OMIM 603482), DPCD (OMIM 616467), FBXW4 (OMIM 608071), exons 1–2 of the TLX1NB gene (OMIM 612734) and dose sensitive genes: TLX1 (OMIM 186770), LBX1 (OMIM 604255), POLL (OMIM 606343) and FGF8 (OMIM 600483). Duplications of this region have been reported in patients with limb defects (SHFM3; OMIM 246560). Duplication in Xq27.1 involves the dose sensitive SOX3 gene (OMIM 313430). Duplications of this region have been reported in patients with polyhormonal hypopituitarism (OMIM 312000), sex-linked intellectual disability with isolated growth hormone deficiency (OMIM 300123) and neural tube defects [38,39,40]. Parental microarray analysis has shown that the aberrations were inherited from a normal mother.
The aCGH method allows the detection of the entire CNV spectrum, from aneuploidy to very small submicroscopic aberrations, including pathogenic, likely pathogenic, and also those of unknown clinical significance (VOUS). These changes were defined as VOUS in our test group based on the following criteria: CNVs that do not have a clearly defined effect on the clinical phenotype at the time of publication of the test results; includes a gene or genes that have an unknown effect on the identified fetal defect (Table 8). The VOUS were detected in 57 cases, which is 0.7% of all tested fetuses, or 9.8% of fetuses diagnosed with CNV (Table 2). The detection rate of VOUS CNVs in our study was lower than that of a previous multicenter study (1.6%) by Wapner R.J. et al. [15], than that of another cohort of 5026 pregnancies (4.6%) showed in work by Wang J.C. et al., 2019 [41]. These differences could be caused by different aberration reporting criteria and discrepancies in the interpretation of the results.
Table 8.
Variants of unknown significance and the genes within CNV, which may be responsible for abnormal phenotype.
The VOUS interpretation remains the biggest challenge in the prenatal diagnostics. Therefore, a detailed understanding of clinically relevant genetic variants is very important. Including them in the literature together with the clinical description of the fetal defects can provide the basis for the future classification of variants as pathogenic or benign. Unfortunately, only association studies within the family can determine their exact pathogenic potential. As the potential benefits must be weighed against the possible risks, it is now recommended that only pathogenic or likely pathogenic CNV be disclosed to parents. Showing aberrations with VOUS status in the results can cause a lot of stress and anxiety for parents. Therefore, in all cases where VOUS were detected, according to informed consent, the parents were not informed about the variant and therefore the parental origin of the detected aberration could not be tested.
In conclusion, our studies have shown that aCGH is a very useful method in the diagnosis of fetal defects in a pre-selected high-risk population. Careful and individualized counselling before and after genetic testing is necessary due to the relatively high risk of obtaining outcomes of uncertain clinical relevance, such as copy number changes associated with highly variable expression or incomplete penetrance.
Author Contributions
Conceptualization and writing: K.K.; clinical data acquisition: N.B.-W., A.B., M.G., M.D. (Marzena Dębska)., J.C., A.K.-K., P.W. (Paweł Własienko), S.K., B.M., T.I., A.K.-C., T.R., A.N.-B., A.R., A.J.-T., A.K. (Anna Kruczek), E.K., G.P., J.L., J.Z., K.O., T.S., P.W. (Piotr Węgrzyn), M.W., M.S. (Maria Sąsiadek) and E.O.; cytogenetic studies: K.K., M.B.-G., M.S. (Marta Smyk), I.P., J.B., M.K., B.W.-K., J.D., M.D. (Marta Deperas), A.Ł., D.G., M.N., D.D., A.K. (Agata Kozar), K.Z. and K.J.-D.; investigation: R.M.; supervision: B.A.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study was retrospective, and patients were diagnosed as part of routine diagnostics.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Submission of the microarray data to databases may be problematic for ethical reasons, but the data are available upon request to the corresponding author in compliance with EU GDPR.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Luo, X.; Zhu, H.; Wang, L.; Xiao, B.; Fan, Y.; Ye, H.; Ying, X.; Qiu, W.; Zhang, H.; Han, L.; et al. Chromosomal microarray analysis in fetuses with high-risk prenatal indications: A retrospective study in China. Taiwan. J. Obstet. Gynecol. 2021, 60, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Ding, Y.; Song, X.; Mao, J.; Liu, M.; Liu, Y.; Huang, C.; Zhang, Q.; Wang, T. Clinical Utility of SNP Array Analysis in Prenatal Diagnosis: A Cohort Study of 5000 Pregnancies. Front. Genet. 2020, 11, 571219. [Google Scholar] [CrossRef] [PubMed]
- Robson, S.C.; Chitty, L.S.; Morris, S.; Verhoef, T.; Ambler, G.; Wellesley, D.G.; Graham, R.; Leader, C.; Fisher, J.; Crolla, J.A. Evaluation of Array Comparative Genomic Hybridisation in prenatal diagnosis of fetal anomalies: A multicentre cohort study with cost analysis and assessment of patient, health professional and commissioner preferences for array comparative genomic hybridisation. Effic. Mech. Eval. 2017, 4, 1–104. [Google Scholar]
- Stosic, M.; Levy, B.; Wapner, R. The Use of Chromosomal Microarray Analysis in Prenatal Diagnosis. Obstet. Gynecol. Clin. N. Am. 2018, 45, 55–68. [Google Scholar] [CrossRef]
- Mademont-Soler, I.; Morales, C.; Soler, A.; Martínez-Crespo, J.M.; Shen, Y.; Margarit, E.; Clusellas, N.; Obón, M.; Wu, B.L.; Sánchez, A. Prenatal diagnosis of chromosomal abnormalities in fetuses with abnormal cardiac ultrasound findings: Evaluation of chromosomal microarray-based analysis. Ultrasound Obstet. Gynecol. 2013, 41, 375–382. [Google Scholar] [CrossRef]
- Committee on Genetics and the Society for Maternal-Fetal Medicine. Committee Opinion No. 682: Microarrays and Next-Generation Sequencing Technology: The Use of Advanced Genetic Diagnostic Tools in Obstetrics and Gynecology. Obstet. Gynecol. 2016, 128, e262–e268. [Google Scholar] [CrossRef]
- Hay, S.B.; Sahoo, T.; Travis, M.K.; Hovanes, K.; Dzidic, N.; Doherty, C.; Strecker, M.N. ACOG and SMFM guidelines for prenatal diagnosis: Is karyotyping really sufficient? Prenat. Diagn. 2018, 38, 184–189. [Google Scholar] [CrossRef]
- Armour, C.M.; Dougan, S.D.; Brock, J.A.; Chari, R.; Chodirker, B.N.; DeBie, I.; Evans, J.A.; Gibson, W.T.; Kolomietz, E.; Nelson, T.N.; et al. Practice guideline: Joint CCMG-SOGC recommendations for the use of chromosomal microarray analysis for prenatal diagnosis and assessment of fetal loss in Canada. J. Med. Genet. 2018, 55, 215–221. [Google Scholar] [CrossRef]
- Silva, M.; de Leeuw, N.; Mann, K.; Schuring-Blom, H.; Morgan, S.; Giardino, D.; Rack, K.; Hastings, R. European guidelines for constitutional cytogenomic analysis. Eur. J. Hum. Genet. 2019, 27, 1–16. [Google Scholar] [CrossRef]
- Song, T.; Wan, S.; Li, Y.; Xu, Y.; Dang, Y.; Zheng, Y.; Li, C.; Zheng, J.; Chen, B.; Zhang, J. Detection of copy number variants using chromosomal microarray analysis for the prenatal diagnosis of congenital heart defects with normal karyotype. J. Clin. Lab. Anal. 2019, 33, e22630. [Google Scholar] [CrossRef]
- Lee, M.Y.; Won, H.S.; Han, Y.J.; Ryu, H.M.; Lee, D.E.; Jeong, B.D. Clinical value of chromosomal microarray analysis in prenatally diagnosed dextro-transposition of the great arteries. J. Matern. Fetal Neonatal Med. 2020, 33, 1480–1485. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.L.; Li, R.; Fu, F.; Pan, M.; Han, J.; Yang, X.; Zhang, Y.L.; Li, F.T.; Liao, C. Chromosome microarray analysis in the investigation of children with congenital heart disease. BMC Pediatr. 2017, 17, 117. [Google Scholar] [CrossRef] [PubMed]
- Ciaccio, C.; Fontana, L.; Milani, D.; Tabano, S.; Miozzo, M.; Esposito, S. Fragile X syndrome: A review of clinical and molecular diagnoses. Ital. J. Pediatr. 2017, 43, 39. [Google Scholar] [CrossRef]
- Wapner, R.J.; Martin, C.L.; Levy, B.; Ballif, B.C.; Eng, C.M.; Zachary, J.M.; Savage, M.; Platt, L.D.; Saltzman, D.; Grobman, W.A.; et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N. Engl. J. Med. 2012, 367, 2175–2184. [Google Scholar] [CrossRef] [PubMed]
- Srebniak, M.I.; Diderich, K.E.; Joosten, M.; Govaerts, L.C.; Knijnenburg, J.; de Vries, F.A.; Boter, M.; Lont, D.; Knapen, M.F.; de Wit, M.C.; et al. Prenatal SNP array testing in 1000 fetuses with ultrasound anomalies: Causative, unexpected and susceptibility CNVs. Eur. J. Hum. Genet. 2016, 24, 645–651. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Rosenfeld, J.A.; Dabell, M.P.; Coppinger, J.; Bandholz, A.M.; Ellison, J.W.; Ravnan, J.B.; Torchia, B.S.; Ballif, B.C.; Fisher, A.J. Detection rates of clinically significant genomic alterations by microarray analysis for specific anomalies detected by ultrasound. Prenat. Diagn. 2012, 32, 986–995. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, M.; Chen, L.; Huang, H.; Xu, L. Prenatal diagnosis of BACs-on-Beads assay in 1520 cases from Fujian Province, China. Mol. Genet. Genom. Med. 2020, 8, e1446. [Google Scholar] [CrossRef]
- Breman, A.; Pursley, A.N.; Hixson, P.; Bi, W.; Ward, P.; Bacino, C.A.; Shaw, C.; Lupski, J.R.; Beaudet, A.; Patel, A.; et al. Prenatal chromosomal microarray analysis in a diagnostic laboratory; experience with >1000 cases and review of the literature. Prenat. Diagn. 2012, 32, 351–361. [Google Scholar] [CrossRef]
- Chai, H.; DiAdamo, A.; Grommisch, B.; Xu, F.; Zhou, Q.; Wen, J.; Mahoney, M.; Bale, A.; McGrath, J.; Spencer-Manzon, M.; et al. A Retrospective Analysis of 10-Year Data Assessed the Diagnostic Accuracy and Efficacy of Cytogenomic Abnormalities in Current Prenatal and Pediatric Settings. Front. Genet. 2019, 10, 1162. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Dabell, M.P.; Fisher, A.J.; Coppinger, J.; Bandholz, A.M.; Ellison, J.W.; Ravnan, J.B.; Torchia, B.S.; Ballif, B.C.; Rosenfeld, J.A. Experience with microarray-based comparative genomic hybridization for prenatal diagnosis in over 5000 pregnancies. Prenat. Diagn. 2012, 32, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Lovrecic, L.; Remec, Z.I.; Volk, M.; Rudolf, G.; Writzl, K.; Peterlin, B. Clinical utility of array comparative genomic hybridisation in prenatal setting. BMC Med. Genet. 2016, 17, 81. [Google Scholar] [CrossRef] [PubMed]
- De Wit, M.C.; Srebniak, M.I.; Govaerts, L.C.; Van Opstal, D.; Galjaard, R.J.; Go, A.T. Additional value of prenatal genomic array testing in fetuses with isolated structural ultrasound abnormalities and a normal karyotype: A systematic review of the literature. Ultrasound Obstet. Gynecol. 2014, 43, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Sagi-Dain, L.; Maya, I.; Reches, A.; Frumkin, A.; Grinshpun-Cohen, J.; Segel, R.; Manor, E.; Khayat, M.; Tenne, T.; Banne, E.; et al. Chromosomal Microarray Analysis Results From Pregnancies With Various Ultrasonographic Anomalies. Obstet. Gynecol. 2018, 132, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Hureaux, M.; Guterman, S.; Hervé, B.; Till, M.; Jaillard, S.; Redon, S.; Valduga, M.; Coutton, C.; Missirian, C.; Prieur, F.; et al. Chromosomal microarray analysis in fetuses with an isolated congenital heart defect: A retrospective, nationwide, multicenter study in France. Prenat. Diagn. 2019, 39, 464–470. [Google Scholar] [CrossRef]
- Li, S.; Han, X.; Wang, Y.; Chen, S.; Niu, J.; Qian, Z.; Li, P.; Jin, L.; Xu, C. Chromosomal microarray analysis in fetuses with congenital anomalies of the kidney and urinary tract: A prospective cohort study and meta-analysis. Prenat. Diagn. 2019, 39, 165–174. [Google Scholar] [CrossRef]
- Hu, T.; Zhang, Z.; Wang, J.; Li, Q.; Zhu, H.; Lai, Y.; Wang, H.; Liu, S. Prenatal diagnosis of chromosomal aberrations by chromosomal microarray analysis in fetuses with ultrasound anomalies in the urinary system. Prenat. Diagn. 2019, 39, 1096–1106. [Google Scholar] [CrossRef]
- Bernier, R.; Steinman, K.J.; Reilly, B.; Wallace, A.S.; Sherr, E.H.; Pojman, N.; Mefford, H.C.; Gerdts, J.; Earl, R.; Hanson, E.; et al. Simons VIP consortium. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet. Med. 2016, 18, 341–349. [Google Scholar] [CrossRef]
- Upadhyai, P.; Amiri, E.F.; Guleria, V.S.; Bielas, S.L.; Girisha, K.M.; Shukla, A. Recurrent 1q21.1 deletion syndrome: Report on variable expression, nonpenetrance and review of literature. Clin. Dysmorphol. 2020, 29, 127–131. [Google Scholar] [CrossRef]
- Egloff, M.; Hervé, B.; Quibel, T.; Jaillard, S.; Le Bouar, G.; Uguen, K.; Saliou, A.H.; Valduga, M.; Perdriolle, E.; Coutton, C.; et al. Diagnostic yield of chromosomal microarray analysis in fetuses with isolated increased nuchal translucency: A French multicenter study. Ultrasound Obstet. Gynecol. 2018, 52, 715–721. [Google Scholar] [CrossRef]
- Chen, C.P.; Chang, S.Y.; Lin, C.J.; Chern, S.R.; Wu, P.S.; Chen, S.W.; Lai, S.T.; Chuang, T.Y.; Chen, W.L.; Yang, C.W.; et al. Prenatal diagnosis of a familial 5p14.3-p14.1 deletion encompassing CDH18, CDH12, PMCHL1, PRDM9 and CDH10 in a fetus with congenital heart disease on prenatal ultrasound. Taiwan. J. Obstet. Gynecol. 2018, 57, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Faivre, L.; Viot, G.; Prieur, M.; Turleau, C.; Gosset, P.; Romana, S.; Munnich, A.; Vekemans, M.; Cormier-Daire, V. Apparent Sotos syndrome (cerebral gigantism) in a child with trisomy 20p11.2-p12.1 mosaicism. Am. J. Med. Genet. 2000, 91, 273–276. [Google Scholar] [CrossRef]
- Li, R.; Fu, F.; Zhang, Y.L.; Li, D.Z.; Liao, C. Prenatal diagnosis of 17q12 duplication and deletion syndrome in two fetuses with congenital anomalies. Taiwan. J. Obstet. Gynecol. 2014, 53, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Nagamani, S.C.; Erez, A.; Shen, J. Clinical spectrum associated with recurrent genomic rearrangements in chromosome 17q12. Eur. J. Hum. Genet. 2010, 18, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, P.; Sen, P.; Bhatt, S.S.; Storer, M.; Xia, Z.; Bejjani, B.A.; Ou, Z.; Wiszniewska, J.; Driscoll, D.J.; Maisenbacher, M.K.; et al. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am. J. Hum. Genet. 2009, 84, 780–791, Erratum in Am. J. Hum. Genet. 2009, 85, 537. [Google Scholar] [CrossRef]
- Yu, S.; Shao, L.; Kilbride, H.; Zwick, D.L. Haploinsufficiencies of FOXF1 and FOXC2 genes associated with lethal alveolar capillary dysplasia and congenital heart disease. Am. J. Med. Genet. A 2010, 152A, 1257–1262. [Google Scholar] [CrossRef]
- Zufferey, F.; Martinet, D.; Osterheld, M.C.; Niel-Bütschi, F.; Giannoni, E.; Schmutz, N.B.; Xia, Z.; Beckmann, J.S.; Shaw-Smith, C.; Stankiewicz, P.; et al. 16q24.1 microdeletion in a premature newborn: Usefulness of array-based comparative genomic hybridization in persistent pulmonary hypertension of the newborn. Pediatr. Crit. Care Med. 2011, 12, e427–e432. [Google Scholar] [CrossRef]
- Dimitrov, B.I.; de Ravel, T.; Van Driessche, J.; de Die-Smulders, C.; Toutain, A.; Vermeesch, J.R.; Fryns, J.P.; Devriendt, K.; Debeer, P. Distal limb deficiencies, micrognathia syndrome, and syndromic forms of split hand foot malformation (SHFM) are caused by chromosome 10q genomic rearrangements. J. Med. Genet. 2010, 47, 103–111. [Google Scholar] [CrossRef]
- Bauters, M.; Frints, S.G.; Van Esch, H.; Spruijt, L.; Baldewijns, M.M.; de Die-Smulders, C.E.; Fryns, J.P.; Marynen, P.; Froyen, G. Evidence for increased SOX3 dosage as a risk factor for X-linked hypopituitarism and neural tube defects. Am. J. Med. Genet. A 2014, 164A, 1947–1952. [Google Scholar] [CrossRef]
- Hureaux, M.; Ben Miled, S.; Chatron, N.; Coussement, A.; Bessières, B.; Egloff, M.; Mechler, C.; Stirnemann, J.; Tsatsaris, V.; Barcia, G.; et al. SOX3 duplication: A genetic cause to investigate in fetuses with neural tube defects. Prenat. Diagn. 2019, 39, 1026–1034. [Google Scholar] [CrossRef]
- Wang, J.C.; Radcliff, J.; Coe, S.J.; Mahon, L.W. Effects of platforms, size filter cutoffs, and targeted regions of cytogenomic microarray on detection of copy number variants and uniparental disomy in prenatal diagnosis: Results from 5026 pregnancies. Prenat. Diagn. 2019, 39, 137–156. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).