1. Introduction
Precision medicine aims to provide individually tailored cancer treatment by considering an individual’s genetic makeup, genomic makeup and clinical information. Emerging Next Generation Sequencing (NGS) techniques and large-scale cancer screening data helps in achieving this goal [
1,
2]. Databases such as the Cancer Cell Line Encyclopedia (CCLE) [
2] provides public access to genomic data over 1000 cancer cell lines by RNA sequencing (RNA-seq; 1019 cell lines), whole-exome sequencing (326 cell lines), whole-genome sequencing (329 cell lines), and reverse-phase protein array (RPPA; 899 cell lines). The Cancer Therapeutics Response Portal (CTRP;
http://portals.broadinstitute.org/ctrp/, accessed on 1 June 2019) [
3,
4] quantitatively measured the sensitivity of 481 small-molecule probes and drugs. An important step of this process is to use cancer cell line models to simulate mixed tissue and predict his/her drug response [
5].
Accuracy in drug response prediction is of utmost importance in this regard. Over the years various models have been developed for this purpose [
6,
7,
8,
9]. Contemporary models are based on a varied range of techniques such as regression methods, Bayesian inference methods, matrix factorization methods and deep learning methods. Some of these methods use only gene expression data while some use combination of other omics data such as mutation, copy number variation, methylation and so on for response prediction. Prediction models by gene expression profiles show the best performance in all kinds of omics analysis [
10]. The detailed analysis and comparison of different methods can be found in a paper by Chen and Zhang [
11].
These models, however, only focus on a single model [
12] trained over large datasets. Without recognition of the weekly predictive local embedding data contribution, will cause them to make incorrect decisions in facing to outliers and errors. Outliers’ and errors’ in turn, causes these models to be incapable of capturing the dataset’s true variance, thus distorting model complexity [
13]. To deal with high-dimensional genomics data, a promising strategy is to find an effective low-dimensional subspace of the original data and cluster samples in the reduced subspace [
14], and then do a localize regression.
However, it is hard to identify the homology features which one contribution to drug response prediction for increasingly heterogeneous datasets comprised of multi-omics data collected from overlapping latent low-dimensional subpopulations. Principal Component Analysis (PCA) can generate statistically uncorrelated principal components (PC) while retaining as much as possible of the variation present in the original data set. PCA has been used previously to delineate homogenous regions by PCs regression and applying in all kinds of fields such as temperature [
15], hydrology [
16], risk [
17], and animal health [
18], but have not applied in drug response prediction.
Machine learning nonlinear regression Support Vector Regression (SVR) was first introduced by Vapnik [
19] and has been a highly effective and suitable method for regression [
20]; the problem of regression is to find a function surface in high dimension that approximates mapping from an input domain (low dimension) to real values based on a training sample. However, most existing SVR learning algorithms are limited to the parameters selection and slow learning for high-throughput features and large samples [
21,
22].
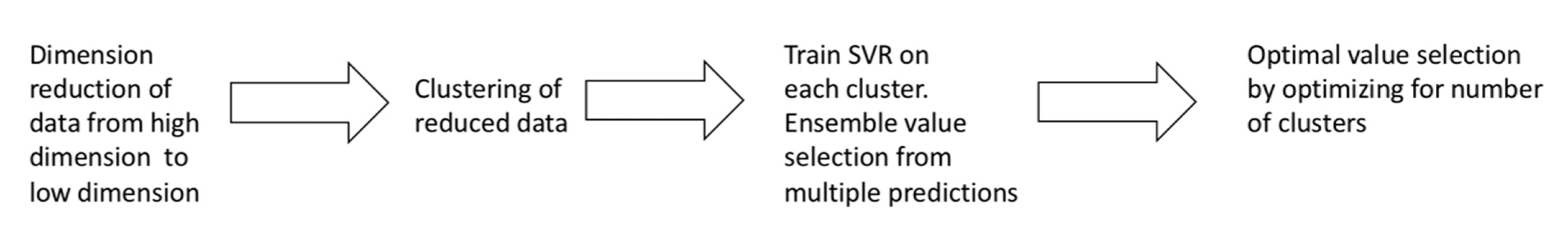
To address these challenges, a novelty k-means Ensemble Support Vector Regression (kESVR) is developed to predict each drug response values for single patient based on cell-line gene expression data. kESVR’s origin stems from previous work interval SVR [
22], where we separated a global nonlinear SVR predictor into interval subspaces and ran a SVR in each interval subspace. However, kESVR is different to the interval SVR in that it constructs a local SVR regression in each principal component embedding subspaces, where the K-means algorithm clusters these homogenous regions and then predict associated drug response. The prediction process function by repeatedly running the local SVR learning algorithm on various distributions’ clustering over the whole training data, then comparing the regression value produced by these local SVR learners to obtain a single regression value with the best performance in accuracy and output. The last step used a boosting strategy as literature [
23] mentioned to obtain the high accuracy of any local SVR learning algorithm.
The kESVR is a blend of supervised and unsupervised learning methods while being an entirely data driven model. It utilizes embedded clustering (PCA and k-means clustering) and local regression (Support Vector Regression) to predict drug response and obtain the global optimal value with the smallest mean squared error (MSE) while overcoming missing data and outliers’ noise. In contrast to classical 17 machine learning models [
11] that estimate a single, complex model (or only a few complex models), our results show that kESVR model with PCA-compressed features make the training and validation more efficient in both model accuracy and computational costs, outperforming other machine learning methods in generalization performance.
4. Discussion
kESVR is a data driven model. That is, it does not require any external input of parameter values. kESVR calculates all of its parameters (, , parameters of individual SVRs) from the input data directly. This makes this model highly robust.
kESVR uses PCA in its first step. This leads to the creation of the reduced dataset
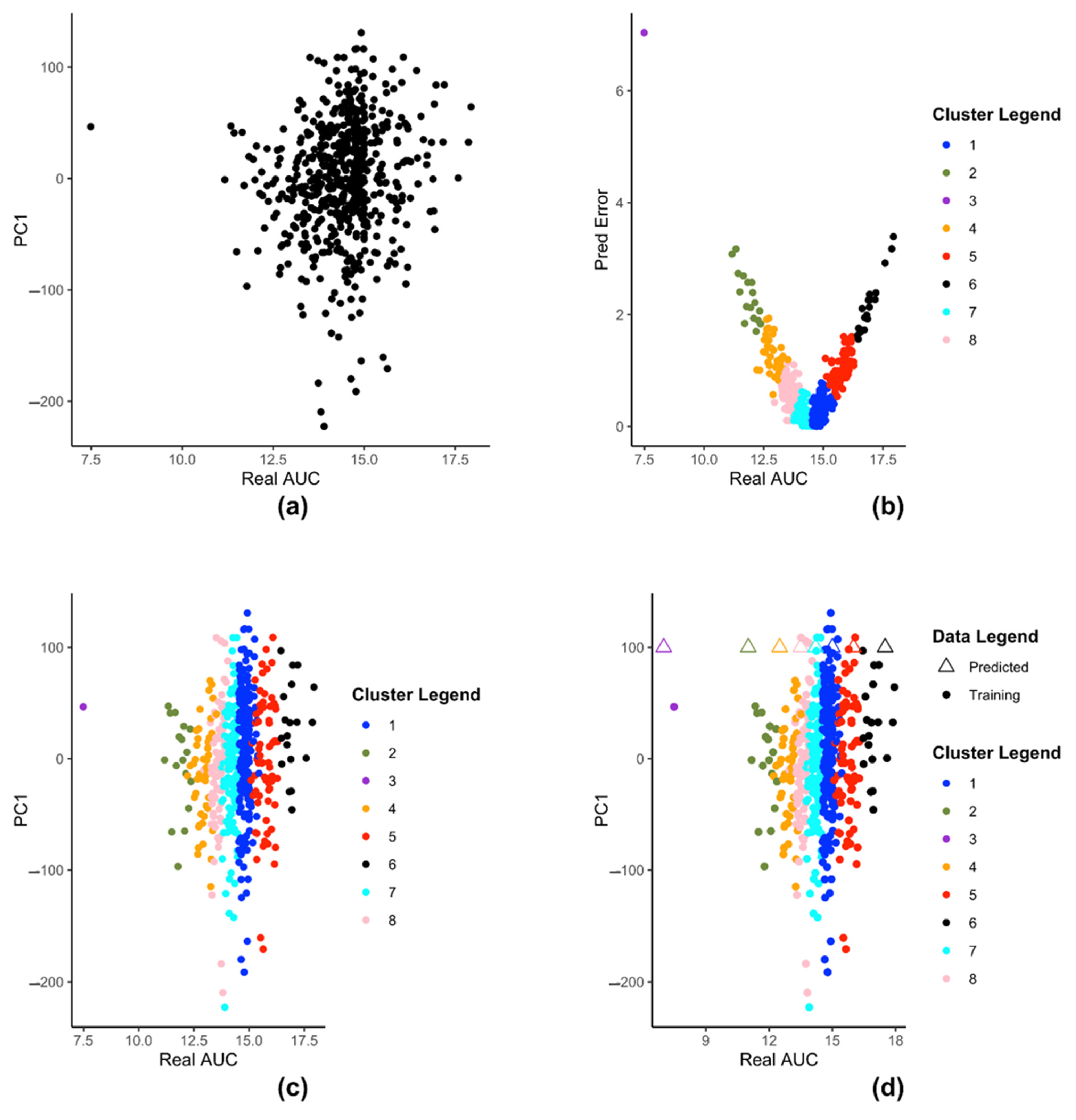
(Equation (2)). At this step, there are several principal components to choose from, for our model creation. The decision to use the first principal component for this step stems from the fact that the first principal component retains the maximum percentage of variation in the reduced data set. Graphically this means, it gives a better visualization/separation (
Figure 2a) of the distinct data points on the 2-D plot that is used in the subsequent steps.
The embedded clustering step employs K-means clustering on the dataset
(Equation (4)) in order to cluster the cell-lines into groups. We use K-means for two reasons, firstly we plan to cluster cell-lines that produce similar prediction errors and secondly since
is a set of 2-tuple data. These clusters then train separate SVRs to reduce the prediction errors. Typically, K-means method itself is prone to producing different clusters each time due to the randomized nature of its initiation. However, in this particular scenario, we observe that even after running multiple times, with this data K-means always produce the same set of centroids. Traditionally, optimal value of
is determined by using metrics such as Silhouette value [
34] or Calinski-Harabasz index [
35]. However, such methods are not applicable in case of kESVR as our main objective is not to determine how well the data-points are clustered. Clustering is an intermediate step, that plays an important role in the performance of kESVR. That is, the performance metric MSE is dependent on the choice of
. In that respect the choice of value of
, is data driven. As our final objective is to minimize the value of MSE (Equation (8)), kESVR loops through different values of
, and selects the optimal one that gives the lowest MSE value.
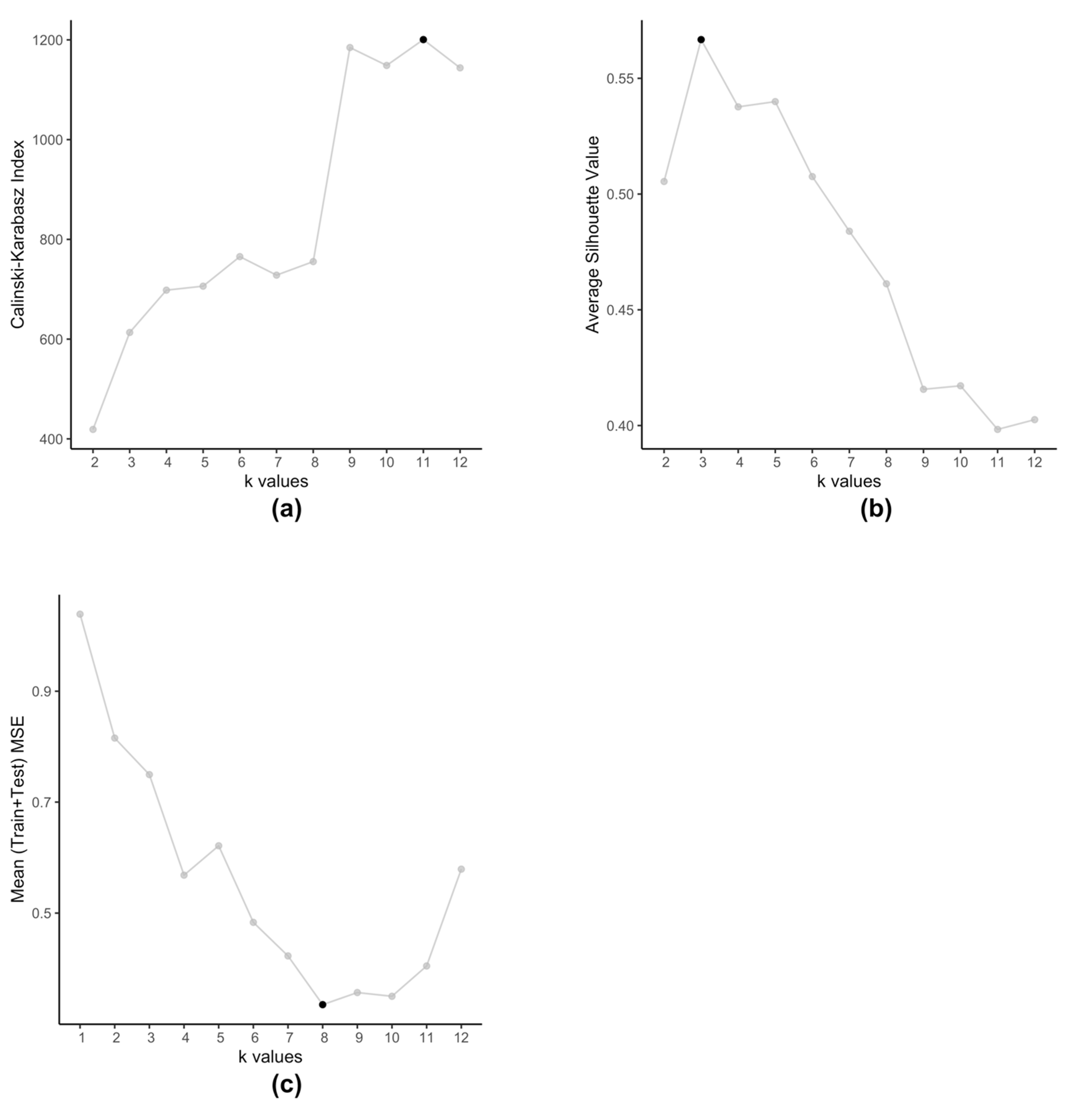
Figure 5 illustrates how the optimal value of
varies according to metric used for evaluation. Here we use the model for drug zebularine.
Figure 5a uses the Calinski-Harabasz index,
Figure 5b uses the average Silhouette value and
Figure 5c uses the average (Training+Testing) MSE values to get optimal
for drug zebularine. Accordingly, the suggested optimal value of
turn out to be 11, 3 and 8 respectively. From
Table 6, we known that only
gives the lowest average MSE value (best kESVR performance). So
is selected as the optimal value.
Computation of neighbors for any data-point using
(Equation (6)) is dependent on the value of
. We use the clustering information of the reduced data
to calculate the best value of
. Our model kESVR is data-driven. Being an ensemble method, its performance is dependent on the size/volume of the training data being used on each individual SVR. That is, some clusters in
can be denser than others. Depending on the value of
, that density of a cluster can influence the overall performance of kESVR.
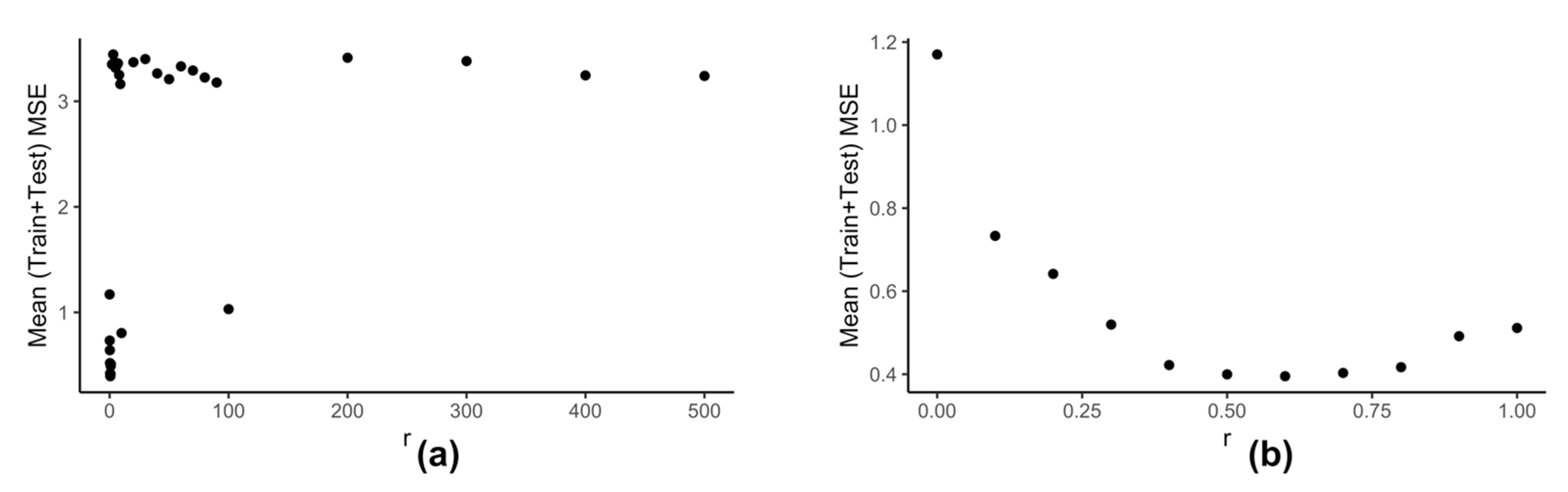
Figure 6 shows how the performance of kESVR varies with
for drug zebularine.
We can see from
Figure 6a that as the value of
increases the performance of our model deteriorates. This observation is intuitive: if a cluster is very dense (more training instances) and the value of
is sufficiently large, then that cluster can end up dominating the prediction value selection process and hence the overall performance. The best performance is given when
is small. This is shown is
Figure 6b. Keeping these things in mind we calculate
as follows: We calculate the x-axis distance (AUC values) between the two farthest points for each cluster. We sort these distances in ascending order and select
to be smallest value. This is done in order to be fair in comparing the number of neighbors for each cluster. If in a particular prediction instance, a value of
results in zero neighbors in all clusters, we simply select the next higher value in the sorted list, and redo the neighbor selection process for that instance.
kESVR uses an ensemble approach to return the final prediction value from potential predicted values. It uses a maximum score (Equation (7)) approach wherein the potential predicted value with the highest score is selected as a final prediction value. We empirically test a total of five different approaches before settling on the score one. The four other approaches are:
(i) select the prediction value whose data-point has the maximum average Spearman correlation value with all cell-lines in the training set of its parent SVR.
(ii) return a weighted average of the prediction values using the scores of the corresponding data-points as weights.
(iii) select the prediction value whose data-point has the maximum number of neighbors (using ).
(iv) return a weighted average of the prediction data-points using the number of neighbors (from ) of the corresponding data-points as weights.
Our experimental comparisons show that the maximum score approach gives the best accuracy performance i.e. lowest MSE values.
Lastly feature selection plays an important role in the performance of our model kESVR. Intuitively, features indicate those genes that play a crucial role in the functioning of a drug. Expression (high/low) of these feature genes, affects how a cell-line/patient responds to the drug. We use genes that are known to be target genes for the drugs (available from public databases) as our feature genes. This contributes to the improved performance of kESVR over other drug response prediction methods.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}