
ATM: Main Features, Signaling Pathways, and Its Diverse Roles in DNA Damage Response, Tumor Suppression, and Cancer Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
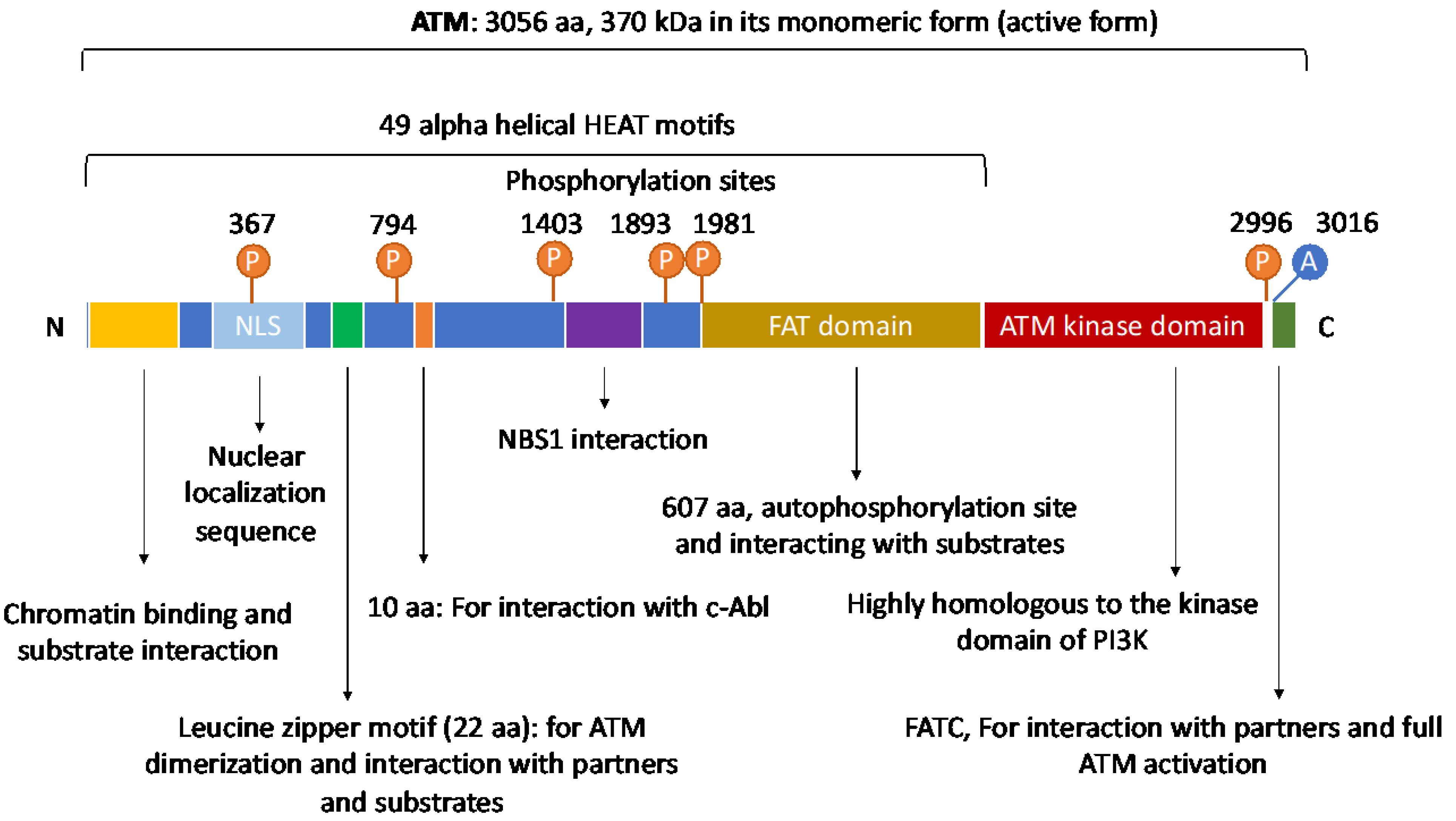
2. The Structure and Domain Mapping of ATM
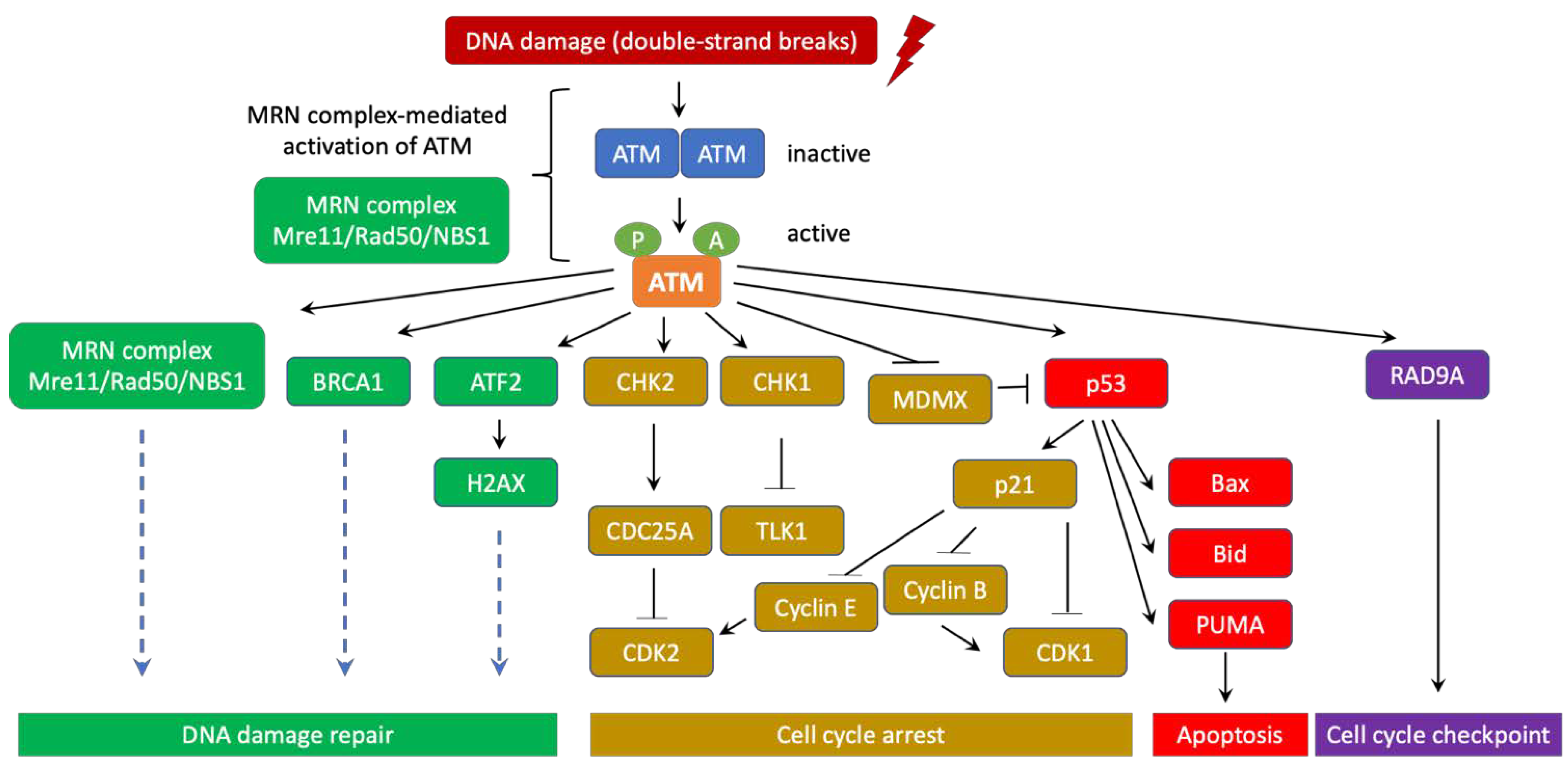
3. ATM’s role in DNA Damage Response and Ataxia Telangiectasia
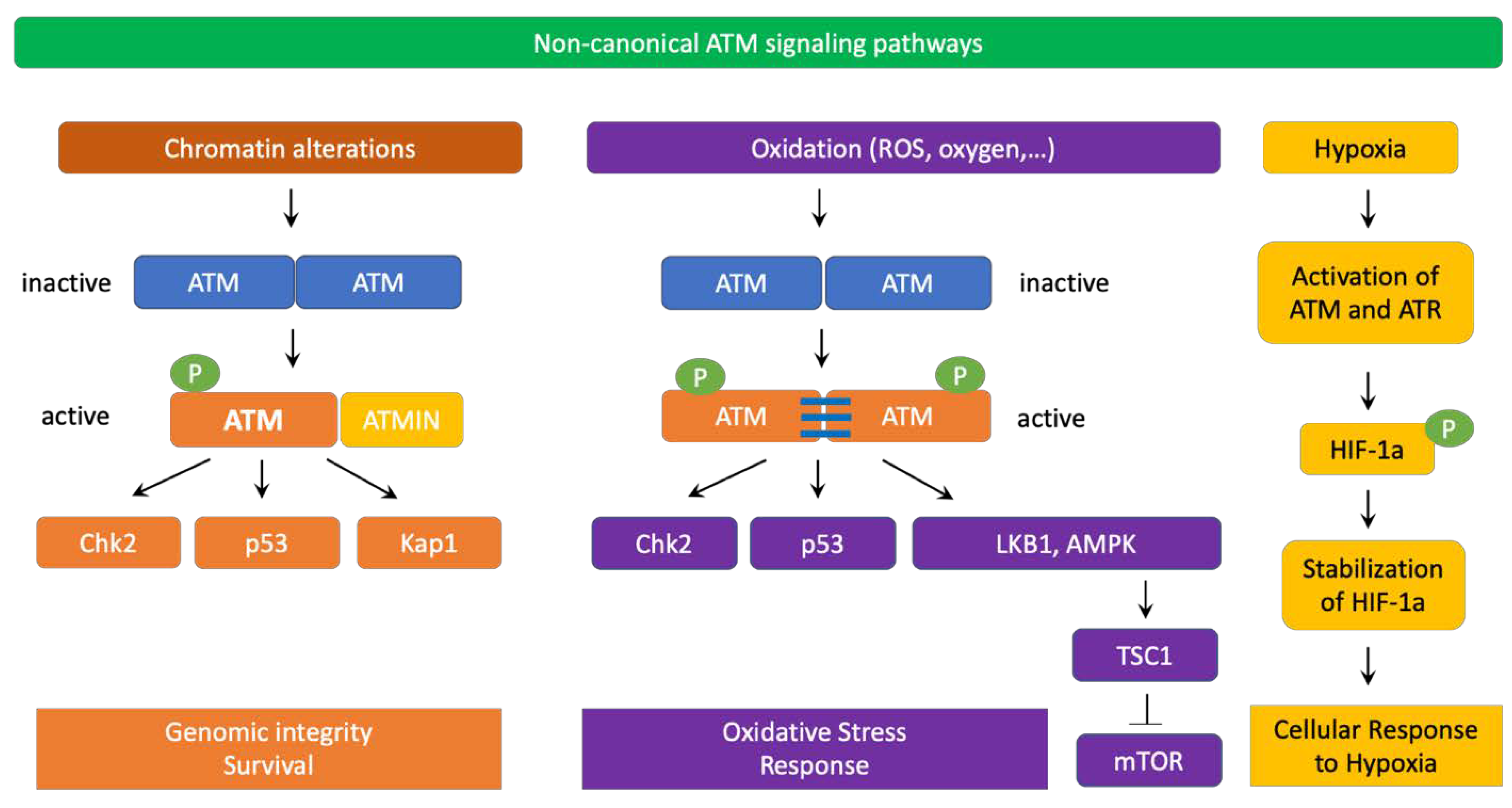
4. ATM Signaling Pathways
5. ATM Signaling in Cancer
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; A Tagle, D.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair 2004, 3, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, C.A.; Cortez, D. Common mechanisms of PIKK regulation. DNA Repair 2009, 8, 1004–1008. [Google Scholar] [CrossRef]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef]
- Bosotti, R.; Isacchi, A.; Sonnhammer, E.L. FAT: A novel domain in PIK-related kinases. Trends Biochem. Sci. 2000, 25, 225–227. [Google Scholar] [CrossRef]
- Jiang, X.; Sun, Y.; Chen, S.; Roy, K.; Price, B.D. The FATC Domains of PIKK Proteins Are Functionally Equivalent and Participate in the Tip60-dependent Activation of DNA-PKcs and ATM. J. Biol. Chem. 2006, 281, 15741–15746. [Google Scholar] [CrossRef]
- Fernandes, N.; Sun, Y.; Chen, S.; Paul, P.; Shaw, R.J.; Cantley, L.C.; Price, B.D. DNA Damage-induced Association of ATM with Its Target Proteins Requires a Protein Interaction Domain in the N Terminus of ATM. J. Biol. Chem. 2005, 280, 15158–15164. [Google Scholar] [CrossRef]
- Andrade, M.A.; Petosa, C.; I O’Donoghue, S.; Müller, C.W.; Bork, P. Comparison of ARM and HEAT protein repeats. J. Mol. Biol. 2001, 309, 1–18. [Google Scholar] [CrossRef]
- Piazza, I.; Rutkowska, A.; Ori, A.; Walczak, M.; Metz, J.; Pelechano, V.; Beck, M.; Haering, C.H. Association of condensin with chromosomes depends on DNA binding by its HEAT-repeat subunits. Nat. Struct. Mol. Biol. 2014, 21, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Rubinson, E.H.; Gowda, A.S.P.; Spratt, T.E.; Gold, B.; Eichman, B.F. An Unprecedented Nucleic Acid Capture Mechanism for Excision of DNA Damage. Nature 2010, 468, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Dupré, A.; Boyer-Chatenet, L.; Gautier, J. Two-step activation of ATM by DNA and the Mre11–Rad50–Nbs1 complex. Nat. Struct. Mol. Biol. 2006, 13, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef]
- Kanu, N.; Behrens, A. ATMIN defines an NBS1-independent pathway of ATM signalling. EMBO J. 2007, 26, 2933–2941. [Google Scholar] [CrossRef] [PubMed]
- Bowen, C.; Ju, J.-H.; Lee, J.-H.; Paull, T.T.; Gelmann, E.P. Functional activation of ATM by the prostate cancer suppressor NKX3.1. Cell Rep. 2013, 4, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F. The Mre11 complex and ATM: A two-way functional interaction in recognising and signaling DNA double strand breaks. DNA Repair 2004, 3, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Scott, S.; Gueven, N.; Kozlov, S.; Peng, C.; Chen, P. Functional consequences of sequence alterations in the ATM gene. DNA Repair 2004, 3, 1197–1205. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Direct Activation of the ATM Protein Kinase by the Mre11/Rad50/Nbs1 Complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Berkovich, E.; Monnat, R.J., Jr.; Kastan, M.B. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 2007, 9, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.-Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. ATM-Dependent and -Independent Dynamics of the Nuclear Phosphoproteome After DNA Damage. Sci. Signal. 2010, 3, rs3. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Swift, M.; Morrell, D.; Cromartie, E.; Chamberlin, A.R.; Skolnick, M.H.; Bishop, D.T. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am. J. Hum. Genet. 1986, 39, 573–583. [Google Scholar] [PubMed]
- Álvarez-Quilón, A.; Serrano-Benítez, A.; Lieberman, J.A.; Quintero, C.; Sánchez-Gutiérrez, D.; Escudero, L.M.; Cortés-Ledesma, F. ATM specifically mediates repair of double-strand breaks with blocked DNA ends. Nat. Commun. 2014, 5, 3347. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J. ATM and ataxia telangiectasia. EMBO Rep. 2004, 5, 772–776. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Gatti, R.A.; Berkel, I.; Boder, E.; Braedt, G.; Charmley, P.; Concannon, P.; Ersoy, F.; Foroud, T.; Jaspers, N.G.J.; Lange, K.; et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22–23. Nature 1988, 336, 577–580. [Google Scholar] [CrossRef]
- Taylor, A.M.; Byrd, P.J. Molecular pathology of ataxia telangiectasia. J. Clin. Pathol. 2005, 58, 1009–1015. [Google Scholar] [CrossRef]
- Chun, H.H.; A Gatti, R. Ataxia–telangiectasia, an evolving phenotype. DNA Repair 2004, 3, 1187–1196. [Google Scholar] [CrossRef]
- Renwick, A.; The Breast Cancer Susceptibility Collaboration (UK); Thompson, D.; Seal, S.; Kelly, P.; Chagtai, T.; Ahmed, M.; North, B.; Jayatilake, H.; Barfoot, R.; et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 873–875. [Google Scholar] [CrossRef] [PubMed]
- Cremona, C.A.; Behrens, A. ATM signalling and cancer. Oncogene 2014, 33, 3351–3360. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, X.; Zhang, L.; Wu, C.-Y.; Rezaeian, A.H.; Chan, C.-H.; Li, J.-M.; Wang, J.; Gao, Y.; Han, F.; et al. Skp2 E3 Ligase Integrates ATM Activation and Homologous Recombination Repair by Ubiquitinating NBS1. Mol. Cell 2012, 46, 351–361. [Google Scholar] [CrossRef] [PubMed]
- So, S.; Davis, A.J.; Chen, D.J. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J. Cell Biol. 2009, 187, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef]
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The RIDDLE Syndrome Protein Mediates a Ubiquitin-Dependent Signaling Cascade at Sites of DNA Damage. Cell 2009, 136, 420–434. [Google Scholar] [CrossRef]
- Kolas, N.K.; Chapman, R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-Damage Response by the RNF8 Ubiquitin Ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef]
- Mattiroli, F.; Vissers, J.H.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.; Sixma, T.K. RNF168 Ubiquitinates K13-15 on H2A/H2AX to Drive DNA Damage Signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef]
- Gudjonsson, T.; Altmeyer, M.; Savic, V.; Toledo, L.; Dinant, C.; Grøfte, M.; Bartkova, J.; Poulsen, M.; Oka, Y.; Bekker-Jensen, S.; et al. TRIP12 and UBR5 Suppress Spreading of Chromatin Ubiquitylation at Damaged Chromosomes. Cell 2012, 150, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, E.; Mock, C.S.; Bhan, I.; Loewer, A.; Lahav, G. Recurrent Initiation: A Mechanism for Triggering p53 Pulses in Response to DNA Damage. Mol. Cell 2008, 30, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006, 8, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Noon, A.T.; Jeggo, P.A. The impact of heterochromatin on DSB repair. Biochem. Soc. Trans. 2009, 37, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.-Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-Dependent Monoubiquitylation of Histone H2B for Timely Repair of DNA Double-Strand Breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Penicud, K.; Behrens, A. DMAP1 is an essential regulator of ATM activity and function. Oncogene 2014, 33, 525–531. [Google Scholar] [CrossRef]
- Kim, Y.-C.; Gerlitz, G.; Furusawa, T.; Catez, F.; Nussenzweig, A.; Oh, K.-S.; Kraemer, K.H.; Shiloh, Y.; Bustin, M. Activation of ATM depends on chromatin interactions occurring before induction of DNA damage. Nat. Cell Biol. 2008, 11, 92–96. [Google Scholar] [CrossRef]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-Dependent Chromatin Changes Silence Transcription In cis to DNA Double-Strand Breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef]
- Ginjala, V.; Nacerddine, K.; Kulkarni, A.; Oza, J.; Hill, S.J.; Yao, M.; Citterio, E.; Van Lohuizen, M.; Ganesan, S. BMI1 Is Recruited to DNA Breaks and Contributes to DNA Damage-Induced H2A Ubiquitination and Repair. Mol. Cell. Biol. 2011, 31, 1972–1982. [Google Scholar] [CrossRef]
- Ismail, I.H.; Andrin, C.; McDonald, D.; Hendzel, M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol. 2010, 191, 45–60. [Google Scholar] [CrossRef]
- Loizou, J.I.; Sancho, R.; Kanu, N.; Bolland, D.J.; Yang, F.; Rada, C.; Corcoran, A.E.; Behrens, A. ATMIN Is Required for Maintenance of Genomic Stability and Suppression of B Cell Lymphoma. Cancer Cell 2011, 19, 587–600. [Google Scholar] [CrossRef]
- Zhang, T.; Penicud, K.; Bruhn, C.; Loizou, J.I.; Kanu, N.; Wang, Z.-Q.; Behrens, A. Competition between NBS1 and ATMIN Controls ATM Signaling Pathway Choice. Cell Rep. 2012, 2, 1498–1504. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kanu, N.; Penicud, K.; Hristova, M.; Wong, B.; Irvine, E.; Plattner, F.; Raivich, G.; Behrens, A. The ATM Cofactor ATMIN Protects against Oxidative Stress and Accumulation of DNA Damage in the Aging Brain. J. Biol. Chem. 2010, 285, 38534–38542. [Google Scholar] [CrossRef]
- Burdak-Rothkamm, S.; Rothkamm, K.; Prise, K.M. ATM Acts Downstream of ATR in the DNA Damage Response Signaling of Bystander Cells. Cancer Res. 2008, 68, 7059–7065. [Google Scholar] [CrossRef] [PubMed]
- Tresini, M.; Warmerdam, D.O.; Kolovos, P.; Snijder, L.; Vrouwe, M.G.; Demmers, J.A.; van IJcken, W.F.J.; Grosveld, F.G.; Medema, R.H.; Hoeijmakers, J.H.; et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 2015, 523, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM Activation by Oxidative Stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef]
- Alexander, A.; Cai, S.-L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.-L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef]
- Cam, H.; Easton, J.B.; High, A.; Houghton, P.J. mTORC1 Signaling under Hypoxic Conditions Is Controlled by ATM-Dependent Phosphorylation of HIF-1α. Mol. Cell 2010, 40, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Hammond, E.M.; Kaufmann, M.R.; Giaccia, A.J. Oxygen sensing and the DNA-damage response. Curr. Opin. Cell Biol. 2007, 19, 680–684. [Google Scholar] [CrossRef]
- Pires, I.M.; Bencokova, Z.; Milani, M.; Folkes, L.K.; Li, J.-L.; Stratford, M.R.; Harris, A.L.; Hammond, E.M. Effects of Acute versus Chronic Hypoxia on DNA Damage Responses and Genomic Instability. Cancer Res. 2010, 70, 925–935. [Google Scholar] [CrossRef]
- Bencokova, Z.; Kaufmann, M.R.; Pires, I.M.; Lecane, P.S.; Giaccia, A.J.; Hammond, E.M. ATM Activation and Signaling under Hypoxic Conditions. Mol. Cell. Biol. 2008, 29, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Olcina, M.M.; Foskolou, I.P.; Anbalagan, S.; Senra, J.M.; Pires, I.M.; Jiang, Y.; Ryan, A.J.; Hammond, E.M. Replication Stress and Chromatin Context Link ATM Activation to a Role in DNA Replication. Mol. Cell 2013, 52, 758–766. [Google Scholar] [CrossRef]
- Rezaeian, A.-H.; Li, C.-F.; Wu, C.-Y.; Zhang, X.; DeLacerda, J.; You, M.J.; Han, F.; Cai, Z.; Jeong, Y.S.; Jin, G.; et al. A hypoxia-responsive TRAF6–ATM–H2AX signalling axis promotes HIF1α activation, tumorigenesis and metastasis. Nat. Cell Biol. 2017, 19, 38–51. [Google Scholar] [CrossRef]
- Stagni, V.; Ferri, A.; Cirotti, C.; Barilà, D. ATM Kinase-Dependent Regulation of Autophagy: A Key Player in Senescence? Front. Cell Dev. Biol. 2021, 8, 599048. [Google Scholar] [CrossRef] [PubMed]
- Stagni, V.; Cirotti, C.; Barilà, D. Ataxia-Telangiectasia Mutated Kinase in the Control of Oxidative Stress, Mitochondria, and Autophagy in Cancer: A Maestro with a Large Orchestra. Front. Oncol. 2018, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V. DNA Damage Response and Autophagy: A Meaningful Partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Walker, C.L. Differential localization of ATM is correlated with activation of distinct downstream signaling pathways. Cell Cycle 2010, 9, 3709–3710. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Kim, J.; Walker, C.L. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 2010, 6, 672–673. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, M.; Strappazzon, F.; Arisi, I.; Brandi, R.; D’Onofrio, M.; Sambucci, M.; Manic, G.; Vitale, I.; Barilà, D.; Stagni, V. ATM kinase sustains breast cancer stem-like cells by promoting ATG4C expression and autophagy. Oncotarget 2017, 8, 21692–21709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.L.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.-H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef]
- Tripathi, D.N.; Zhang, J.; Jing, J.; Dere, R.; Walker, C.L. A new role for ATM in selective autophagy of peroxisomes (pexophagy). Autophagy 2016, 12, 711–712. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. ATM: Expanding roles as a chief guardian of genome stability. Exp. Cell Res. 2014, 329, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. Mitochondria at the crossroads of ATM-mediated stress signaling and regulation of reactive oxygen species. Redox Biol. 2020, 32, 101511. [Google Scholar] [CrossRef] [PubMed]
- Liang, N.; He, Q.; Liu, X.; Sun, H. Multifaceted roles of ATM in autophagy: From nonselective autophagy to selective autophagy. Cell Biochem. Funct. 2019, 37, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Valentin-Vega, Y.A.; MacLean, K.H.; Tait-Mulder, J.; Milasta, S.; Steeves, M.; Dorsey, F.C.; Cleveland, J.L.; Green, D.R.; Kastan, M.B. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 2012, 119, 1490–1500. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, S.; Xu, H.; Li, X.; Guan, Y.; Yi, F.; Zhou, T.; Jiang, B.; Bai, N.; Ma, M.; et al. ATM—CHK 2-Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. EMBO J. 2020, 39, e103111. [Google Scholar] [CrossRef]
- Qi, Y.; Qiu, Q.; Gu, X.; Tian, Y.; Zhang, Y. ATM mediates spermidine-induced mitophagy via PINK1 and Parkin regulation in human fibroblasts. Sci. Rep. 2016, 6, 24700. [Google Scholar] [CrossRef]
- Song, L.; Lin, C.; Wu, Z.; Gong, H.; Zeng, Y.; Wu, J.; Li, M.; Li, J. miR-18a Impairs DNA Damage Response through Downregulation of Ataxia Telangiectasia Mutated (ATM) Kinase. PLoS ONE 2011, 6, e25454. [Google Scholar] [CrossRef]
- Rezaeian, A.-H.; Khanbabaei, H.; Calin, G.A. Therapeutic Potential of the miRNA-ATM Axis in the Management of Tumor Radioresistance. Cancer Res. 2019, 80, 139–150. [Google Scholar] [CrossRef]
- Le Guezennec, X.; Bulavin, D.V. WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem. Sci. 2010, 35, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, K.; Coppola, D.; Rawal, B.; Chen, Y.A.; Lawrence, H.R.; Engelman, R.W.; Lawrence, N.J.; Mahajan, N.P. Ack1-mediated Androgen Receptor Phosphorylation Modulates Radiation Resistance in Castration-resistant Prostate Cancer. J. Biol. Chem. 2012, 287, 22112–22122. [Google Scholar] [CrossRef] [PubMed]
- Ripka, S.; Neesse, A.; Riedel, J.; Bug, E.; Aigner, A.; Poulsom, R.; Fulda, S.; Neoptolemos, J.; Greenhalf, W.; Barth, P.; et al. CUX1: Target of Akt signalling and mediator of resistance to apoptosis in pancreatic cancer. Gut 2010, 59, 1101–1110. [Google Scholar] [CrossRef]
- Vadnais, C.; Davoudi, S.; Afshin, M.; Harada, R.; Dudley, R.; Clermont, P.-L.; Drobetsky, E.; Nepveu, A. CUX1 transcription factor is required for optimal ATM/ATR-mediated responses to DNA damage. Nucleic Acids Res. 2012, 40, 4483–4495. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, N.; Xiao, T.Z.; Rosenthal, K.A.; Siddiqui, I.A.; Thiyagarajan, S.; Smart, B.; Meng, Q.; Zuleger, C.L.; Mukhtar, H.; Kenney, S.C.; et al. MAGE-C2 Promotes Growth and Tumorigenicity of Melanoma Cells, Phosphorylation of KAP1, and DNA Damage Repair. J. Investig. Dermatol. 2013, 133, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin Remodeling Underlies the Senescence-Associated Secretory Phenotype of Tumor Stromal Fibroblasts That Supports Cancer Progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2010, 30, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Krüger, A.; Grüning, N.-M.; Wamelink, M.M.; Kerick, M.; Kirpy, A.; Parkhomchuk, D.; Bluemlein, K.; Schweiger, M.-R.; Soldatov, A.; Lehrach, H.; et al. The Pentose Phosphate Pathway Is a Metabolic Redox Sensor and Regulates Transcription During the Antioxidant Response. Antioxid. Redox Signal. 2011, 15, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Krüger, A.; Ralser, M. ATM Is a Redox Sensor Linking Genome Stability and Carbon Metabolism. Sci. Signal. 2011, 4, pe17. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, T.; Takahashi, F.; Chiba, N.; Brachtel, E.; Takahashi, M.; Godin-Heymann, N.; Gross, K.W.; Vivanco, M.D.M.; Wijendran, V.; Shioda, T.; et al. HOXB9, a gene overexpressed in breast cancer, promotes tumorigenicity and lung metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1100–1105. [Google Scholar] [CrossRef]
- Wang, N.; Eckert, K.A.; Zomorrodi, A.R.; Xin, P.; Pan, W.; Shearer, D.A.; Weisz, J.; Maranus, C.D.; Clawson, G.A. Down-Regulation of HtrA1 Activates the Epithelial-Mesenchymal Transition and ATM DNA Damage Response Pathways. PLoS ONE 2012, 7, e39446. [Google Scholar] [CrossRef]
- Chiba, N.; Comaills, V.; Shiotani, B.; Takahashi, F.; Shimada, T.; Tajima, K.; Winokur, D.; Hayashida, T.; Willers, H.; Brachtel, E.; et al. Homeobox B9 induces epithelial-to-mesenchymal transition-associated radioresistance by accelerating DNA damage responses. Proc. Natl. Acad. Sci. USA 2012, 109, 2760–2765. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phan, L.M.; Rezaeian, A.-H. ATM: Main Features, Signaling Pathways, and Its Diverse Roles in DNA Damage Response, Tumor Suppression, and Cancer Development. Genes 2021, 12, 845. https://doi.org/10.3390/genes12060845
Phan LM, Rezaeian A-H. ATM: Main Features, Signaling Pathways, and Its Diverse Roles in DNA Damage Response, Tumor Suppression, and Cancer Development. Genes. 2021; 12(6):845. https://doi.org/10.3390/genes12060845
Chicago/Turabian StylePhan, Liem Minh, and Abdol-Hossein Rezaeian. 2021. "ATM: Main Features, Signaling Pathways, and Its Diverse Roles in DNA Damage Response, Tumor Suppression, and Cancer Development" Genes 12, no. 6: 845. https://doi.org/10.3390/genes12060845
APA StylePhan, L. M., & Rezaeian, A.-H. (2021). ATM: Main Features, Signaling Pathways, and Its Diverse Roles in DNA Damage Response, Tumor Suppression, and Cancer Development. Genes, 12(6), 845. https://doi.org/10.3390/genes12060845