
Whole Genome Sequencing Applied to Pathogen Source Tracking in Food Industry: Key Considerations for Robust Bioinformatics Data Analysis and Reliable Results Interpretation


Abstract
1. Introduction
2. Materials and Methods
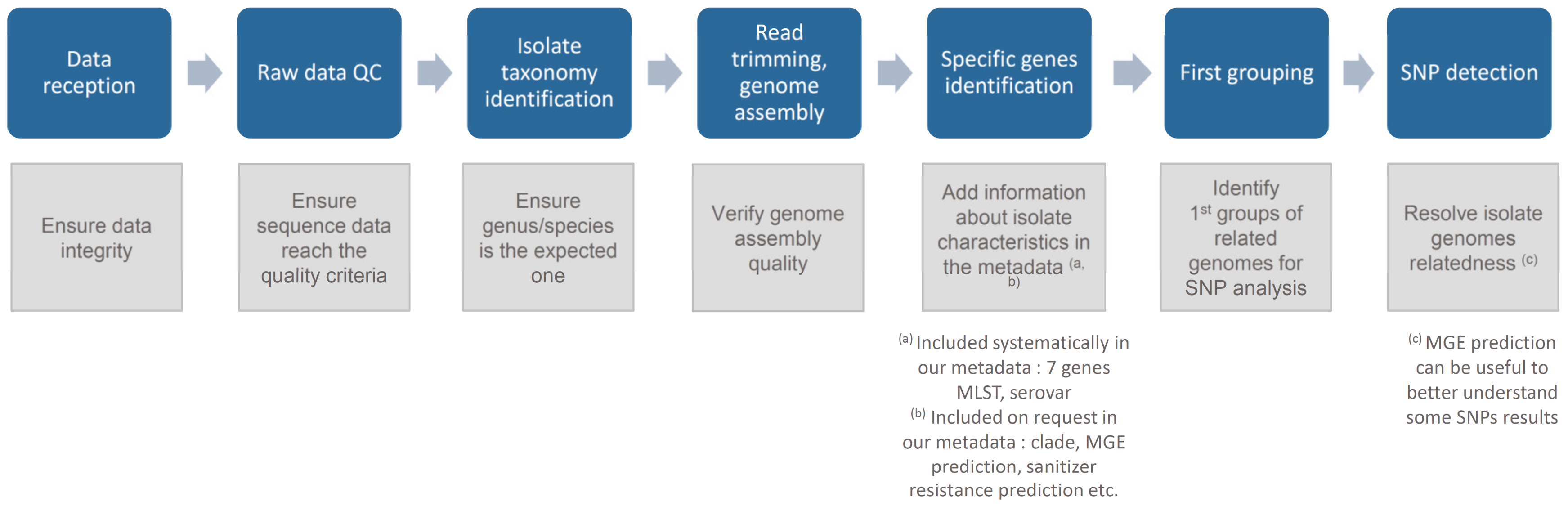
Workflow
3. Metadata
4. Results
5. Discussion
5.1. Turnaround Time
5.2. cg/wgMLST versus SNP
5.3. Standardization and Accreditation
5.4. Internal Genome Database and Metadata Management
5.5. Analysis Reproducibility and Repeatability
5.6. Needed Expertise and Knowledge
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rantsiou, K.; Kathariou, S.; Winkler, A.; Skandamis, P.; Saint-Cyr, M.J.; Rouzeau-Szynalski, K.; Amézquita, A. Next generation microbiological risk assessment: Opportunities of whole genome sequencing (WGS) for foodborne pathogen surveillance, source tracking and risk assessment. Int. J. Food Microbiol. 2018, 287, 3–9. [Google Scholar] [CrossRef]
- Rouzeau-Szynalski, K.; Barretto, C.; Fournier, C.; Moine, D.; Gimonet, J.; Baert, L. Whole genome sequencing used in an industrial context reveals a Salmonella laboratory cross-contamination. Int. J. Food Microbiol. 2019, 298, 39–43. [Google Scholar] [CrossRef]
- EFSA Panel on Biological Hazards (EFSA BIOHAZ Panel); Koutsoumanis, K.; Allende, A.; Alvarez-Ordóñez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; Davies, R.; De Cesare, A.; Hilbert, F.; et al. Whole genome sequencing and metagenomics for outbreak investigation, source attribution and risk assessment of food-borne microorganisms. EFSA J. 2019, 17, e05898. [Google Scholar] [CrossRef] [PubMed]
- Hurley, D.; Luque-Sastre, L.; Parker, C.T.; Huynh, S.; Eshwar, A.K.; Van Nguyen, S.; Andrews, N.; Moura, A.; Fox, E.M.; Jordan, K.; et al. Whole-genome sequencing-based characterization of 100 Listeria monocytogenes isolates collected from food processing environments over a four-year period. mSphere 2019, 4, 1–14. [Google Scholar] [CrossRef]
- Wang, S.; Weller, D.L.; Falardeau, J.; Strawn, L.K.; Mardones, F.O.; Adell, A.; Switt, A.I.M. Food safety trends: From globalization of whole genome sequencing to application of new tools to prevent foodborne diseases. Trends Food Sci. Technol. 2016, 57, 188–198. [Google Scholar] [CrossRef]
- Brown, E.; Dessai, U.; McGarry, S.; Gerner-Smidt, P. Use of whole-genome sequencing for food safety and public health in the United States. Foodborne Pathog. Dis. 2019, 16, 441–450. [Google Scholar] [CrossRef]
- Allard, M.W.; Strain, E.; Melka, D.; Bunning, K.; Musser, S.M.; Brown, E.W.; Timme, R. Practical value of food pathogen traceability through building a whole-genome sequencing network and database. J. Clin. Microbiol. 2016, 54, 1975–1983. [Google Scholar] [CrossRef]
- Alegbeleye, O.O.; Sant’Ana, A.S. Pathogen subtyping tools for risk assessment and management of produce-borne outbreaks. Curr. Opin. Food Sci. 2020, 32, 83–89. [Google Scholar] [CrossRef]
- Jackson, B.R.; Tarr, C.; Strain, E.; Jackson, K.A.; Conrad, A.; Carleton, H.; Katz, L.S.; Stroika, S.; Gould, L.H.; Mody, R.K.; et al. Implementation of nationwide real-time whole-genome sequencing to enhance listeriosis outbreak detection and investigation. Clin. Infect. Dis. 2016, 63, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, D.; Kajitani, R.; Gotoh, Y.; Katahira, K.; Okuno, M.; Ogura, Y.; Hayashi, T.; Itoh, T. Evaluation of SNP calling methods for closely related bacterial isolates and a novel high-accuracy pipeline: BactSNP. Microb. Genom. 2019, 5, e000261. [Google Scholar] [CrossRef]
- Kwong, J.C.; Stafford, R.; Strain, E.; Stinear, T.; Seemann, T.; Howden, B.P. Sharing is caring: International sharing of data enhances genomic surveillance of Listeria monocytogenes. Clin. Infect. Dis. 2016, 63, 846–848. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.K.; Wang, Q.; Bag, S.K.; Franklin, N.; Shadbolt, C.T.; Howard, P.; Fearnley, E.J.; Quinn, H.E.; Sintchenko, V.; Hope, K.G. Epidemiology and whole genome sequencing of an ongoing point-source Salmonella Agona outbreak associated with sushi consumption in western Sydney, Australia 2015. Epidemiol. Infect. 2017, 145, 2062–2071. [Google Scholar] [CrossRef] [PubMed]
- Van Walle, I.; Guerra, B.; Borges, V.; Carriço, J.A.; Cochrane, G.; Dallman, T.; Franz, E.; Karpíšková, R.; Litrup, E.; Mistou, M.-Y.; et al. EFSA and ECDC technical report on the collection and analysis of whole genome sequencing data from food-borne pathogens and other relevant microorganisms isolated from human, animal, food, feed and food/feed environmental samples in the joint ECDC-EFSA molecular typing database. EFSA Support. Publ. 2019, EN-1337, 1–92. [Google Scholar] [CrossRef]
- Ferguson, B. Adoption of WGS: What is Going On? Food Safety Magazine. 2020. Available online: https://www.foodsafetymagazine.com/magazine-archive1/augustseptember-2020/adoption-of-wgs-what-is-going-on/ (accessed on 16 December 2020).
- Klijn, A.D.; Akins-Lewenthal, B.; Jagadeesan, L.; Baert, A.; Winkler, C.B.; Amézquita, A. The Benefits and Barriers of Whole-Genome Sequencing for Pathogen Source Tracking: A Food Industry Perspective. Food Safety Magazine. 2020. Available online: https://www.foodsafetymagazine.com/magazine-archive1/junejuly-2020/the-benefits-and-barriers-of-whole-genome-sequencing-for-pathogen-source-tracking-a-food-industry-perspective/ (accessed on 14 December 2020).
- Davis, S.; Pettengill, J.B.; Luo, Y.; Payne, J.; Shpuntoff, A.; Rand, H.; Strain, E. CFSAN SNP Pipeline: An automated method for constructing SNP matrices from next-generation sequence data. PeerJ Comput. Sci. 2015, 1, e20. [Google Scholar] [CrossRef]
- Kwong, J.C.; Mercoulia, K.; Tomita, T.; Easton, M.; Li, H.Y.; Bulach, D.M.; Stinear, T.P.; Seemann, T.; Howden, B.P. Prospective Whole-Genome Sequencing Enhances National Surveillance of Listeria monocytogenes. J. Clin. Microbiol. 2016, 54, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Bogaerts, B.; Winand, R.; Fu, Q.; Van Braekel, J.; Ceyssens, P.-J.; Mattheus, W.; Bertrand, S.; De Keersmaecker, S.C.J.; Roosens, N.H.C.; Vanneste, K. Validation of a Bioinformatics Workflow for Routine Analysis of Whole-Genome Sequencing Data and Related Challenges for Pathogen Typing in a European National Reference Center: Neisseria meningitidis as a Proof-of-Concept. Front. Microbiol. 2019, 10, 362. [Google Scholar] [CrossRef] [PubMed]
- Lepuschitz, S.; Weinmaier, T.; Mrazek, K.; Beisken, S.; Weinberger, J.; Posch, A.E. Analytical Performance Validation of Next-Generation Sequencing Based Clinical Microbiology Assays Using a K-mer Analysis Workflow. Front. Microbiol. 2020, 11, 1883. [Google Scholar] [CrossRef] [PubMed]
- Portmann, A.-C.; Fournier, C.; Gimonet, J.; Ngom-Bru, C.; Barretto, C.; Baert, L. A Validation Approach of an End-to-End Whole Genome Sequencing Workflow for Source Tracking of Listeria monocytogenes and Salmonella enterica. Front. Microbiol. 2018, 9, 446. [Google Scholar] [CrossRef]
- Andrews, S. Fastqc: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 17 December 2020).
- Den Bakker, H.; van Heus, P.; Zhang, S. Rapid Confirmation of Salmonella Spp. and Subspp. From Sequence Data. 2018. Available online: https://github.com/hcdenbakker/SalmID (accessed on 17 December 2020).
- Seeman, T. Rapid 16s rDNA from Isolate Fastq Files. 2017. Available online: https://github.com/tseemann/sixess (accessed on 17 December 2020).
- Schaefer, U.; Gallop, S. K-Mer Based Isolate of Fastq Reads Against Reference Genomes. 2014. Available online: https://github.com/phe-bioinformatics/kmerid (accessed on 17 December 2020).
- Wood, D.E.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Souvorov, A.; Agarwala, R.; Lipman, D.J. SKESA: Strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018, 19, 1–13. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Timme, R.; Wolfgang, W.J.; Balkey, M.; Venkata, S.L.G.; Randolph, R.; Allard, M.W.; Strain, E. Optimizing open data to support one health: Best practices to ensure interoperability of genomic data from bacterial pathogens. One Health Outlook 2020, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Seeman, T. Scan Contig Files Against Pubmlst Typing Schemes. 2018. Available online: https://github.com/tseemann/mlst (accessed on 16 December 2020).
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef]
- Gupta, A.; Jordan, I.K.; Rishishwar, L. stringMLST: A fast k-mer based tool for multilocus sequence typing. Bioinformatics 2017, 33, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, C.E.; Kruczkiewicz, P.; Laing, C.R.; Lingohr, E.J.; Gannon, V.P.J.; Nash, J.H.E.; Taboada, E.N. The Salmonella in silico typing resource (SISTR): An open web-accessible tool for rapidly typing and subtyping draft Salmonella genome assemblies. PLoS ONE 2016, 11, e0147101. [Google Scholar] [CrossRef]
- Zhang, S.; Bakker, H.C.D.; Li, S.; Chen, J.; Dinsmore, B.A.; Lane, C.; Lauer, A.C.; Fields, P.I.; Deng, X. SeqSero2: Rapid and improved Salmonella serotype determination using whole-genome sequencing data. Appl. Environ. Microbiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Carroll, L.M.; Kovac, J.; Miller, R.A.; Wiedmann, M. Rapid, high-throughput identification of anthrax-causing and emetic Bacillus cereus group genome assemblies via BTyper, a computational tool for virulence-based classification of Bacillus cereus group isolates by using nucleotide sequencing data. Appl. Environ. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Diep, B.; Barretto, C.; Portmann, A.-C.; Fournier, C.; Karczmarek, A.; Voets, G.; Li, S.; Deng, X.; Klijn, A. Salmonella serotyping; Comparison of the traditional method to a microarray-based method and an in silico platform using whole genome sequencing data. Front. Microbiol. 2019, 10, 2554. [Google Scholar] [CrossRef]
- Silva, M.; Machado, M.P.; Silva, D.N.; Rossi, M.; Moran-Gilad, J.; Santos, S.; Ramirez, M.; Carriço, J.A. chewBBACA: A complete suite for gene-by-gene schema creation and strain identification. Microb. Genom. 2018, 4, e000166. [Google Scholar] [CrossRef]
- Starikova, E.V.; Tikhonova, P.O.; Prianichnikov, N.A.; Rands, C.M.; Zdobnov, E.M.; Ilina, E.N.; Govorun, V.M. Phigaro: High-throughput prophage sequence annotation. Bioinformatics 2020, 36, 3882–3884. [Google Scholar] [CrossRef] [PubMed]
- Reis-Cunha, J.L.; Bartholomeu, D.C.; Manson, A.L.; Earl, A.M.; Cerqueira, G.C. ProphET, prophage estimation tool: A stand-alone prophage sequence prediction tool with self-updating reference database. PLoS ONE 2019, 14, e0223364. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Nash, J.H.E. MOB-suite: Software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 2018, 4, e000206. [Google Scholar] [CrossRef] [PubMed]
- Pightling, A.W.; Petronella, N.; Pagotto, F. Choice of reference sequence and assembler for alignment of Listeria monocytogenes short-read sequence data greatly influences rates of error in SNP analyses. PLoS ONE 2014, 9, e104579. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, S.; Baert, L.; Jagadeesan, B.; Ngom-Bru, C.; Griswold, T.; Katz, L.S.; Carleton, H.A.; Deng, X. Implications of mobile genetic elements for Salmonella enterica single-nucleotide polymorphism subtyping and source tracking investigations. Appl. Environ. Microbiol. 2019, 85, 1–12. [Google Scholar] [CrossRef]
- Bowers, R.M.; The Genome Standards Consortium; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef]
- Yilmaz, P.; Kottmann, R.; Field, D.; Knight, R.; Cole, J.R.; Amaralzettler, L.A.; Gilbert, J.A.; Karsch-Mizrachi, I.; Johnston, A.; Cochrane, G.; et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat. Biotechnol. 2011, 29, 415–420. [Google Scholar] [CrossRef] [PubMed]
- EFSA. The food classification and description system FoodEx 2 (revision 2). EFSA Support. Publ. 2015, 12. [Google Scholar] [CrossRef]
- Ireland, J.D.; Møller, A. LanguaL Food Description: A Learning Process. Eur. J. Clin. Nutr. 2010, 64, S44–S48. [Google Scholar] [CrossRef]
- Lambert, D.; Pightling, A.; Griffiths, E.; Van Domselaar, G.; Evans, P.; Berthelet, S.; Craig, D.; Chandry, P.S.; Stones, R.; Brinkman, F.; et al. Baseline practices for the application of genomic data supporting regulatory food safety. J. AOAC Int. 2017, 100, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Coipan, C.E.; Dallman, T.J.; Brown, D.; Hartman, H.; Van Der Voort, M.; Berg, R.R.V.D.; Palm, D.; Kotila, S.; Van Wijk, T.; Franz, E. Concordance of SNP- and allele-based typing workflows in the context of a large-scale international Salmonella Enteritidis outbreak investigation. Microb. Genom. 2020, 6, e000318. [Google Scholar] [CrossRef]
- Gona, F.; Comandatore, F.; Battaglia, S.; Piazza, A.; Trovato, A.; Lorenzin, G.; Cichero, P.; Biancardi, A.; Nizzero, P.; Moro, M.; et al. Comparison of core-genome MLST, coreSNP and PFGE methods for Klebsiella pneumoniae cluster analysis. Microb. Genom. 2020, 6, mgen000347. [Google Scholar] [CrossRef]
- Blanc, D.S.; Magalhães, B.; Koenig, I.; Senn, L.; Grandbastien, B. Comparison of Whole Genome (wg-) and Core Genome (cg-) MLST (BioNumericsTM) Versus SNP Variant Calling for Epidemiological Investigation of Pseudomonas aeruginosa. Front. Microbiol. 2020, 11, 1729. [Google Scholar] [CrossRef]
- Henri, C.; Leekitcharoenphon, P.; Carleton, H.A.; Radomski, N.; Kaas, R.S.; Mariet, J.-F.; Felten, A.; Aarestrup, F.M.; Smidt, P.G.; Roussel, S.; et al. An assessment of different genomic approaches for inferring phylogeny of Listeria monocytogenes. Front. Microbiol. 2017, 8, 2351. [Google Scholar] [CrossRef]
- Pearce, M.E.; Alikhan, N.-F.; Dallman, T.J.; Zhou, Z.; Grant, K.; Maiden, M.C. Comparative analysis of core genome MLST and SNP typing within a European Salmonella serovar Enteritidis outbreak. Int. J. Food Microbiol. 2018, 274, 1–11. [Google Scholar] [CrossRef]
- Tsang, A.K.L.; Lee, H.H.; Yiu, S.-M.; Lau, S.K.P.; Woo, P.C.Y. Failure of phylogeny inferred from multilocus sequence typing to represent bacterial phylogeny. Sci. Rep. 2017, 7, 4536. [Google Scholar] [CrossRef]
- Timme, R.E.; Rand, H.; Leon, M.S.; Hoffmann, M.; Strain, E.; Allard, M.; Roberson, D.; Baugher, J.D. GenomeTrakr proficiency testing for foodborne pathogen surveillance: An exercise from 2015. Microb. Genom. 2018, 4, e000185. [Google Scholar] [CrossRef] [PubMed]
- ECDC. Proficiency Test for Listeria monocytogenes Whole Genome Assembly; ECDC: Solna, Sweden, 2019. [Google Scholar] [CrossRef]
- Lau, K.A.; da Silva, A.G.; Ballard, S.A.; Theis, T.; Gray, J.; Rawlinson, W.D. Proficiency Testing for bacterial whole genome sequencing in assuring the quality of microbiology diagnostics in clinical and public health laboratories. bioRxiv 2020. [Google Scholar] [CrossRef]
- Pruden, A.; Larsson, D.G.J.; Amézquita, A.; Collignon, P.; Brandt, K.K.; Graham, D.W.; Lazorchak, J.M.; Suzuki, S.; Silley, P.; Snape, J.R.; et al. Management options for reducing the release of antibiotics and antibiotic resistance genes to the environment. Environ. Health Perspect. 2013, 121, 878–885. [Google Scholar] [CrossRef]
- Nijsingh, N.; Munthe, C.; Larsson, D.G.J. Managing pollution from antibiotics manufacturing: Charting actors, incentives and disincentives. Environ. Health 2019, 18, 1–18. [Google Scholar] [CrossRef]
- Fahimipour, A.K.; Ben Mamaar, S.; McFarland, A.G.; Blaustein, R.A.; Chen, J.; Glawe, A.J.; Kline, J.; Green, J.L.; Halden, R.U.; Wymelenberg, K.V.D.; et al. Antimicrobial chemicals associate with microbial function and antibiotic resistance indoors. mSystems 2018, 3, e00200-18. [Google Scholar] [CrossRef]
- Kampf, G. Biocidal agents used for disinfection can enhance antibiotic resistance in gram-negative species. Antibiotics 2018, 7, 110. [Google Scholar] [CrossRef]
- Ellington, M.; Ekelund, O.; Aarestrup, F.; Canton, R.; Doumith, M.; Giske, C.; Grundman, H.; Hasman, H.; Holden, M.; Hopkins, K.; et al. The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: Report from the EUCAST Subcommittee. Clin. Microbiol. Infect. 2017, 23, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Oniciuc, E.A.; Likotrafiti, E.; Alvarez-Molina, A.; Prieto, M.; Santos, J.A.; Alvarez-Ordóñez, A. The present and future of whole genome sequencing (WGS) and whole metagenome sequencing (WMS) for surveillance of antimicrobial resistant microorganisms and antimicrobial resistance genes across the food chain. Genes 2018, 9, 268. [Google Scholar] [CrossRef] [PubMed]
- Pietzka, A.; Allerberger, F.; Murer, A.; Lennkh, A.; Stöger, A.; Rosel, A.C.; Huhulescu, S.; Maritschnik, S.; Springer, B.; Lepuschitz, S.; et al. Whole genome sequencing based surveillance of L. monocytogenes for early detection and investigations of listeriosis outbreaks. Front. Public Health 2019, 7, 139. [Google Scholar] [CrossRef] [PubMed]
- Higgins, P.G.; Prior, K.; Harmsen, D.; Seifert, H. Development and evaluation of a core genome multilocus typing scheme for whole-genome sequence-based typing of Acinetobacter baumannii. PLoS ONE 2017, 12, e0179228. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Stage | Evaluated Software | Parameters Considered to Ensure High-Quality Data | Examples of QC Evaluation |
|---|---|---|---|
| Raw reads quality control (QC) | FASTQC [21] |
| GC content deviating from expected indicates a possible contamination or sample mislabeling. |
| Isolate identification | SalmID [22] Sixess [23] KmerID [24] Kraken [25] |
| A relatively high number of unclassified reads has been associated with plasmid/phage presence, or with a contamination. |
| Read quality trim and removal | Trimmomatic [26] |
| A large number of discarded reads was related to poor quality of the sequencing run. |
| Genome assembly | SPAdes [27] Skesa [28] | Assessed with QUAST [29]
| High number of contigs, or deviation of the expected genome size is an indication of low sequenced genome quality [30]. |
| Sequence typing | mlst [31] MLST-CGE [32] stringMLST [33] | - 7 genes MLST composition | Lack of predicted MLST points to low assembly quality. |
| Salmonella serovar prediction Bacillus clade prediction | Sistr [34] SeqSero2 [35] BTyper [36] | Serovar/clade prediction [37] | Lack of predicted serovar points to low assembly quality. |
| First grouping (cg/wgMLST) | chewBBACA [38] | Number of uncalled loci in the genome | A high number of loci from the profile not found in the genome indicates low assembly quality, contamination, or misidentification of the species. |
| SNP calling | CFSAN SNP pipeline [16] |
| A large number of missing positions suggest an inappropriate choice of the reference. |
| Mobile Genetic Elements (MGE) identification such as phages or plasmids | Phigaro [39] ProphET [40] MOB-Suite [41] |
| Eventually, SNP analyses are run after MGE removal/masking to confirm relatedness. |
| Stage | Software |
|---|---|
| Data integrity | md5sum |
| Raw reads quality control (QC) | FastQC |
| Isolate identification | Kraken |
| Read quality trim and removal | Trimmomatic |
| Genome assembly | SKESA |
| Sequence typing | mlst |
| Bacillus clade prediction | btyper |
| Salmonella serovar prediction | SeqSero2 |
| First grouping (cg/wgMLST) | chewBBACA |
| SNP calling | cfsan_snp_pipeline |
| Mobile Genetic Elements (MGE) identification such as phages or plasmids | MOB-Suite |
| Phigaro | |
| ProphET |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barretto, C.; Rincón, C.; Portmann, A.-C.; Ngom-Bru, C. Whole Genome Sequencing Applied to Pathogen Source Tracking in Food Industry: Key Considerations for Robust Bioinformatics Data Analysis and Reliable Results Interpretation. Genes 2021, 12, 275. https://doi.org/10.3390/genes12020275
Barretto C, Rincón C, Portmann A-C, Ngom-Bru C. Whole Genome Sequencing Applied to Pathogen Source Tracking in Food Industry: Key Considerations for Robust Bioinformatics Data Analysis and Reliable Results Interpretation. Genes. 2021; 12(2):275. https://doi.org/10.3390/genes12020275
Chicago/Turabian StyleBarretto, Caroline, Cristian Rincón, Anne-Catherine Portmann, and Catherine Ngom-Bru. 2021. "Whole Genome Sequencing Applied to Pathogen Source Tracking in Food Industry: Key Considerations for Robust Bioinformatics Data Analysis and Reliable Results Interpretation" Genes 12, no. 2: 275. https://doi.org/10.3390/genes12020275
APA StyleBarretto, C., Rincón, C., Portmann, A.-C., & Ngom-Bru, C. (2021). Whole Genome Sequencing Applied to Pathogen Source Tracking in Food Industry: Key Considerations for Robust Bioinformatics Data Analysis and Reliable Results Interpretation. Genes, 12(2), 275. https://doi.org/10.3390/genes12020275