Evolutionary History of the Risk of SNPs for Diffuse-Type Gastric Cancer in the Japanese Population

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Strategy of Analysis
2.2. Human SNP Data
2.3. FST
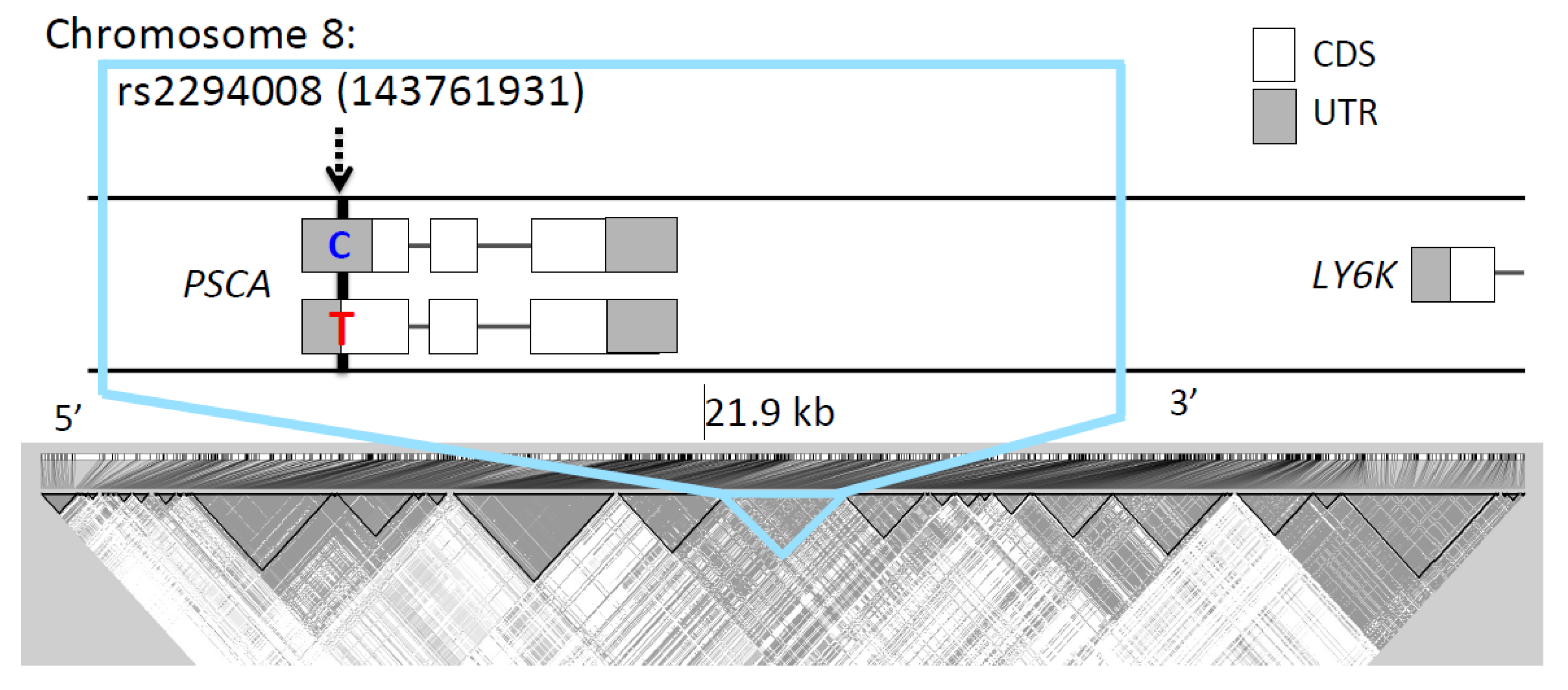
2.4. Linkage Disequilibrium Analysis
2.5. Neutrality Tests
2.5.1. Haplotype-Based Tests (EHH, nSL, and H12)
2.5.2. Site Frequency Spectrum-Based Tests (Tajima’s D and Fay and Wu’s H)
2.5.3. Nucleotide Diversity (π)
2.5.4. Two-Dimensional Site Frequency Spectrum (2D SFS)
2.5.5. Application of Population Branch Statistics (PBS)
2.6. Forward Simulation Using Japanese Demographic Model
2.6.1. Demographic Parameters in Simulations
2.6.2. Investigating Possible Causes of Large FST
2.7. Analysis using Ancient DNA Sequences from the Jomon People
3. Results
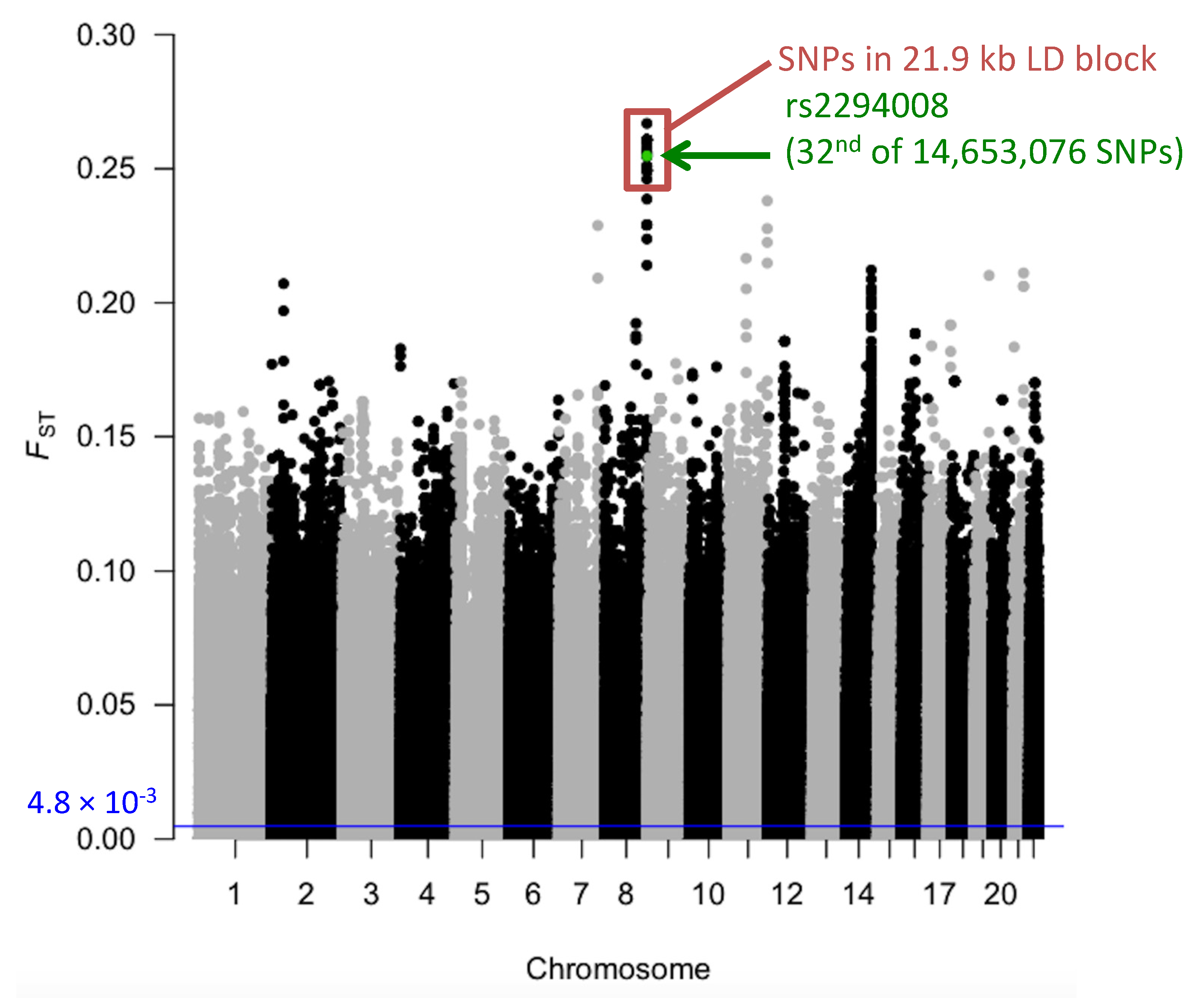
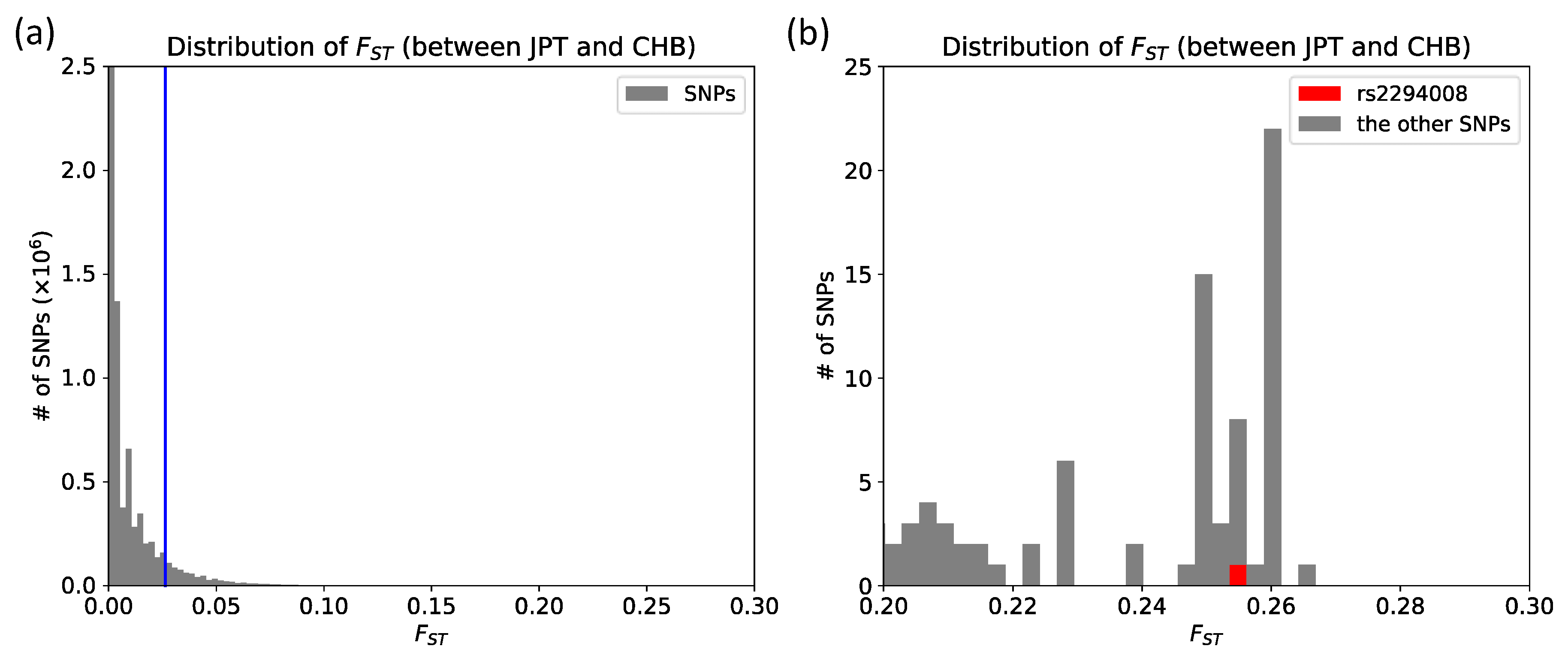
3.1. The T Allele at rs2294008 is Highly Differentiated between JPT and CHB
3.2. Exploring the Signal of Natural Selection Acting on rs2294008
3.3. Two-Dimensional Site Frequency Spectrum (2D SFS)
3.3.1. Detection of Positive Selection in CHB, but not in JPT
3.3.2. Selection Mode in CHB and JPT
3.3.3. History of Natural Selection in JPT and CHB
3.4. Examination of Whether Genetic Drift Can Explain the High FST Using Forward Simulation
3.5. Phylogenic Position of Jomon Haplotypes in the Network of Extant JPT and CHB
4. Discussion
- (i)
- Selection operated on the C allele (the non-risk allele) in the common ancestor of the Han Chinese and the Jomon people. The mode of positive selection in the Japanese is complex; selection on the A-G subhaplotype ceased or relaxed at some point along the Japanese lineage, but ongoing selection occurred on the C-A suphaplotype. Relaxation or cessation of positive selection on the A-G subhaplotype may have led to low frequency of the C allele in the extant JPT.
- (ii)
- The ancestral population (the Jomon people) had a high T allele frequency, which led to a high T allele frequency in the extant Japanese, even though the Jomon people experienced admixture with immigrant Yayoi farmers. These factors result in the large T/C allele frequency difference between JPT and CHB.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Laurén, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- Crew, K.D.; Neugut, A.I. Epidemiology of gastric cancer. World J. Gastroenterol. 2006, 12, 354–362. [Google Scholar] [CrossRef]
- Henson, D.E.; Dittus, C.; Younes, M.; Nguyen, H.; Albores-Saavedra, J. Differential trends in the intestinal and diffuse types of gastric carcinoma in the United States, 1973–2000: Increase in the signet ring cell type. Arch. Pathol. Lab. Med. 2004, 128, 765–770. [Google Scholar]
- Miyahara, R.; Niwa, Y.; Matsuura, T.; Maeda, O.; Ando, T.; Ohmiya, N.; Itoh, A.; Hirooka, Y.; Goto, H. Prevalence and prognosis of gastric cancer detected by screening in a large Japanese population: Data from a single institute over 30 years. J. Gastroenterol. Hepatol. 2007, 22, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- The Study Group of Millennium Genome Project for Cancer; Sakamoto, H.; Yoshimura, K.; Saeki, N.; Katai, H.; Shimoda, T.; Matsuno, Y.; Saito, D.; Sugimura, H.; Tanioka, F.; et al. Genetic variation in PSCA is associated with susceptibility to diffuse-type gastric cancer. Nat. Genet. 2008, 40, 730–740. [Google Scholar] [CrossRef]
- Song, H.-R.; Kim, H.N.; Piao, J.-M.; Kweon, S.-S.; Choi, J.-S.; Bae, W.-K.; Chung, I.J.; Park, Y.-K.; Kim, S.-H.; Choi, Y.-D.; et al. Association of a common genetic variant in prostate stem-cell antigen with gastric cancer susceptibility in a Korean population. Mol. Carcino. 2011, 50, 871–875. [Google Scholar] [CrossRef]
- Park, B.; Yang, S.; Lee, J.; Woo, H.D.; Choi, I.J.; Kim, Y.W.; Ryu, K.W.; Kim, Y.-I.; Kim, J. Genome-Wide Association of Genetic Variation in the PSCA Gene with Gastric Cancer Susceptibility in a Korean Population. Cancer Res. Treat. 2019, 51, 748–757. [Google Scholar] [CrossRef]
- Turdikulova, S.; Dalimova, D.; Abdurakhimov, A.; Adilov, B.; Navruzov, S.; Yusupbekov, A.; Djuraev, M.; Abdujapparov, S.; Egamberdiev, D.; Mukhamedov, R. Association of rs2294008 and rs9297976 Polymorphisms in PSCA Gene with Gastric Cancer Susceptibility in Uzbekistan. Cent. Asian J. Glob. Health 2016, 5, 227. [Google Scholar] [CrossRef]
- Lochhead, P.; Frank, B.; Hold, G.L.; Rabkin, C.S.; Ng, M.T.H.; Vaughan, T.L.; Risch, H.A.; Gammon, M.D.; Lissowska, J.; Weck, M.N.; et al. Genetic Variation in the Prostate Stem Cell Antigen Gene and Upper Gastrointestinal Cancer in White Individuals. Gastroenterology 2011, 140, 435–441. [Google Scholar] [CrossRef]
- Sala, N.; Muñoz, X.; Travier, N.; Agudo, A.; Duell, E.J.; Moreno, V.; Overvad, K.; Tjønneland, A.; Boutron-Ruault, M.C.; Clavel-Chapelon, F.; et al. Prostate stem-cell antigen gene is associated with diffuse and intestinal gastric cancer in Caucasians: Results from the EPIC-EURGAST study. Int. J. Cancer 2012, 130, 2417–2427. [Google Scholar] [CrossRef]
- García-González, M.A.; Bujanda, L.; Quintero, E.; Santolaria, S.; Benito, R.; Strunk, M.; Sopena, F.; Thomson, C.; Perez-Aisa, A.; Nicolás-Pérez, D.; et al. Association ofPSCArs2294008 gene variants with poor prognosis and increased susceptibility to gastric cancer and decreased risk of duodenal ulcer disease. Int. J. Cancer 2015, 137, 1362–1373. [Google Scholar] [CrossRef]
- Tanikawa, C.; Urabe, Y.; Matsuo, K.; Kubo, M.; Takahashi, A.; Ito, H.; Tajima, K.; Kamatani, N.; Nakamura, Y.; Matsuda, K. A genome-wide association study identifies two susceptibility loci for duodenal ulcer in the Japanese population. Nat. Genet. 2012, 44, 430–434. [Google Scholar] [CrossRef] [PubMed]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Yang, J.-H.; Chiang, C.W.; Hsiung, C.-N.; Wu, P.-E.; Chang, L.-C.; Chu, H.-W.; Chang, J.; Song, I.-W.; Yang, S.-L.; et al. Population structure of Han Chinese in the modern Taiwanese population based on 10,000 participants in the Taiwan Biobank project. Hum. Mol. Genet. 2016, 25, 5321–5331. [Google Scholar] [CrossRef]
- Bae, J.S.; Cheong, H.S.; Kim, J.-O.; Lee, S.O.; Kim, E.-M.; Lee, H.W.; Kim, S.; Kim, J.-W.; Cui, T.; Inoue, I.; et al. Identification of SNP markers for common CNV regions and association analysis of risk of subarachnoid aneurysmal hemorrhage in Japanese population. Biochem. Biophys. Res. Commun. 2008, 373, 593–596. [Google Scholar] [CrossRef]
- Hudson, R.R.; Slatkin, M.; Maddison, W.P. Estimation of levels of gene flow from DNA sequence data. Genetics 1992, 132, 583–589. [Google Scholar] [PubMed]
- Bhatia, G.; Patterson, N.; Sankararaman, S.; Escott-Price, V. Estimating and interpreting FST: The impact of rare variants. Genome Res. 2013, 23, 1514–1521. [Google Scholar] [CrossRef]
- Takahata, N.; Satta, Y.; Klein, Y. Polymorphism and balancing selection at major histocompatibility complex loci. Genetics. 1992, 130, 925–938. [Google Scholar]
- Turner, S.D. Qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open Source Softw. 2018, 3, 1–2. [Google Scholar] [CrossRef]
- Hartl, D.L.; Clark, A.G. Principles of Population Genetics, 4th ed.; Sinauer Associates: Sunderland, MA, USA, 2007; Chapter 2; pp. 45–88. ISBN 978-0-87893-308-2. [Google Scholar]
- Barrett, J.C.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinfomatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The Structure of Haplotype Blocks in the Human Genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Reich, D.E.; Higgins, J.M.; Levine, H.Z.P.; Richter, D.J.; Schaffner, S.F.; Gabriel, S.B.; Platko, J.V.; Patterson, N.J.; McDonald, G.J.; et al. Detecting recent positive selection in the human genome from haplotype structure. Nature 2002, 419, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Admetlla, A.; Liang, M.; Korneliussen, T.S.; Nielsen, R. On detecting incomplete soft or hard selective sweeps using haplotype structure. Mol. Boil. Evol. 2014, 31, 1275–1291. [Google Scholar] [CrossRef]
- Garud, N.R.; Messer, P.W.; Buzbas, E.O.; Petrov, D.A. Recent Selective Sweeps in North American Drosophila melanogaster Show Signatures of Soft Sweeps. PLoS Genet. 2015, 11, e1005004. [Google Scholar] [CrossRef] [PubMed]
- Szpiech, Z.A.; Hernandez, R.D. Selscan: An Efficient Multithreaded Program to Perform EHH-Based Scans for Positive Selection. Mol. Boil. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Fay, J.; Wu, C.-I. Hitchhiking Under Positive Darwinian Selection. Genetics 2000, 155, 1405–1413. [Google Scholar]
- Zeng, K.; Fu, Y.-X.; Shi, S.; Wu, C.-I. Statistical Tests for Detecting Positive Selection by Utilizing High-Frequency Variants. Genetics 2006, 174, 1431–1439. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Boil. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Nei, M.; Li, W.-H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Satta, Y.; Zheng, W.; Nishiyama, K.V.; Iwasaki, R.L.; Hayakawa, T.; Fujito, N.T.; Takahata, N. Two-dimensional site frequency spectrum for detecting, classifying and dating incomplete selective sweeps. Genes Genet. Syst. 2019, 94, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Fujito, N.T.; Satta, Y.; Hayakawa, T.; Takahata, N. A new inference method for detecting an ongoing selective sweep. Genes Genet. Syst. 2018, 93, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.R. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 2002, 18, 337–338. [Google Scholar] [CrossRef]
- Schaffner, S.F.; Foo, C.; Gabriel, S.; Reich, D.; Daly, M.J.; Altshuler, D. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005, 15, 1576–1583. [Google Scholar] [CrossRef]
- Yi, X.; Liang, Y.; Huerta-Sanchez, E.; Jin, X.; Cuo, Z.X.P.; Pool, J.; Xu, X.; Jiang, H.; Vinckenbosch, N.; Korneliussen, T.; et al. Sequencing of 50 Human Exomes Reveals Adaptation to High Altitude. Science 2010, 329, 75–78. [Google Scholar] [CrossRef]
- Cavalli-Sforza, L.L. Human diversity. In Proceedings of the 12th International Congress on Genetics, Tokyo, Japan, 19–28 August 1968; pp. 405–416. [Google Scholar]
- Ewens, W.J. Mathematical Population Genetics 1. Biomathematics; Springer: New York, NY, USA, 1979; Volume 9. [Google Scholar]
- Hanihara, K. Dual Structure Model for the Population History of the Japanese. Japan Rev. 1991, 2, 1–33. [Google Scholar] [CrossRef][Green Version]
- Habu, J. Ancient Jomon of Japan; Cambridge University Press: Cambridge, UK, 2004; ISBN 0521776708. [Google Scholar]
- Jinam, T.A.; Kanzawa-Kiriyama, H.; Inoue, I.; Tokunaga, K.; Omoto, K.; Saitou, N. Unique characteristics of the Ainu population in Northern Japan. J. Hum. Genet. 2015, 60, 565–571. [Google Scholar] [CrossRef]
- Nakagome, S.; Sato, T.; Ishida, H.; Hanihara, T.; Yamaguchi, T.; Kimura, R.; Mano, S.; Oota, H. The Asian DNA Repository Consortium. Model-Based Verification of Hypotheses on the Origin of Modern Japanese Revisited by Bayesian Inference Based on Genome-Wide SNP Data. Mol. Boil. Evol. 2015, 32, 1533–1543. [Google Scholar] [CrossRef]
- Kanzawa-Kiriyama, H.; Kryukov, K.; Jinam, T.A.; Hosomichi, K.; Saso, A.; Suwa, G.; Ueda, S.; Yoneda, M.; Tajima, A.; Shinoda, K.-I.; et al. A partial nuclear genome of the Jomons who lived 3000 years ago in Fukushima, Japan. J. Hum. Genet. 2016, 62, 213–221. [Google Scholar] [CrossRef]
- Kanzawa-Kiriyama, H.; Jinam, T.A.; Kawai, Y.; Sato, T.; Hosomichi, K.; Tajima, A.; Adachi, N.; Matsumura, H.; Kryukov, K.; Saitou, N.; et al. Late Jomon male and female genome sequences from the Funadomari site in Hokkaido, Japan. Anthr. Sci. 2019, 127, 83–108. [Google Scholar] [CrossRef]
- Fenner, J.N. Cross-cultural estimation of the human generation interval for use in genetics-based population divergence studies. Am. J. Phys. Anthr. 2005, 128, 415–423. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Method 2020, 17, 261–272. [Google Scholar] [CrossRef]
- McColl, H.; Racimo, F.; Vinner, L.; Demeter, F.; Gakuhari, T.; Moreno-Mayar, J.V.; Van Driem, G.; Wilken, U.G.; Seguin-Orlando, A.; Castro, C.D.L.F.; et al. The prehistoric peopling of Southeast Asia. Science 2018, 361, 88–92. [Google Scholar] [CrossRef]
- Available online: https://www.fluxus-engineering.com/index.htm (accessed on 1 July 2020).
- Bandelt, H.-J.; Forster, P.; Röhl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Sikora, M.; Seguin-Orlando, A.; Sousa, V.C.; Albrechtsen, A.; Korneliussen, T.; Ko, A.; Rasmussen, S.; Dupanloup, I.; Nigst, P.; Bosch, M.D.; et al. Ancient genomes show social and reproductive behavior of early Upper Paleolithic foragers. Science 2017, 358, 659–662. [Google Scholar] [CrossRef]
- Wakeley, J.; Alicar, N. Gene genealogies in a metapopulation. Genetics 2001, 159, 893–905. [Google Scholar]
- Przeworski, M. The signature of positive selection at randomly chosen loci. Genetics 2002, 160, 1179–1189. [Google Scholar]
- Nakayama, K.; Ohashi, J.; Watanabe, K.; Munkhtulga, L.; Iwamoto, S. Evidence for Very Recent Positive Selection in Mongolians. Mol. Boil. Evol. 2017, 34, 1936–1946. [Google Scholar] [CrossRef]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2018, 47, D745–D751. [Google Scholar] [CrossRef]
- Sato, T.; Nakagome, S.; Watanabe, C.; Yamaguchi, K.; Kawaguchi, A.; Koganebuchi, K.; Haneji, K.; Yamaguchi, T.; Hanihara, T.; Yamamoto, K.; et al. Genome-Wide SNP Analysis Reveals Population Structure and Demographic History of the Ryukyu Islanders in the Southern Part of the Japanese Archipelago. Mol. Boil. Evol. 2014, 31, 2929–2940. [Google Scholar] [CrossRef] [PubMed]
- Japanese Archipelago Human Population Genetics Consortium; Jinam, T.; Nishida, N.; Hirai, M.; Kawamura, S.; Oota, H.; Umetsu, K.; Kimura, R.; Ohashi, J.; Tajima, A.; et al. The history of human populations in the Japanese Archipelago inferred from genome-wide SNP data with a special reference to the Ainu and the Ryukyuan populations. J. Hum. Genet. 2012, 57, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, D.; Chung, Y.-J.; Xu, S. Genetic structure, divergence and admixture of Han Chinese, Japanese and Korean populations. Hereditas 2018, 155, 19. [Google Scholar] [CrossRef] [PubMed]
- Fujio, S. History of Yayoi Period; Kodansha: Tokyo, Japan, 2015; ISBN 9784062883306. (In Japanese) [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | JPT | CHB | ||
|---|---|---|---|---|
| Core Region | 143755915- 143770914 | 143755876- 143771875 | ||
| n # | 208 (C = 77, T = 131) | 206 (C = 155, T = 51) | ||
| S † | 91 | 88 | ||
| Tested Allele | C | T | C | T |
| Allele Frequency | 0.370 | 0.630 | 0.752 | 0.248 |
| Fc § | 0.167 (0.834) | 0.833 (>0.999) | 0.352 × 10−1 (0.223 × 10−2) ** | 0.869 (>0.999) |
| Gc0 | 9.60 (0.693) | 31.84 (0.975) | 1.84 (0.167 × 10−2) ** | 25.13 (>0.999) |
| Lc0 | 0.178 × 10−1 (0.708) | 0.259 (>0.999) | 0.565 × 10−2 (>0.167 × 10−2) ** | 0.130 (>0.999) |
| G*c0 | 22.50 | 46.23 | 5.00 | 30.69 |
| γ*(10) | 0.500 | 0.700 | 0.000 | 0.962 |
| imax | 40 | 130 | 8 | 50 |
| i*max | 0 | 29 | 75 | 21 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwasaki, R.L.; Ishiya, K.; Kanzawa-Kiriyama, H.; Kawai, Y.; Gojobori, J.; Satta, Y. Evolutionary History of the Risk of SNPs for Diffuse-Type Gastric Cancer in the Japanese Population. Genes 2020, 11, 775. https://doi.org/10.3390/genes11070775
Iwasaki RL, Ishiya K, Kanzawa-Kiriyama H, Kawai Y, Gojobori J, Satta Y. Evolutionary History of the Risk of SNPs for Diffuse-Type Gastric Cancer in the Japanese Population. Genes. 2020; 11(7):775. https://doi.org/10.3390/genes11070775
Chicago/Turabian StyleIwasaki, Risa L., Koji Ishiya, Hideaki Kanzawa-Kiriyama, Yosuke Kawai, Jun Gojobori, and Yoko Satta. 2020. "Evolutionary History of the Risk of SNPs for Diffuse-Type Gastric Cancer in the Japanese Population" Genes 11, no. 7: 775. https://doi.org/10.3390/genes11070775
APA StyleIwasaki, R. L., Ishiya, K., Kanzawa-Kiriyama, H., Kawai, Y., Gojobori, J., & Satta, Y. (2020). Evolutionary History of the Risk of SNPs for Diffuse-Type Gastric Cancer in the Japanese Population. Genes, 11(7), 775. https://doi.org/10.3390/genes11070775