Mitochondrial PCK2 Missense Variant in Shetland Sheepdogs with Paroxysmal Exercise-Induced Dyskinesia (PED)


 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal Selection
2.3. Clinical Examinations
2.4. Laboratory Examinations
2.5. Cardiac Examinations
2.6. Muscle Examinations and Histopathology
2.7. Additional Diagnostic Examinations
2.8. Autoantibodies
2.9. Fibroblast Culture
2.10. Tryptophan Content of Therapeutic Diet
2.11. Whole Genome Sequencing of Two Affected Shetland Sheepdogs
2.12. Variant Calling
2.13. Gene Analysis
2.14. Sanger Sequencing
3. Results
3.1. Clinical Examinations and Family History
3.2. Laboratory Examinations
3.3. Additonal Examinations
3.4. Histopathology
3.5. Clinical Management
3.6. Outcome
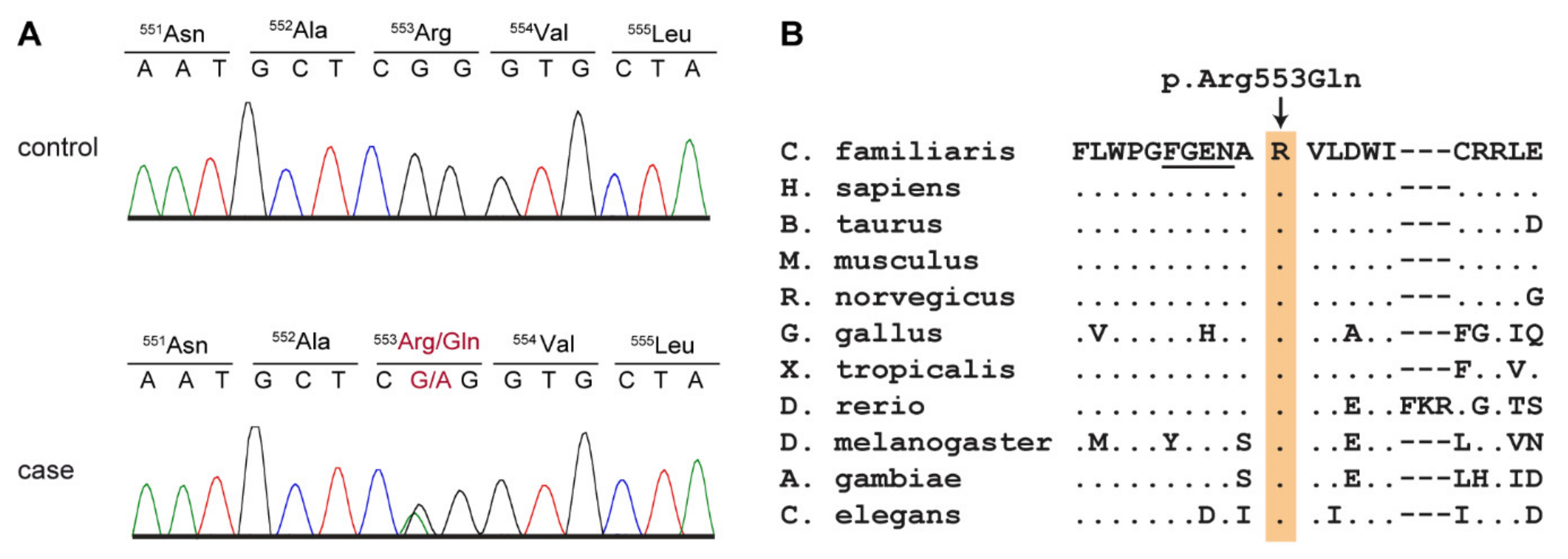
3.7. Genetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xu, Z.; Lim, C.K.; Tan, L.C.S.; Tan, E.-K. Paroxysmal Movement Disorders: Recent Advances. Curr. Neurol. Neurosci. Rep. 2019, 19, 48. [Google Scholar] [CrossRef]
- Weber, Y.G.; Storch, A.; Wuttke, T.V.; Brockmann, K.; Kempfle, J.; Maljevic, S.; Margari, L.; Kamm, C.; Schneider, S.A.; Huber, S.M.; et al. GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J. Clin. Investig. 2008, 118, 2157–2168. [Google Scholar] [CrossRef]
- Weber, Y.G.; Kamm, C.; Suls, A.; Kempfle, J.; Kotschet, K.; Schüle, R.; Wuttke, T.V.; Maljevic, S.; Liebrich, J.; Gasser, T.; et al. Paroxysmal choreoathetosis/spasticity (DYT9) is caused by a GLUT1 defect. Neurology 2011, 77, 959–964. [Google Scholar] [CrossRef]
- Seidner, G.; Alvarez, M.G.; Yeh, J.-I.; O’Driscoll, K.R.; Klepper, J.; Stump, T.S.; Wang, N.; Spinner, N.B.; Birnbaum, M.; De Vivo, D.C. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat. Genet. 1998, 18, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, H.; Ohye, T.; Takahashi, E.-I.; Seki, N.; Hori, T.-A.; Segawa, M.; Nomura, Y.; Endo, K.; Tanaka, H.; Tsuji, S.; et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat. Genet. 1994, 8, 236–242. [Google Scholar] [CrossRef]
- Méneret, A.; Roze, E. Paroxysmal movement disorders: An update. Rev. Neurol. 2016, 172, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Urkasemsin, G.; Olby, N.J. Canine Paroxysmal Movement Disorders. Vet. Clin. Small Anim. Pract. 2014, 44, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Meyers, K.M.; Padgett, G.A.; Dickson, W.M. The Genetic Basis of a Kinetic Disorder of Scottish Terrier Dogs. J. Hered. 1970, 61, 189–192. [Google Scholar] [CrossRef]
- Geiger, K.M.; Klopp, L.S. Use of a selective serotonin reuptake inhibitor for treatment of episodes of hypertonia and kyphosis in a young adult Scottish Terrier. J. Am. Vet. Med. Assoc. 2009, 235, 168–171. [Google Scholar] [CrossRef]
- Urkasemsin, G.; Olby, N.J. Clinical characteristics of Scottie Cramp in 31 cases. J. Small Anim. Pract. 2015, 56, 276–280. [Google Scholar] [CrossRef]
- Penderis, J.; Franklin, R. Dyskinesia in an adult bichon frise. J. Small Anim. Pract. 2001, 42, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Lowrie, M.; Varejão, A.S.P. Paroxysmal dyskinesia in the bichon frise. Vet. Rec. 2018, 182, 578. [Google Scholar] [CrossRef] [PubMed]
- Black, V.L.; Garosi, L.; Lowrie, M.; Harvey, R.J.; Gale, J. Phenotypic characterisation of canine epileptoid cramping syndrome in the Border terrier. J. Small Anim. Pract. 2013, 55, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Stassen, Q.E.M.; Koskinen, L.; Van Steenbeek, F.; Seppälä, E.; Jokinen, T.; Prins, P.; Bok, H.; Zandvliet, M.M.; Vos-Loohuis, M.; Leegwater, P.; et al. Paroxysmal Dyskinesia in Border Terriers: Clinical, Epidemiological, and Genetic Investigations. J. Vet. Intern. Med. 2017, 31, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Lowrie, M.; Garden, O.; Hadjivassiliou, M.; Sanders, D.; Powell, R.; Garosi, L. Characterization of Paroxysmal Gluten-Sensitive Dyskinesia in Border Terriers Using Serological Markers. J. Vet. Intern. Med. 2018, 32, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, I.; Chandler, K.E.; Franklin, R.J.M. A movement disorder in boxer pups. Vet. Rec. 1999, 144, 179–180. [Google Scholar] [CrossRef]
- Packer, R.; Patterson, E.; Taylor, J.; Coates, J.; Schnabel, R.D.; O’Brien, D. Characterization and Mode of Inheritance of a Paroxysmal Dyskinesia in Chinook Dogs. J. Vet. Intern. Med. 2010, 24, 1305–1313. [Google Scholar] [CrossRef]
- Harcourt-Brown, T. Anticonvulsant responsive, episodic movement disorder in a German shorthaired pointer. J. Small Anim. Pract. 2008, 49, 405–407. [Google Scholar] [CrossRef]
- Shelton, G.D. Muscle pain, cramps and hypertonicity. Vet. Clin. Small Anim. Pract. 2004, 34, 1483–1496. [Google Scholar] [CrossRef]
- Polidoro, D.; Van Ham, L.; Santens, P.; Cornelis, I.; Charalambous, M.; Broeckx, B.J.G.; Bhatti, S.F.M. Phenotypic characterization of paroxysmal dyskinesia in Maltese dogs. J. Vet. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Kolicheski, A.L.; Johnson, G.S.; Mhlanga-Mutangadura, T.; Taylor, J.F.; Schnabel, R.D.; Kinoshita, T.; Murakami, Y.; O’Brien, D.P. A homozygous PIGN missense mutation in Soft-Coated Wheaten Terriers with a canine paroxysmal dyskinesia. Neurogenetics 2016, 18, 39–47. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Garosi, L.S.; Platt, S.R.; Shelton, G.D. Hypertonicity in Cavalier King Charles spaniel. J. Vet. Intern. Med. 2002, 16, 330. [Google Scholar]
- Forman, O.P.; Penderis, J.; Hartley, C.; Hayward, L.J.; Ricketts, S.L.; Mellersh, C.S. Parallel Mapping and Simultaneous Sequencing Reveals Deletions in BCAN and FAM83H Associated with Discrete Inherited Disorders in a Domestic Dog Breed. PLoS Genet. 2012, 8, e1002462. [Google Scholar] [CrossRef] [PubMed]
- Crippa, L.; Ferro, E.; Melloni, E.; Brambilla, P.; Cavalletti, E. Echocardiographic parameters and indices in the normal Beagle dog. Lab. Anim. 1992, 26, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.H.; Lehmkuhl, L.B.; Bonagura, J.D.; Beall, M.J. Retrospective analysis of the clinical utility of ambulatory electrocardiographic (Holter) recordings in syncopal dogs: 44 cases (1991–1995). J. Vet. Intern. Med. 1999, 13, 111–122. [Google Scholar]
- Platt, S.R.; Olby, N.J. BSAVA Manual of Canine and Feline Neurology, 4th ed.; British Small Animal Veterinary Association: Quedgeley, UK, 2014; pp. 47–55. [Google Scholar]
- Brauer, C.; Kästner, S.B.; Rohn, K.; Schenk, H.C.; Tünsmeyer, J.; Tipold, A. Electroencephalographic recordings in dogs suffering from idiopathic and symptomatic epilepsy: Diagnostic value of interictal short time EEG protocols supplemented by two activation techniques. Vet. J. 2012, 193, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Das, A.M.; Byrd, D.J.; Brodehl, J. Regulation of the mitochondrial ATP-synthase in human fibroblasts. Clin. Chim. Acta 1994, 231, 61–68. [Google Scholar] [CrossRef]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Aguirre, G.; André, C.; Bannasch, D.; Becker, D.; Davis, B.; Ekenstedt, K.; Faller, K.; et al. A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.E.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Holyoak, T.; Sullivan, S.M.; Nowak, T. Structural Insights into the Mechanism of PEPCK Catalysis. Biochemistry 2006, 45, 8254–8263. [Google Scholar] [CrossRef]
- Chakravarty, K.; Cassuto, H.; Reshef, L.; Hanson, R.W. Factors That Control the Tissue-Specific Transcription of the Gene for Phosphoenolpyruvate Carboxykinase-C. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 129–154. [Google Scholar] [CrossRef]
- Quinn, P.G.; Yeagley, D. Insulin regulation of PEPCK gene expression: A model for rapid and reversible modulation. Curr. Drug Targets Immuneendocr. Metab. Disord. 2005, 5, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Kibbey, R.G. The mitochondrial isoform of phosphoenolpyruvate carboxykinase (PEPCK-M) and glucose homeostasis: Has it been overlooked? Biochim. Biophys. 2014, 1840, 1313–1330. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Pasquel, F.; Turcu, A.; Pongratz, R.L.; Roden, M.; Cline, G.W.; Shulman, G.I.; Kibbey, R.G. Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvate carboxykinase links anaplerosis and mitochondrial GTP with insulin secretion. J. Biol. Chem. 2009, 284, 26578–26590. [Google Scholar] [CrossRef]
- Santra, S.; Cameron, J.M.; Shyr, C.; Zhang, L.; Drogemoller, B.; Ross, C.; Wasserman, W.W.; Wevers, R.A.; Rodenburg, R.J.; Gupte, G.; et al. Cytosolic phosphoenolpyruvate carboxykinase deficiency presenting with acute liver failure following gastroenteritis. Mol. Genet. Metab. 2016, 118, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Vieira, P.; Cameron, J.; Rahikkala, E.; Keski-Filppula, R.; Zhang, L.-H.; Santra, S.; Matthews, A.; Myllynen, P.; Nuutinen, M.; Moilanen, J.; et al. Novel homozygous PCK1 mutation causing cytosolic phosphoenolpyruvate carboxykinase deficiency presenting as childhood hypoglycemia, an abnormal pattern of urine metabolites and liver dysfunction. Mol. Genet. Metab. 2017, 120, 337–341. [Google Scholar] [CrossRef]
- Adams, D.R.; Yuan, H.; Holyoak, T.; Arajs, K.H.; Hakimi, P.; Markello, T.C.; Wolfe, L.A.; Vilboux, T.; Burton, B.K.; Fajardo, K.F.; et al. Three rare diseases in one Sib pair: RAI1, PCK1, GRIN2B mutations associated with Smith-Magenis Syndrome, cytosolic PEPCK deficiency and NMDA receptor glutamate insensitivity. Mol. Genet. Metab. 2014, 113, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Clayton, P.T.; Hyland, K.; Brand, M.; Leonard, J.V. Mitochondrial phosphoenolpyruvate carboxykinase deficiency. Eur. J. Nucl. Med. Mol. Imaging 1986, 145, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.V.; Hyland, K.; Furukawa, N.; Clayton, P.T. Mitochondrial phosphoenolpyruvate carboxykinase deficiency. Eur. J. Nucl. Med. Mol. Imaging 1991, 150, 198–199. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.H.; Taylor, J.; Sherwood, W.G. The Genetic Heterogeneity of Lactic Acidosis: Occurrence of Recognizable Inborn Errors of Metabolism in a Pediatric Population with Lactic Acidosis. Pediatr. Res. 1980, 14, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- GnomAD–Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org/ (accessed on 2 June 2020).
- Lorenz, M.D.; Coates, J.; Kent, M. Handbook of Veterinary Neurology-E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2010; pp. 209–211. [Google Scholar]
- Nakahata, K.; Uzuka, Y.; Matsumoto, H.; Gotoh, N.; Sasaki, K. Hyperkinetic involuntary movements in a young Shetland sheepdog. J. Am. Anim. Hosp. Assoc. 1992, 28, 347–348. [Google Scholar]
- Pons, R.; Collins, A.; Rotstein, M.; Engelstad, K.; De Vivo, D.C. The spectrum of movement disorders in Glut-1 deficiency. Mov. Disord. 2010, 25, 275–281. [Google Scholar] [CrossRef]
- Yip, J.; Geng, X.; Shen, J.; Ding, Y. Cerebral Gluconeogenesis and Diseases. Front. Pharmacol. 2017, 7, 1547. [Google Scholar] [CrossRef]
- Thöny, B.; Auerbach, G.; Blau, N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem. J. 2000, 347 Pt 1, 1–16. [Google Scholar] [CrossRef]
- Escós, M.; Latorre, P.; Hidalgo, J.; Hurtado-Guerrero, R.; Carrodeguas, J.A.; López-Buesa, P. Kinetic and functional properties of human mitochondrial phosphoenolpyruvate carboxykinase. Biochem. Biophys. Rep. 2016, 7, 124–129. [Google Scholar] [CrossRef][Green Version]
- Maalouf, M.A.; Rho, J.M.; Mattson, M.P. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Rev. 2009, 59, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Anheim, M.; Maillart, E.; Vuillaumier-Barrot, S.; Flamand-Rouvière, C.; Pineau, F.; Ewenczyk, C.; Riant, F.; Apartis, E.; Roze, E. Excellent response to acetazolamide in a case of paroxysmal dyskinesias due to GLUT1-deficiency. J. Neurol. 2010, 258, 316–317. [Google Scholar] [CrossRef] [PubMed]
- Mayorandan, S.; Meyer, U.; Hartmann, H.; Das, A. Glycogen storage disease type III: Modified Atkins diet improves myopathy. Orphanet J. Rare Dis. 2014, 9, 196. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.M.; Kossoff, E.H.; Hartman, A. The Ketogenic Diet: One Decade Later. Pediatrics 2007, 119, 535–543. [Google Scholar] [CrossRef]
- Keller, U.; Cherrington, A.D.; Liljenquist, J.E. Ketone body turnover and net hepatic ketone production in fasted and diabetic dogs. Am. J. Physiol. Metab. 1978, 235, E238. [Google Scholar] [CrossRef] [PubMed]
- Packer, R.M.; Shihab, N.K.; Torres, B.B.J.; Volk, H.A. Responses to successive anti-epileptic drugs in canine idiopathic epilepsy. Vet. Rec. 2015, 176, 203. [Google Scholar] [CrossRef] [PubMed]
- Schaechter, J.D.; Wurtman, R.J. Serotonin release varies with brain tryptophan levels. Brain Res. 1990, 532, 203–210. [Google Scholar] [CrossRef]
- Putman, P.; Roelofs, K. Effects of single cortisol administrations on human affect reviewed: Coping with stress through adaptive regulation of automatic cognitive processing. Psychoneuroendocrinology 2011, 36, 439–448. [Google Scholar] [CrossRef]
- Okabe, K.; Yaku, K.; Tobe, K.; Nakagawa, T. Implications of altered NAD metabolism in metabolic disorders. J. Biomed. Sci. 2019, 26, 34. [Google Scholar] [CrossRef]
- Royaux, E.; Bhatti, S.; Harvey, R.J.; Garosi, L.; Shelton, G.D.; Van Ham, L. Acetazolamide-responsive paroxysmal dyskinesia in a 12-week-old female golden retriever dog. Vet. Q. 2015, 36, 1–12. [Google Scholar] [CrossRef]
- O’Brien, D.; Kolicheski, A.; Packer, R.; Thomovsky, S.; Taylor, J.; Schnabel, R.; Berg, J.; Vasquez, L.; Johnson, G. Paroxysmal non-kinesogenic dyskinesia in soft coated Wheaten terriers is associated with a missense mutation in PIGN and responds to acetazolamide therapy. J. Vet. Intern. Med. 2015, 29, 1267. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diet | Dry Matter | Tryptophan | Clinical Response |
|---|---|---|---|
| normal | 921 g/kg | 1.52 g/kg | worsening of clinical signs |
| gluten-free | 950 g/kg | 1.65 g/kg | improved clinical signs |
| seafood gluten- and grain free | 951 g/kg | 2.97 g/kg | markedly improved clinical signs |
| Filtering Step | Heterozygous Variants | Homozygous Variants |
|---|---|---|
| Case-specific variants | 1030 | 36 |
| Case-specific protein-changing variants | 10 | 0 |
| Gene | Protein | Variant |
|---|---|---|
| C7 | complement C7 | p.Glu799* |
| CDH24 | cadherin 24 | p.Asp669Asn |
| CNNM1 | cyclin and CBS domain divalent metal cation transport mediator 1 | p.Asp170Tyr |
| LOC485317 | p.Lys270Asn | |
| LOC106559343 | p.Ala179Gly | |
| LOC111097338 | p.Arg182Trp | |
| OR2A14 | olfactory receptor family 2 subfamily A member 14 | p.Ser56Arg |
| OR2A14 | olfactory receptor family 2 subfamily A member 14 | p.Leu54Gln |
| PCK2 | phosphoenolpyruvate carboxykinase 2, mitochondrial | p.Arg553Gln |
| TM9SF1 | transmembrane 9 superfamily member 1 | p.His362Tyr |
| Dogs | G/G | G/A |
|---|---|---|
| Cases Shetland Sheepdogs (n = 4) | 0 | 4 |
| Controls Shetland Sheepdogs (n = 117) | 117 | 0 |
| Controls other breeds (n = 515) 1 | 515 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nessler, J.; Hug, P.; Mandigers, P.J.J.; Leegwater, P.A.J.; Jagannathan, V.; Das, A.M.; Rosati, M.; Matiasek, K.; Sewell, A.C.; Kornberg, M.; et al. Mitochondrial PCK2 Missense Variant in Shetland Sheepdogs with Paroxysmal Exercise-Induced Dyskinesia (PED). Genes 2020, 11, 774. https://doi.org/10.3390/genes11070774
Nessler J, Hug P, Mandigers PJJ, Leegwater PAJ, Jagannathan V, Das AM, Rosati M, Matiasek K, Sewell AC, Kornberg M, et al. Mitochondrial PCK2 Missense Variant in Shetland Sheepdogs with Paroxysmal Exercise-Induced Dyskinesia (PED). Genes. 2020; 11(7):774. https://doi.org/10.3390/genes11070774
Chicago/Turabian StyleNessler, Jasmin, Petra Hug, Paul J. J. Mandigers, Peter A. J. Leegwater, Vidhya Jagannathan, Anibh M. Das, Marco Rosati, Kaspar Matiasek, Adrian C. Sewell, Marion Kornberg, and et al. 2020. "Mitochondrial PCK2 Missense Variant in Shetland Sheepdogs with Paroxysmal Exercise-Induced Dyskinesia (PED)" Genes 11, no. 7: 774. https://doi.org/10.3390/genes11070774
APA StyleNessler, J., Hug, P., Mandigers, P. J. J., Leegwater, P. A. J., Jagannathan, V., Das, A. M., Rosati, M., Matiasek, K., Sewell, A. C., Kornberg, M., Hoffmann, M., Wolf, P., Fischer, A., Tipold, A., & Leeb, T. (2020). Mitochondrial PCK2 Missense Variant in Shetland Sheepdogs with Paroxysmal Exercise-Induced Dyskinesia (PED). Genes, 11(7), 774. https://doi.org/10.3390/genes11070774