Holding All the Cards—How Fanconi Anemia Proteins Deal with Replication Stress and Preserve Genomic Stability


{kind=link}
{kind=link}
{kind=link}
Abstract
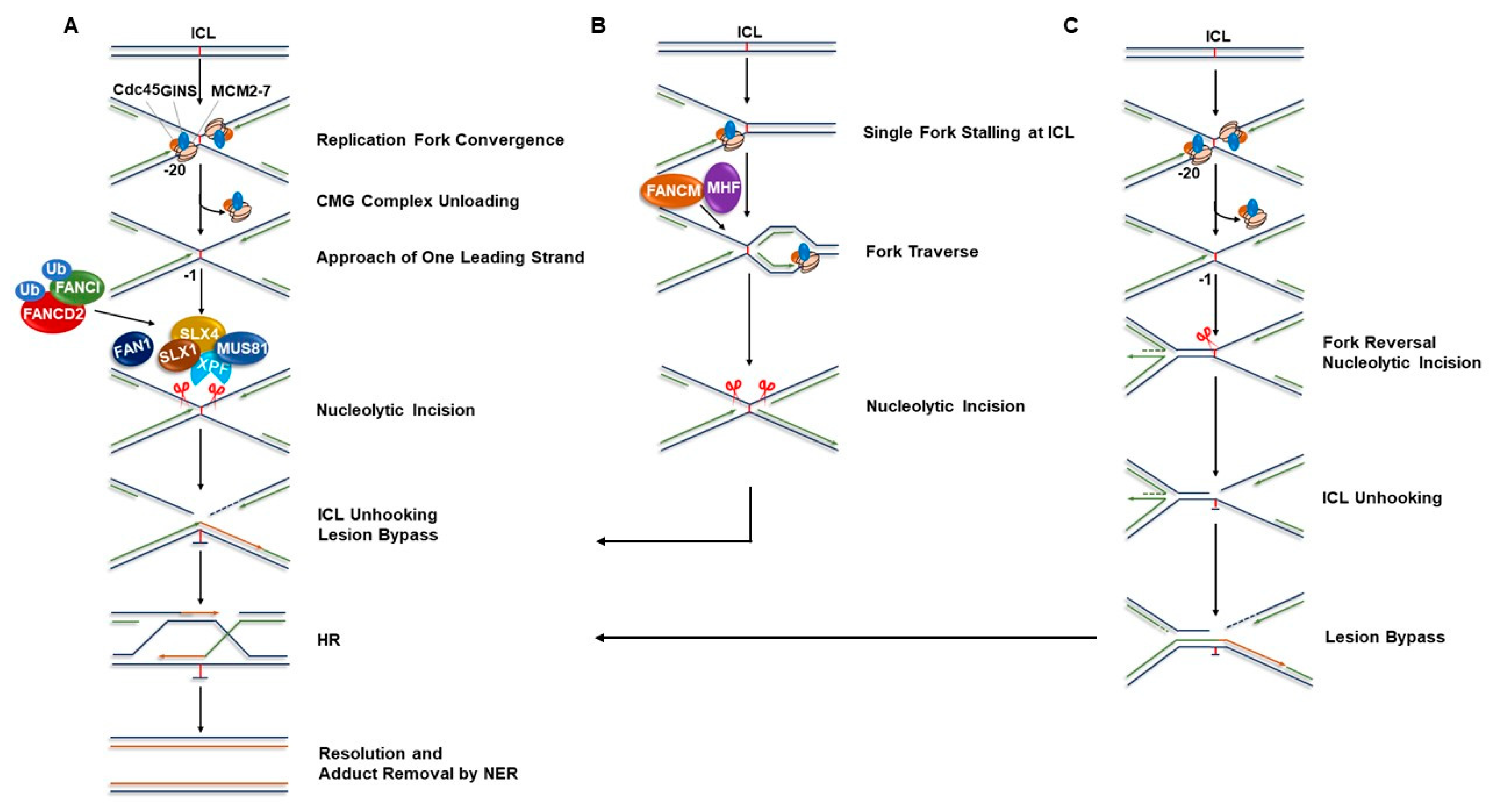
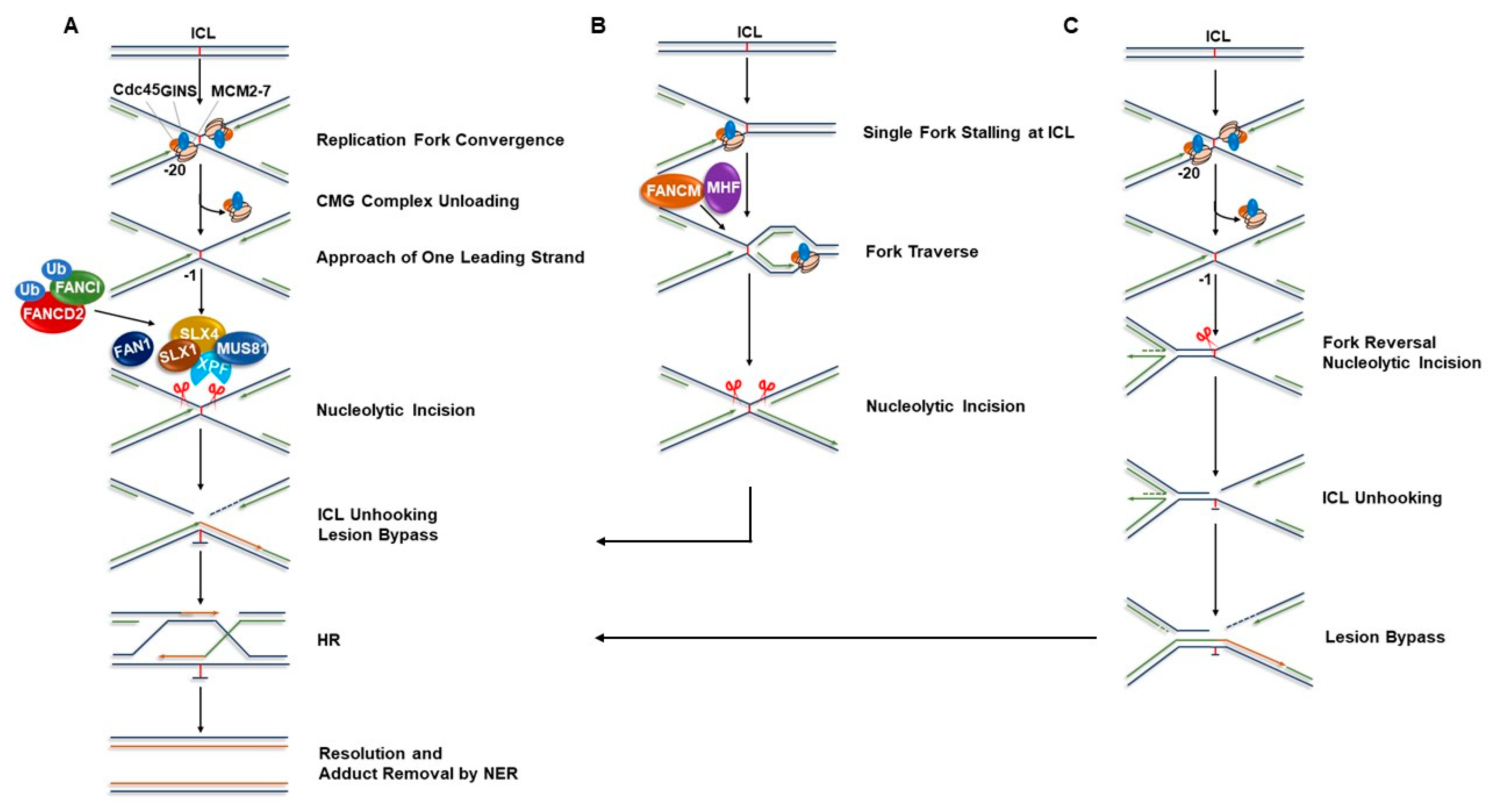
:1. FA Pathway and Models of Interstrand Cross-link Repair
2. Functional Involvement of FA Proteins with Players in Other DNA Repair Pathways
2.1. Nucleotide Excision Repair
2.2. Double-Strand Break Repair
2.3. Mismatch Repair
3. Involvement of the FA Pathway in Replication Fork Stabilization
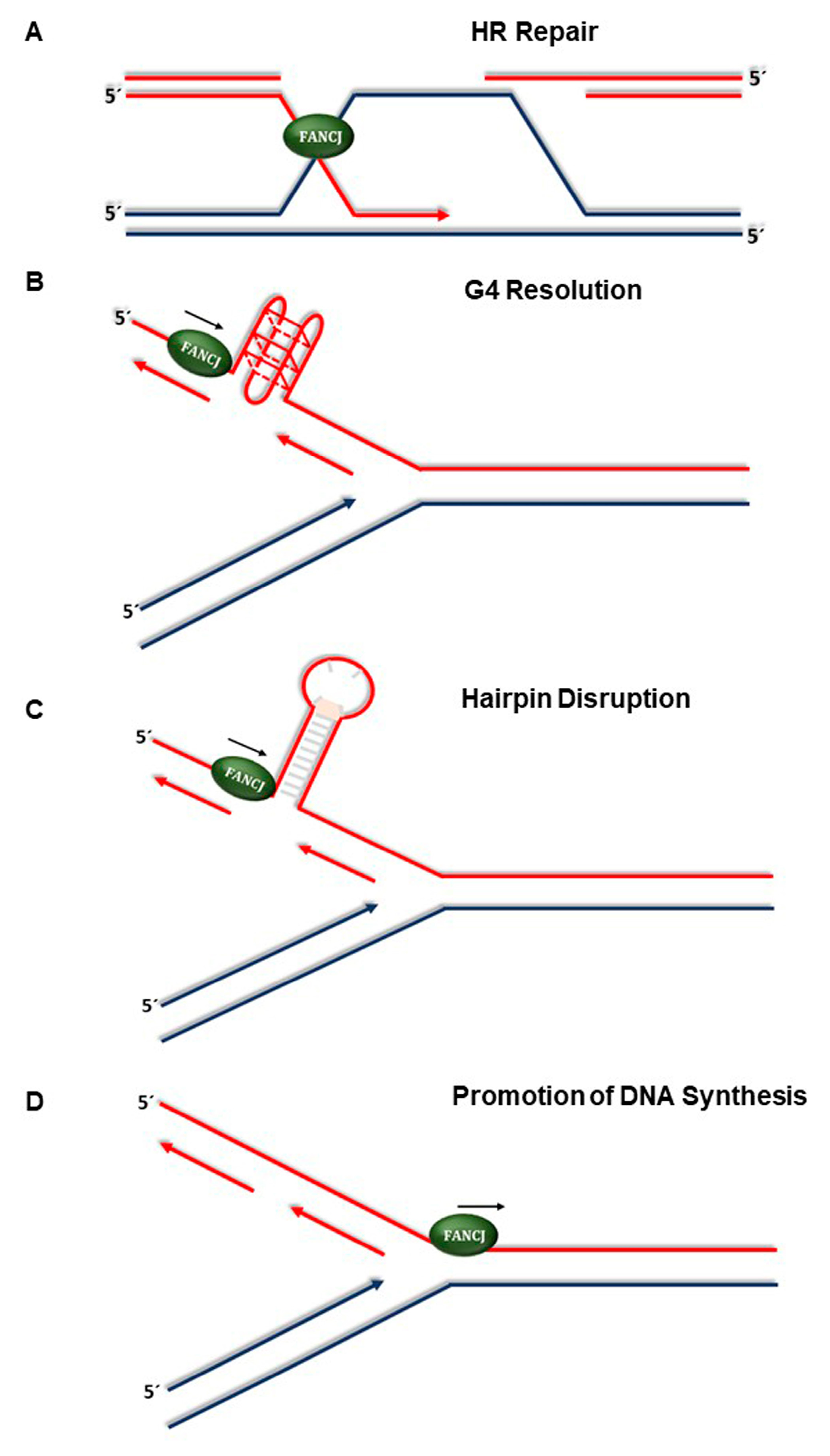
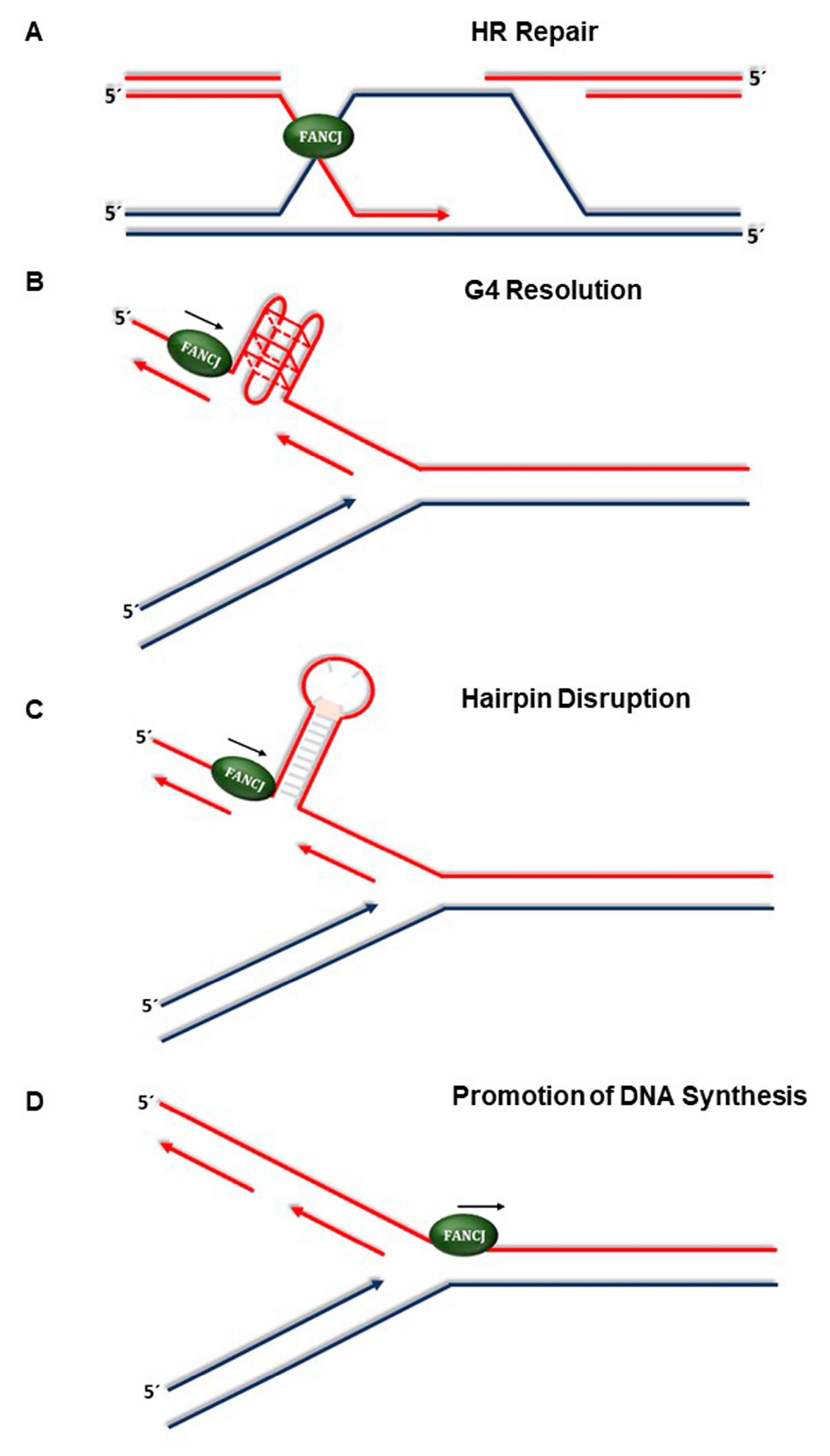
4. New Developments in Understanding FANCJ’s Role in the Replication Stress Response
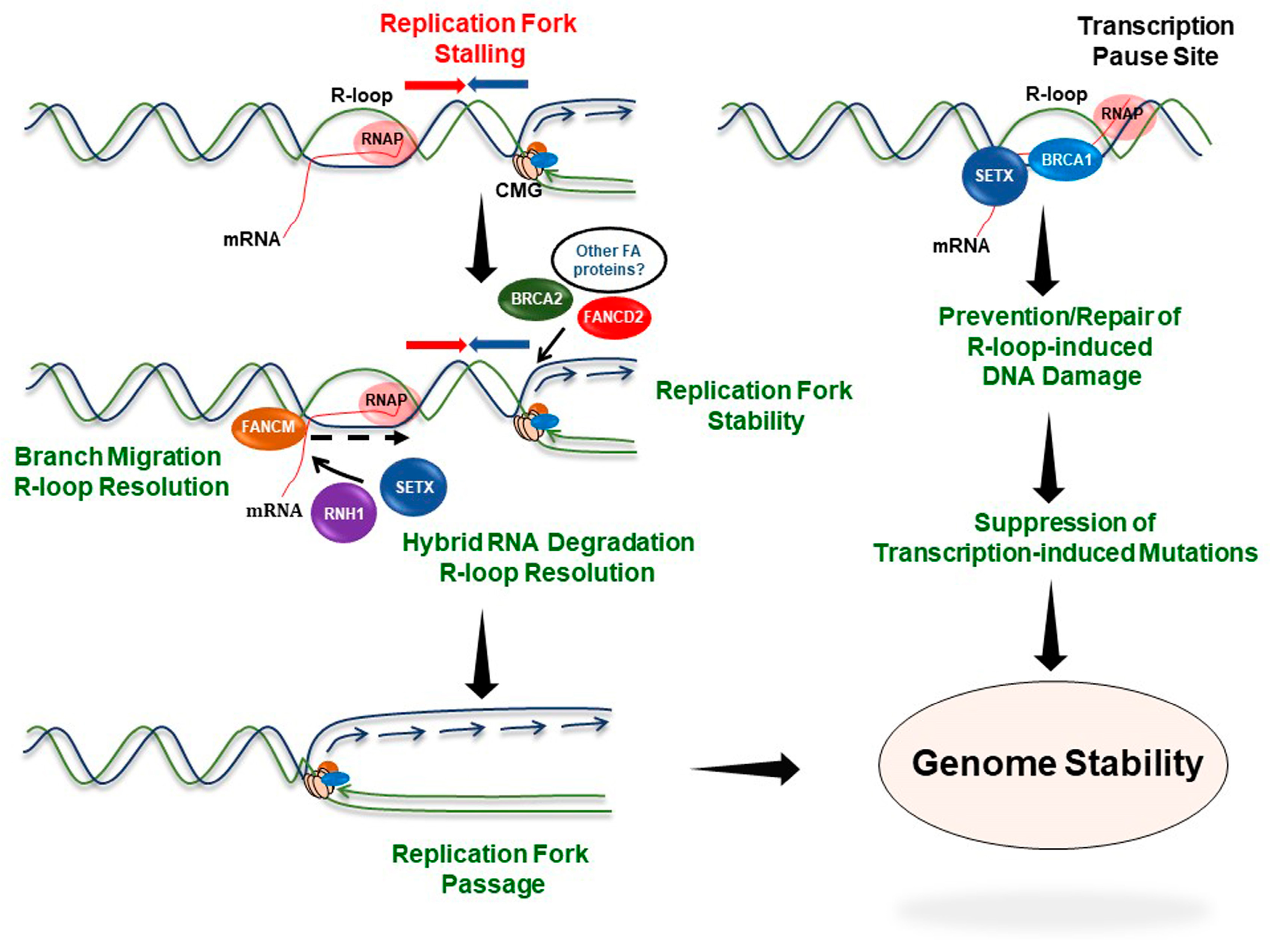
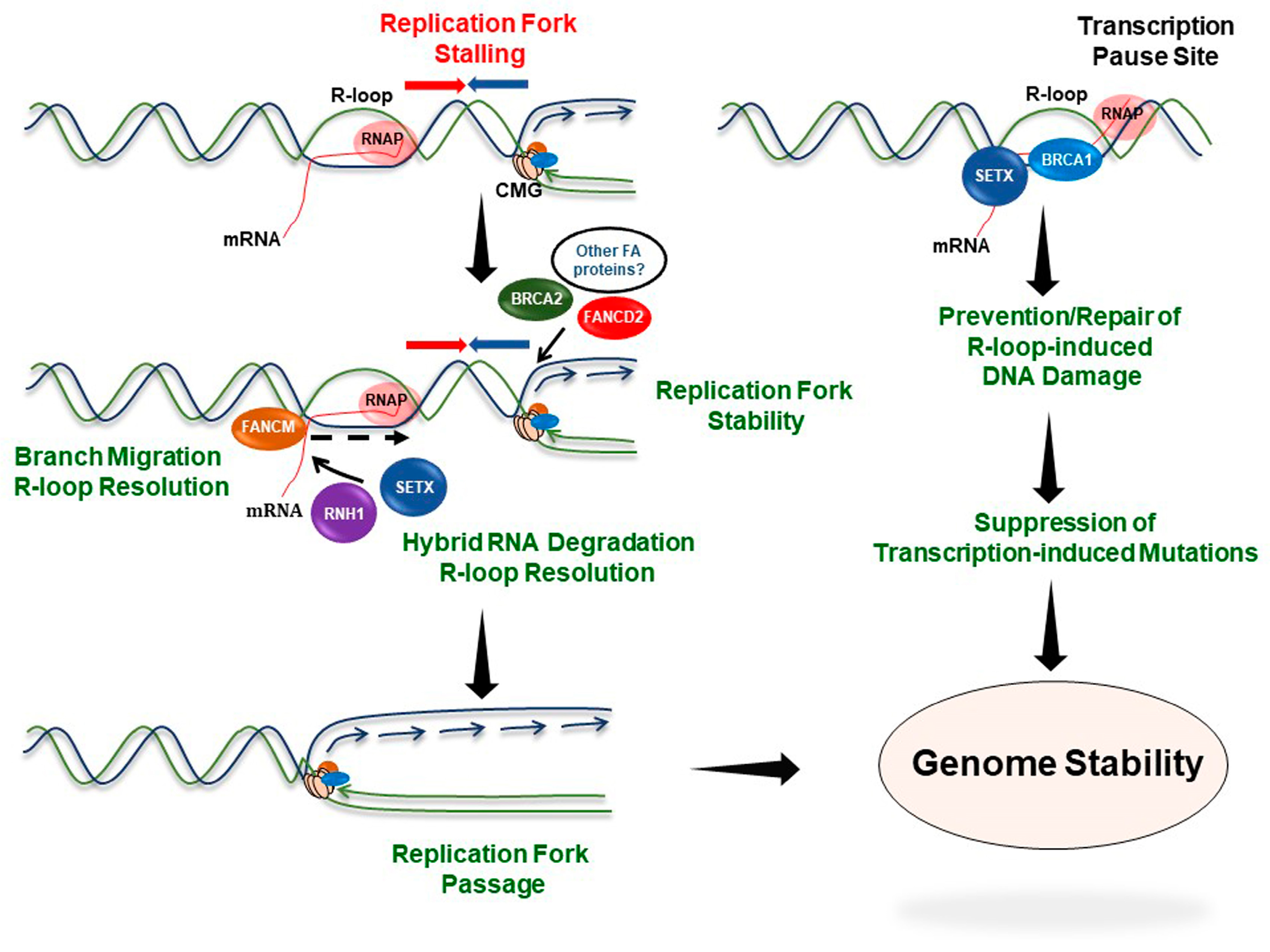
5. Involvement of FA Proteins in the Response to R-Loop-Induced Replication Stress
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kee, Y.; D’Andrea, A.D. Molecular pathogenesis and clinical management of Fanconi Anemia. J. Clin. Investig. 2012, 122, 3799–3806. [Google Scholar] [CrossRef] [PubMed]
- Romick-Rosendale, L.E.; Hoskins, E.E.; Privette Vinnedge, L.M.; Foglesong, G.D.; Brusadelli, M.G.; Potter, S.S.; Komurov, K.; Brugmann, S.A.; Lambert, P.F.; Kimple, R.J.; et al. Defects in the Fanconi Anemia pathway in head and neck cancer cells stimulate tumor cell invasion through DNA-PK and Rac1 signaling. Clin. Cancer Res. 2016, 22, 2062–2073. [Google Scholar] [CrossRef] [PubMed]
- Romick-Rosendale, L.E.; Lui, V.W.; Grandis, J.R.; Wells, S.I. The Fanconi anemia pathway: Repairing the link between DNA damage and squamous cell carcinoma. Mutat. Res. 2013, 743–744, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Bochtler, T.; Frohling, S.; Kramer, A. Role of chromosomal aberrations in clonal diversity and progression of acute myeloid leukemia. Leukemia 2015, 29, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; D’Andrea, A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012, 26, 1393–1408. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Knipscheer, P.; Raschle, M.; Smogorzewska, A.; Enoiu, M.; Ho, T.V.; Scharer, O.D.; Elledge, S.J.; Walter, J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009, 326, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Raschle, M.; Knipscheer, P.; Enoiu, M.; Angelov, T.; Sun, J.; Griffith, J.D.; Ellenberger, T.E.; Scharer, O.D.; Walter, J.C. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 2008, 134, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Clauson, C.; Scharer, O.D.; Niedernhofer, L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harbor Perspect. Biol. 2013, 5, a012732. [Google Scholar] [CrossRef] [PubMed]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Pontel, L.B.; Rosado, I.V.; Burgos-Barragan, G.; Garaycoechea, J.I.; Yu, R.; Arends, M.J.; Chandrasekaran, G.; Broecker, V.; Wei, W.; Liu, L.; et al. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol. Cell 2015, 60, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M., Jr.; Bellani, M.; Liu, Y.; Seidman, M.M. Fanconi Anemia: A DNA repair disorder characterized by accelerated decline of the hematopoietic stem cell compartment and other features of aging. Ageing Res. Rev. 2017, 33, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Cole, R.S.; Sinden, R.R. Repair of cross-linked DNA in Escherichia coli. Basic Life Sci. 1975, 5b, 487–495. [Google Scholar] [PubMed]
- Lin, P.F.; Bardwell, E.; Howard-Flanders, P. Initiation of genetic exchanges in lambda phage—Prophage crosses. Proc. Natl. Acad. Sci. USA 1977, 74, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Sinden, R.R.; Cole, R.S. Repair of cross-linked DNA and survival of Escherichia coli treated with psoralen and light: Effects of mutations influencing genetic recombination and DNA metabolism. J. Bacteriol. 1978, 136, 538–547. [Google Scholar] [PubMed]
- Berardini, M.; Foster, P.L.; Loechler, E.L. DNA polymerase II (polB) is involved in a new DNA repair pathway for DNA interstrand cross-links in Escherichia coli. J. Bacteriol. 1999, 181, 2878–2882. [Google Scholar] [PubMed]
- Berardini, M.; Mackay, W.; Loechler, E.L. Evidence for a recombination-independent pathway for the repair of DNA interstrand cross-links based on a site-specific study with nitrogen mustard. Biochemistry 1997, 36, 3506–3513. [Google Scholar] [CrossRef] [PubMed]
- Daee, D.L.; Myung, K. Fanconi-like crosslink repair in yeast. Genome Integr. 2012, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- McHugh, P.J.; Ward, T.A.; Chovanec, M. A prototypical Fanconi anemia pathway in lower eukaryotes? Cell Cycle 2012, 11, 3739–3744. [Google Scholar] [CrossRef] [PubMed]
- Strathdee, C.A.; Gavish, H.; Shannon, W.R.; Buchwald, M. Cloning of cDNAs for Fanconi’s anaemia by functional complementation. Nature 1992, 356, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Surralles, J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 2015, 33, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Gueiderikh, A.; Rosselli, F.; Neto, J.B.C. A never-ending story: The steadily growing family of the FA and FA-like genes. Genet. Mol. Biol. 2017, 40, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, S.; Wu, Z. Fanconi anemia pathway defects in inherited and sporadic cancers. Transl. Pediatr. 2014, 3, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Kawasoe, Y.; Williams, H.; Coates, E.; Roy, U.; Shi, Y.; Beese, L.S.; Scharer, O.D.; Yan, H.; Gottesman, M.E.; et al. Sensing and Processing of DNA Interstrand Crosslinks by the Mismatch Repair Pathway. Cell Rep. 2017, 21, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Davies, A.A.; Ulrich, H.D.; McHugh, P.J. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. EMBO J. 2006, 25, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.L.; Gottesman, M.E.; Gautier, J. Replication-independent repair of DNA interstrand crosslinks. Mol. Cell 2012, 47, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Enoiu, M.; Jiricny, J.; Scharer, O.D. Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res. 2012, 40, 8953–8964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Walter, J.C. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair 2014, 19, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Lalai, A.S.; Hoeijmakers, J.H. Fanconi anemia (cross)linked to DNA repair. Cell 2005, 123, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 2007, 8, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Long, D.T.; Joukov, V.; Budzowska, M.; Walter, J.C. BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol. Cell 2014, 56, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dewar, J.M.; Budzowska, M.; Motnenko, A.; Cohn, M.A.; Walter, J.C. DNA interstrand cross-link repair requires replication-fork convergence. Nat. Struct. Mol. Biol. 2015, 22, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Berezney, R.; Dubey, D.D.; Huberman, J.A. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 2000, 108, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, S.; Bellani, M.A.; Thazhathveetil, A.K.; Ling, C.; de Winter, J.P.; Wang, Y.; Wang, W.; Seidman, M.M. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol. Cell 2013, 52, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Huang, J.; Yan, Z.; Li, Y.; Ohzeki, M.; Ishiai, M.; Xu, D.; Takata, M.; Seidman, M.; Wang, W. Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discov. 2016, 2, 16047. [Google Scholar] [CrossRef] [PubMed]
- Rohleder, F.; Huang, J.; Xue, Y.; Kuper, J.; Round, A.; Seidman, M.; Wang, W.; Kisker, C. FANCM interacts with PCNA to promote replication traverse of DNA interstrand crosslinks. Nucleic Acids Res. 2016, 44, 3219–3232. [Google Scholar] [CrossRef] [PubMed]
- Mosedale, G.; Niedzwiedz, W.; Alpi, A.; Perrina, F.; Pereira-Leal, J.B.; Johnson, M.; Langevin, F.; Pace, P.; Patel, K.J. The vertebrate Hef ortholog is a component of the Fanconi anemia tumor-suppressor pathway. Nat. Struct. Mol. Biol. 2005, 12, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Meetei, A.R.; Medhurst, A.L.; Ling, C.; Xue, Y.; Singh, T.R.; Bier, P.; Steltenpool, J.; Stone, S.; Dokal, I.; Mathew, C.G.; et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 2005, 37, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Kee, Y.; Gurtan, A.; D’Andrea, A.D. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood 2008, 111, 5215–5222. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Leung, J.W.; Jiang, Y.; Lowery, M.G.; Do, H.; Vasquez, K.M.; Chen, J.; Wang, W.; Li, L. FANCM and FAAP24 maintain genome stability via cooperative as well as unique functions. Mol. Cell 2013, 49, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Mutreja, K.; Krietsch, J.; Hess, J.; Ursich, S.; Berti, M.; Roessler, F.K.; Zellweger, R.; Patra, M.; Gasser, G.; Lopes, M. ATR-mediated global fork slowing and reversal assist fork traverse and prevent chromosomal breakage at DNA interstrand cross-links. Cell Rep. 2018, 24, 2629–2642.e25. [Google Scholar] [CrossRef] [PubMed]
- Semlow, D.R.; Zhang, J.; Budzowska, M.; Drohat, A.C.; Walter, J.C. Replication-dependent unhooking of DNA interstrand cross-Links by the NEIL3 glycosylase. Cell 2016, 167, 498–511.e14. [Google Scholar] [CrossRef] [PubMed]
- Amunugama, R.; Willcox, S.; Wu, R.A.; Abdullah, U.B.; El-Sagheer, A.H.; Brown, T.; McHugh, P.J.; Griffith, J.D.; Walter, J.C. Replication fork reversal during DNA interstrand crosslink repair requires CMG unloading. Cell Rep. 2018, 23, 3419–3428. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.E.; Oshima, J.; Fu, Y.H.; Wijsman, E.M.; Hisama, F.; Alisch, R.; Matthews, S.; Nakura, J.; Miki, T.; Ouais, S.; et al. Positional cloning of the Werner’s syndrome gene. Science 1996, 272, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.T.; Kim, T.; Wagner, J.E.; Conti, B.A.; Lach, F.P.; Huang, A.L.; Molina, H.; Sanborn, E.M.; Zierhut, H.; Cornes, B.K.; et al. A dominant mutation in human RAD51 reveals its function in DNA interstrand crosslink repair independent of homologous recombination. Mol. Cell 2015, 59, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Mukherjee, S.; Yang, Y.; Mori, E.; Bhattacharya, S.; Kobayashi, J.; Yannone, S.M.; Chen, D.J.; Asaithamby, A. Nonenzymatic role for WRN in preserving nascent DNA strands after replication stress. Cell Rep. 2014, 9, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Iannascoli, C.; Palermo, V.; Murfuni, I.; Franchitto, A.; Pichierri, P. The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res. 2015, 43, 9788–9803. [Google Scholar] [CrossRef] [PubMed]
- Scharer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harbor Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- DiGiovanna, J.J.; Kraemer, K.H. Shining a light on xeroderma pigmentosum. J. Investig. Dermatol. 2012, 132, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Saffi, J.; Agnoletto, M.H.; Guecheva, T.N.; Batista, L.F.; Carvalho, H.; Henriques, J.A.; Stary, A.; Menck, C.F.; Sarasin, A. Effect of the anti-neoplastic drug doxorubicin on XPD-mutated DNA repair-deficient human cells. DNA Repair 2010, 9, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Budzowska, M.; Modesti, M.; Maas, A.; Wyman, C.; Essers, J.; Kanaar, R. The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J. 2006, 25, 4921–4932. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, N.G.; Raams, A.; Silengo, M.C.; Wijgers, N.; Niedernhofer, L.J.; Robinson, A.R.; Giglia-Mari, G.; Hoogstraten, D.; Kleijer, W.J.; Hoeijmakers, J.H.; et al. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am. J. Hum. Genet. 2007, 80, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka, I.; Kobertz, W.R.; Ariza, R.R.; Biggerstaff, M.; Essigmann, J.M.; Wood, R.D. Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. J. Biol. Chem. 2000, 275, 26632–26636. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Odijk, H.; Budzowska, M.; van Drunen, E.; Maas, A.; Theil, A.F.; de Wit, J.; Jaspers, N.G.; Beverloo, H.B.; Hoeijmakers, J.H.; et al. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell. Biol. 2004, 24, 5776–5787. [Google Scholar] [CrossRef] [PubMed]
- Prasher, J.M.; Lalai, A.S.; Heijmans-Antonissen, C.; Ploemacher, R.E.; Hoeijmakers, J.H.; Touw, I.P.; Niedernhofer, L.J. Reduced hematopoietic reserves in DNA interstrand crosslink repair-deficient Ercc1-/- mice. EMBO J. 2005, 24, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Schuster, B.; Stoepker, C.; Derkunt, B.; Su, Y.; Raams, A.; Trujillo, J.P.; Minguillon, J.; Ramirez, M.J.; Pujol, R.; et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Klein Douwel, D.; Boonen, R.A.; Long, D.T.; Szypowska, A.A.; Raschle, M.; Walter, J.C.; Knipscheer, P. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell 2014, 54, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.N.; Kobayashi, S.; Tsuda, M.; Kurumizaka, H.; Takata, M.; Kono, K.; Jiricny, J.; Takeda, S.; Hirota, K. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 6492–6496. [Google Scholar] [CrossRef] [PubMed]
- Hodskinson, M.R.; Silhan, J.; Crossan, G.P.; Garaycoechea, J.I.; Mukherjee, S.; Johnson, C.M.; Scharer, O.D.; Patel, K.J. Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF-ERCC1 in DNA crosslink repair. Mol. Cell 2014, 54, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Benitez, A.; Yuan, F.; Nakajima, S.; Wei, L.; Qian, L.; Myers, R.; Hu, J.J.; Lan, L.; Zhang, Y. Damage-dependent regulation of MUS81-EME1 by Fanconi anemia complementation group A protein. Nucleic Acids Res. 2014, 42, 1671–1683. [Google Scholar] [CrossRef] [PubMed]
- Interthal, H.; Heyer, W.D. MUS81 encodes a novel helix-hairpin-helix protein involved in the response to UV- and methylation-induced DNA damage in Saccharomyces cerevisiae. Mol. Gen. Genet. 2000, 263, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Pathania, S.; Nguyen, J.; Hill, S.J.; Scully, R.; Adelmant, G.O.; Marto, J.A.; Feunteun, J.; Livingston, D.M. BRCA1 is required for postreplication repair after UV-induced DNA damage. Mol. Cell 2011, 44, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Kelsall, I.R.; Langenick, J.; MacKay, C.; Patel, K.J.; Alpi, A.F. The Fanconi anaemia components UBE2T and FANCM are functionally linked to nucleotide excision repair. PLoS ONE 2012, 7, e36970. [Google Scholar] [CrossRef] [PubMed]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P. The association between FANCD1/BRCA2 mutations and leukaemia. Br. J. Haematol 2006, 133, 446–448; author reply 448. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.E.; Tolar, J.; Levran, O.; Scholl, T.; Deffenbaugh, A.; Satagopan, J.; Ben-Porat, L.; Mah, K.; Batish, S.D.; Kutler, D.I.; et al. Germline mutations in BRCA2: Shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anemia. Blood 2004, 103, 3226–3229. [Google Scholar] [CrossRef] [PubMed]
- Navarro, S.; Meza, N.W.; Quintana-Bustamante, O.; Casado, J.A.; Jacome, A.; McAllister, K.; Puerto, S.; Surralles, J.; Segovia, J.C.; Bueren, J.A. Hematopoietic dysfunction in a mouse model for Fanconi anemia group D1. Mol. Ther. 2006, 14, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Dorsman, J.C.; Ameziane, N.; de Vries, Y.; Rooimans, M.A.; Sheng, Q.; Pals, G.; Errami, A.; Gluckman, E.; Llera, J.; et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat. Genet. 2007, 39, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.; Schindler, D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Freund, M.; et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2007, 39, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Ameziane, N.; May, P.; Haitjema, A.; van de Vrugt, H.J.; van Rossum-Fikkert, S.E.; Ristic, D.; Williams, G.J.; Balk, J.; Rockx, D.; Li, H.; et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat. Commun. 2015, 6, 8829. [Google Scholar] [CrossRef] [PubMed]
- Domchek, S.M.; Tang, J.; Stopfer, J.; Lilli, D.R.; Hamel, N.; Tischkowitz, M.; Monteiro, A.N.; Messick, T.E.; Powers, J.; Yonker, A.; et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 2013, 3, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.T.; Smogorzewska, A. SnapShot: Fanconi anemia and associated proteins. Cell 2015, 160, 354–354.e1. [Google Scholar] [CrossRef] [PubMed]
- Cantor, S.B.; Bell, D.W.; Ganesan, S.; Kass, E.M.; Drapkin, R.; Grossman, S.; Wahrer, D.C.; Sgroi, D.C.; Lane, W.S.; Haber, D.A.; et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 2001, 105, 149–160. [Google Scholar] [CrossRef]
- Levitus, M.; Waisfisz, Q.; Godthelp, B.C.; de Vries, Y.; Hussain, S.; Wiegant, W.W.; Elghalbzouri-Maghrani, E.; Steltenpool, J.; Rooimans, M.A.; Pals, G.; et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J. Nat. Genet. 2005, 37, 934–935. [Google Scholar] [CrossRef] [PubMed]
- Levran, O.; Attwooll, C.; Henry, R.T.; Milton, K.L.; Neveling, K.; Rio, P.; Batish, S.D.; Kalb, R.; Velleuer, E.; Barral, S.; et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat. Genet. 2005, 37, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Litman, R.; Peng, M.; Jin, Z.; Zhang, F.; Zhang, J.; Powell, S.; Andreassen, P.R.; Cantor, S.B. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 2005, 8, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. The role of BRCA1 in homologous recombination repair in response to replication stress: Significance in tumorigenesis and cancer therapy. Cell Biosci. 2013, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fan, Q.; Ren, K.; Andreassen, P.R. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol. Cancer Res. 2009, 7, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- McHugh, P.J.; Spanswick, V.J.; Hartley, J.A. Repair of DNA interstrand crosslinks: Molecular mechanisms and clinical relevance. Lancet Oncol. 2001, 2, 483–490. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callen, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; Lopez-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 2012, 46, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Duquette, M.L.; Zhu, Q.; Taylor, E.R.; Tsay, A.J.; Shi, L.Z.; Berns, M.W.; McGowan, C.H. CtIP is required to initiate replication-dependent interstrand crosslink repair. PLoS Genet. 2012, 8, e1003050. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Higuera, I.; Taniguchi, T.; Ganesan, S.; Meyn, M.S.; Timmers, C.; Hejna, J.; Grompe, M.; D’Andrea, A.D. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell 2001, 7, 249–262. [Google Scholar] [CrossRef]
- Chen, X.; Wilson, J.B.; McChesney, P.; Williams, S.A.; Kwon, Y.; Longerich, S.; Marriott, A.S.; Sung, P.; Jones, N.J.; Kupfer, G.M. The Fanconi anemia proteins FANCD2 and FANCJ interact and regulate each other’s chromatin localization. J. Biol. Chem. 2014, 289, 25774–25782. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fan, Q.; Ren, K.; Auerbach, A.D.; Andreassen, P.R. FANCJ/BRIP1 recruitment and regulation of FANCD2 in DNA damage responses. Chromosoma 2010, 119, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Murina, O.; von Aesch, C.; Karakus, U.; Ferretti, L.P.; Bolck, H.A.; Hanggi, K.; Sartori, A.A. FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Rep. 2014, 7, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Unno, J.; Itaya, A.; Taoka, M.; Sato, K.; Tomida, J.; Sakai, W.; Sugasawa, K.; Ishiai, M.; Ikura, T.; Isobe, T.; et al. FANCD2 binds CtIP and regulates DNA-end resection during DNA interstrand crosslink repair. Cell Rep. 2014, 7, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Karanja, K.K.; Cox, S.W.; Duxin, J.P.; Stewart, S.A.; Campbell, J.L. DNA2 and EXO1 in replication-coupled, homology-directed repair and in the interplay between HDR and the FA/BRCA network. Cell Cycle 2012, 11, 3983–3996. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.G.; Herceg, Z.; Nakanishi, K.; Demuth, I.; Piccoli, C.; Michelon, J.; Hildebrand, G.; Jasin, M.; Digweed, M.; Wang, Z.Q. The Fanconi anemia group A protein modulates homologous repair of DNA double-strand breaks in mammalian cells. Carcinogenesis 2005, 26, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Yang, Y.G.; Pierce, A.J.; Taniguchi, T.; Digweed, M.; D’Andrea, A.D.; Wang, Z.Q.; Jasin, M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc. Natl. Acad. Sci. USA 2005, 102, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Cavallo, F.; Perrouault, L.; Giovannangeli, C.; Moynahan, M.E.; Barchi, M.; Brunet, E.; Jasin, M. Homology-directed Fanconi anemia pathway cross-link repair is dependent on DNA replication. Nat. Struct. Mol. Biol. 2011, 18, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Westermark, U.K.; Reyngold, M.; Olshen, A.B.; Baer, R.; Jasin, M.; Moynahan, M.E. BARD1 participates with BRCA1 in homology-directed repair of chromosome breaks. Mol. Cell. Biol. 2003, 23, 7926–7936. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef]
- Stark, J.M.; Pierce, A.J.; Oh, J.; Pastink, A.; Jasin, M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell. Biol. 2004, 24, 9305–9316. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M., Jr. DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer 2013, 13, 542–558. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, S.; Lee, L.; Davalos, A.; Schiestl, R.H.; Campisi, J.; Oshima, J. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell 2003, 2, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Saintigny, Y.; Makienko, K.; Swanson, C.; Emond, M.J.; Monnat, R.J., Jr. Homologous recombination resolution defect in Werner syndrome. Mol. Cell. Biol. 2002, 22, 6971–6978. [Google Scholar] [CrossRef] [PubMed]
- Sturzenegger, A.; Burdova, K.; Kanagaraj, R.; Levikova, M.; Pinto, C.; Cejka, P.; Janscak, P. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J. Biol. Chem. 2014, 289, 27314–27326. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.H.; Kusumoto, R.; Opresko, P.L.; Sui, X.; Huang, S.; Nicolette, M.L.; Paull, T.T.; Campisi, J.; Seidman, M.; Bohr, V.A. Collaboration of Werner syndrome protein and BRCA1 in cellular responses to DNA interstrand cross-links. Nucleic Acids Res. 2006, 34, 2751–2760. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Banerjee, T.; Sommers, J.A.; Iannascoli, C.; Pichierri, P.; Shoemaker, R.H.; Brosh, R.M., Jr. Werner syndrome helicase has a critical role in DNA damage responses in the absence of a functional fanconi anemia pathway. Cancer Res. 2013, 73, 5497–5507. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.M.; Yanez, D.A.; Stark, J.M. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 2015, 11, e1004943. [Google Scholar] [CrossRef] [PubMed]
- Benitez, A.; Liu, W.; Palovcak, A.; Wang, G.; Moon, J.; An, K.; Kim, A.; Zheng, K.; Zhang, Y.; Bai, F.; et al. FANCA promotes DNA double-strand break repair by catalyzing single-strand annealing and strand exchange. Mol. Cell 2018, 71, 621–628.e4. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; O’Regan, E.; Brown, R.; Karran, P. Selective recognition of a cisplatin-DNA adduct by human mismatch repair proteins. Nucleic Acids Res. 1997, 25, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Lu, X.; Zhang, X.; Peterson, C.A.; Legerski, R.J. hMutSbeta is required for the recognition and uncoupling of psoralen interstrand cross-links in vitro. Mol. Cell. Biol. 2002, 22, 2388–2397. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Jain, A.; Iyer, R.R.; Modrich, P.L.; Vasquez, K.M. Mismatch repair and nucleotide excision repair proteins cooperate in the recognition of DNA interstrand crosslinks. Nucleic Acids Res. 2009, 37, 4420–4429. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Liu, X.; Li, L.; Legerski, R. Double-strand breaks induce homologous recombinational repair of interstrand cross-links via cooperation of MSH2, ERCC1-XPF, REV3, and the Fanconi anemia pathway. DNA Repair 2007, 6, 1670–1678. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000, 14, 927–939. [Google Scholar] [PubMed]
- Svendsen, J.M.; Smogorzewska, A.; Sowa, M.E.; O’Connell, B.C.; Gygi, S.P.; Elledge, S.J.; Harper, J.W. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 2009, 138, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ghosal, G.; Yuan, J.; Chen, J.; Huang, J. FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 2010, 329, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Smogorzewska, A.; Desetty, R.; Saito, T.T.; Schlabach, M.; Lach, F.P.; Sowa, M.E.; Clark, A.B.; Kunkel, T.A.; Harper, J.W.; Colaiacovo, M.P.; et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell 2010, 39, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Kennedy, R.; Ali, A.M.; Moreau, L.A.; Meetei, A.R.; D’Andrea, A.D.; Chen, C.C. Human MutS and FANCM complexes function as redundant DNA damage sensors in the Fanconi Anemia pathway. DNA Repair 2011, 10, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.A.; Wilson, J.B.; Clark, A.P.; Mitson-Salazar, A.; Tomashevski, A.; Ananth, S.; Glazer, P.M.; Semmes, O.J.; Bale, A.E.; Jones, N.J.; et al. Functional and physical interaction between the mismatch repair and FA-BRCA pathways. Hum. Mol. Genet. 2011, 20, 4395–4410. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Litman, R.; Xie, J.; Sharma, S.; Brosh, R.M., Jr.; Cantor, S.B. The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 2007, 26, 3238–3249. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, E.; Gerrits, B.; Marra, G.; Schlapbach, R.; Jiricny, J. Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J. Biol. Chem. 2007, 282, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Kratz, K.; Schopf, B.; Kaden, S.; Sendoel, A.; Eberhard, R.; Lademann, C.; Cannavo, E.; Sartori, A.A.; Hengartner, M.O.; Jiricny, J. Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 2010, 142, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Xie, J.; Ucher, A.; Stavnezer, J.; Cantor, S.B. Crosstalk between BRCA-Fanconi anemia and mismatch repair pathways prevents MSH2-dependent aberrant DNA damage responses. EMBO J. 2014, 33, 1698–1712. [Google Scholar] [CrossRef] [PubMed]
- Quinet, A.; Lemacon, D.; Vindigni, A. Replication Fork Reversal: Players and Guardians. Mol. Cell 2017, 68, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, J. A game of substrates: Replication fork remodeling and its roles in genome stability and chemo-resistance. Cell Stress 2017, 1, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for stalled replication fork stabilization: New targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018. [Google Scholar] [CrossRef] [PubMed]
- Pasero, P.; Vindigni, A. Nucleases acting at stalled forks: How to reboot the replication program with a few shortcuts. Annu. Rev. Genet. 2017, 51, 477–499. [Google Scholar] [CrossRef] [PubMed]
- Howlett, N.G.; Taniguchi, T.; Durkin, S.G.; D’Andrea, A.D.; Glover, T.W. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet. 2005, 14, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Wilson, J.B.; Medhurst, A.L.; Hejna, J.; Witt, E.; Ananth, S.; Davies, A.; Masson, J.Y.; Moses, R.; West, S.C.; et al. Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum. Mol. Genet. 2004, 13, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Madireddy, A.; Kosiyatrakul, S.T.; Boisvert, R.A.; Herrera-Moyano, E.; Garcia-Rubio, M.L.; Gerhardt, J.; Vuono, E.A.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 facilitates replication through common fragile sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Lachaud, C.; Moreno, A.; Marchesi, F.; Toth, R.; Blow, J.J.; Rouse, J. Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science 2016, 351, 846–849. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Zimmer, J.; Buffa, F.M.; McDermott, U.; Tarsounas, M. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat. Struct. Mol. Biol. 2016, 23, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Kais, Z.; Rondinelli, B.; Holmes, A.; O’Leary, C.; Kozono, D.; D’Andrea, A.D.; Ceccaldi, R. FANCD2 maintains fork stability in BRCA1/2-deficient tumors and promotes alternative end-joining DNA Repair. Cell Rep. 2016, 15, 2488–2499. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Son, M.Y.; Dodds, S.; Hu, L.; Luo, G.; Hasty, P. RECQL5 and BLM exhibit divergent functions in cells defective for the Fanconi anemia pathway. Nucleic Acids Res. 2015, 43, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Cong, K.; Panzarino, N.J.; Nayak, S.; Calvo, J.; Deng, B.; Zhu, L.J.; Morocz, M.; Hegedus, L.; Haracska, L.; et al. Opposing roles of FANCJ and HLTF protect forks and restrain replication during stress. Cell Rep. 2018, 24, 3251–3261. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Reynolds, J.J.; Winczura, A.; Blackford, A.N.; Borel, V.; Miller, E.S.; Zlatanou, A.; Nieminuszczy, J.; Ryan, E.L.; Davies, N.J.; et al. BOD1L Is required to suppress deleterious resection of stressed replication forks. Mol. Cell 2015, 59, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, I.; Sareen, A.; Raghunandan, M.; Sobeck, A. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res. 2013, 41, 6444–6459. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, I.; Stroik, D.R.; Sobeck, A. FANCD2-controlled chromatin access of the Fanconi-associated nuclease FAN1 is crucial for the recovery of stalled replication forks. Mol. Cell. Biol. 2014, 34, 3939–3954. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.E.; Lee, E.H.; Hendrickson, E.A.; Sobeck, A. CtIP mediates replication fork recovery in a FANCD2-regulated manner. Hum. Mol. Genet. 2014, 23, 3695–3705. [Google Scholar] [CrossRef] [PubMed]
- Raghunandan, M.; Chaudhury, I.; Kelich, S.L.; Hanenberg, H.; Sobeck, A. FANCD2, FANCJ and BRCA2 cooperate to promote replication fork recovery independently of the Fanconi Anemia core complex. Cell Cycle 2015, 14, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Lossaint, G.; Larroque, M.; Ribeyre, C.; Bec, N.; Larroque, C.; Decaillet, C.; Gari, K.; Constantinou, A. FANCD2 binds MCM proteins and controls replisome function upon activation of S phase checkpoint signaling. Mol. Cell 2013, 51, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Jones, M.J.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J., 3rd; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2015, 58, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Tonzi, P.; Yin, Y.; Lee, C.W.T.; Rothenberg, E.; Huang, T.T. Translesion polymerase kappa-dependent DNA synthesis underlies replication fork recovery. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Estep, K.N.; Brosh, R.M., Jr. RecQ and Fe-S helicases have unique roles in DNA metabolism dictated by their unwinding directionality, substrate specificity, and protein interactions. Biochem. Soc. Trans. 2018, 46, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Cantor, S.B.; Guillemette, S. Hereditary breast cancer and the BRCA1-associated FANCJ/BACH1/BRIP1. Future Oncol. 2011, 7, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Cantor, S.B.; Nayak, S. FANCJ at the FORK. Mutat. Res. 2016, 788, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M., Jr.; Cantor, S.B. Molecular and cellular functions of the FANCJ DNA helicase defective in cancer and in Fanconi Anemia. Front. Genet. 2014, 5, 372. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Awate, S.; Banerjee, T.; Brosh, R.M. Getting ready for the dance: FANCJ irons out DNA wrinkles. Genes 2016, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Sommers, J.A.; Awate, S.; Bellani, M.A.; Khan, I.; Bradley, L.; King, G.A.; Seol, Y.; Vidhyasagar, V.; Wu, Y.; et al. A minimal threshold of FANCJ helicase activity is required for its response to replication stress or double-strand break repair. Nucleic Acids Res. 2018, 46, 6238–6256. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sommers, J.A.; Suhasini, A.N.; Leonard, T.; Deakyne, J.S.; Mazin, A.V.; Shin-Ya, K.; Kitao, H.; Brosh, R.M., Jr. Fanconi anemia group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein-DNA complexes. Blood 2010, 116, 3780–3791. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Shin-ya, K.; Brosh, R.M., Jr. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol. Cell. Biol. 2008, 28, 4116–4128. [Google Scholar] [CrossRef] [PubMed]
- London, T.B.; Barber, L.J.; Mosedale, G.; Kelly, G.P.; Balasubramanian, S.; Hickson, I.D.; Boulton, S.J.; Hiom, K. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J. Biol. Chem. 2008, 283, 36132–36139. [Google Scholar] [CrossRef] [PubMed]
- Sarkies, P.; Murat, P.; Phillips, L.G.; Patel, K.J.; Balasubramanian, S.; Sale, J.E. FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res. 2012, 40, 1485–1498. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Sommers, J.A.; George, F.; Kuper, J.; Hamon, F.; Shin-ya, K.; Teulade-Fichou, M.P.; Kisker, C.; Brosh, R.M., Jr. Specialization among iron-sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability. J. Biol. Chem. 2013, 288, 28217–28229. [Google Scholar] [CrossRef] [PubMed]
- Castillo Bosch, P.; Segura-Bayona, S.; Koole, W.; van Heteren, J.T.; Dewar, J.M.; Tijsterman, M.; Knipscheer, P. FANCJ promotes DNA synthesis through G-quadruplex structures. EMBO J. 2014, 33, 2521–2533. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.G.; Spies, M. G-quadruplex recognition and remodeling by the FANCJ helicase. Nucleic Acids Res. 2016, 44, 8742–8753. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Helleday, T. DNA REPAIR. Drugging DNA repair. Science 2016, 352, 1178–1179. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Sommers, J.A.; Shoemaker, R.H.; Brosh, R.M., Jr. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Banerjee, T.; Sommers, J.A.; Brosh, R.M., Jr. Targeting an Achilles’ heel of cancer with a WRN helicase inhibitor. Cell Cycle 2013, 12, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.H.; Dexheimer, T.S.; Rosenthal, A.S.; Chu, W.K.; Singh, D.K.; Mosedale, G.; Bachrati, C.Z.; Schultz, L.; Sakurai, M.; Savitsky, P.; et al. A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem. Biol. 2013, 20, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Brosh, R.M., Jr. New insights into DNA helicases as druggable targets for cancer therapy. Front. Mol. Biosci. 2018, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Somyajit, K.; Mishra, A.; Scully, R.; Nagaraju, G. FANCJ helicase controls the balance between short- and long-tract gene conversions between sister chromatids. Nucleic Acids Res. 2017, 45, 8886–8900. [Google Scholar] [CrossRef] [PubMed]
- Barthelemy, J.; Hanenberg, H.; Leffak, M. FANCJ is essential to maintain microsatellite structure genome-wide during replication stress. Nucleic Acids Res. 2017, 45, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Borel, V.; Adelman, C.A.; Schindler, D.; Boulton, S.J. FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi Anemia pathway. Genes Dev. 2015, 29, 2532–2546. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rubio, M.L.; Perez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia pathway protects genome integrity from R-loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gomez-Gonzalez, B. DNA-RNA hybrids: The risks of DNA breakage during transcription. Nat. Struct. Mol. Biol. 2017, 24, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Garcia-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A. The connection between transcription and genomic instability. EMBO J. 2002, 21, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.Y.; Stirling, P.C. Replication fork protection factors controlling R-loop bypass and suppression. Genes 2017, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Barroso, S.I.; Garcia-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Hodson, C.; O’Rourke, J.J.; van Twest, S.; Murphy, V.J.; Dunn, E.; Deans, A.J. FANCM-family branchpoint translocases remove co-transcriptional R-loops. bioRxiv 2018. [Google Scholar] [CrossRef]
- Richard, P.; Manley, J.L. R loops and links to human disease. J. Mol. Biol. 2017, 429, 3168–3180. [Google Scholar] [CrossRef] [PubMed]
- Hills, S.A.; Diffley, J.F. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Datta, A.; Brosh, R.M., Jr. Holding All the Cards—How Fanconi Anemia Proteins Deal with Replication Stress and Preserve Genomic Stability. Genes 2019, 10, 170. https://doi.org/10.3390/genes10020170
Datta A, Brosh RM Jr. Holding All the Cards—How Fanconi Anemia Proteins Deal with Replication Stress and Preserve Genomic Stability. Genes. 2019; 10(2):170. https://doi.org/10.3390/genes10020170
Chicago/Turabian StyleDatta, Arindam, and Robert M. Brosh, Jr. 2019. "Holding All the Cards—How Fanconi Anemia Proteins Deal with Replication Stress and Preserve Genomic Stability" Genes 10, no. 2: 170. https://doi.org/10.3390/genes10020170
APA StyleDatta, A., & Brosh, R. M., Jr. (2019). Holding All the Cards—How Fanconi Anemia Proteins Deal with Replication Stress and Preserve Genomic Stability. Genes, 10(2), 170. https://doi.org/10.3390/genes10020170