De Novo Transcriptome Assembly of Agave H11648 by Illumina Sequencing and Identification of Cellulose Synthase Genes in Agave Species

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and RNA Extraction
2.2. Library Construction and Illumina Sequencing
2.3. De Novo Assembly and Functional Annotation
2.4. Identification and Cloning of CesA Genes
2.5. Phylogenetic Analysis
2.6. Expression Analysis
3. Results
3.1. Illumina Sequencing and De Novo Assembly
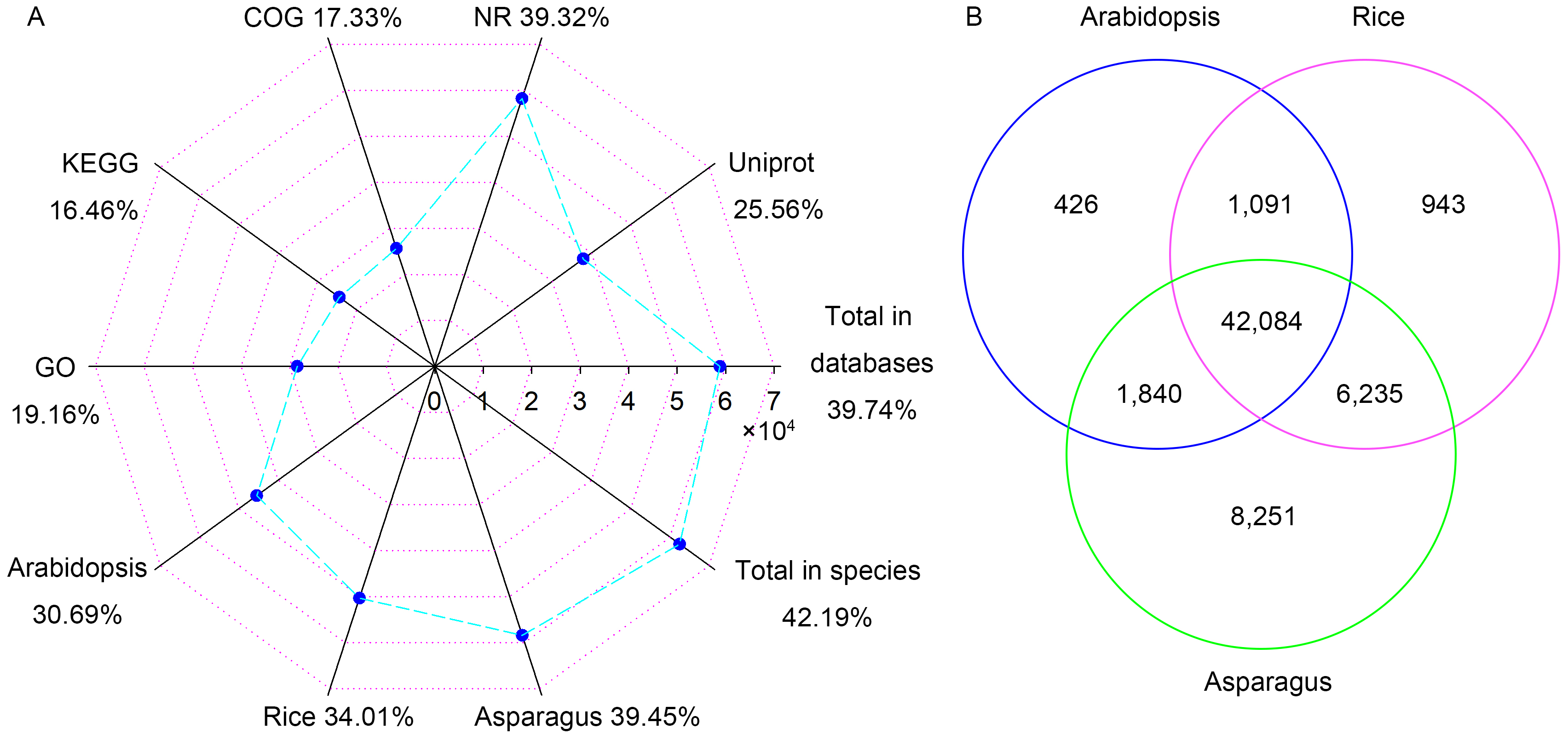
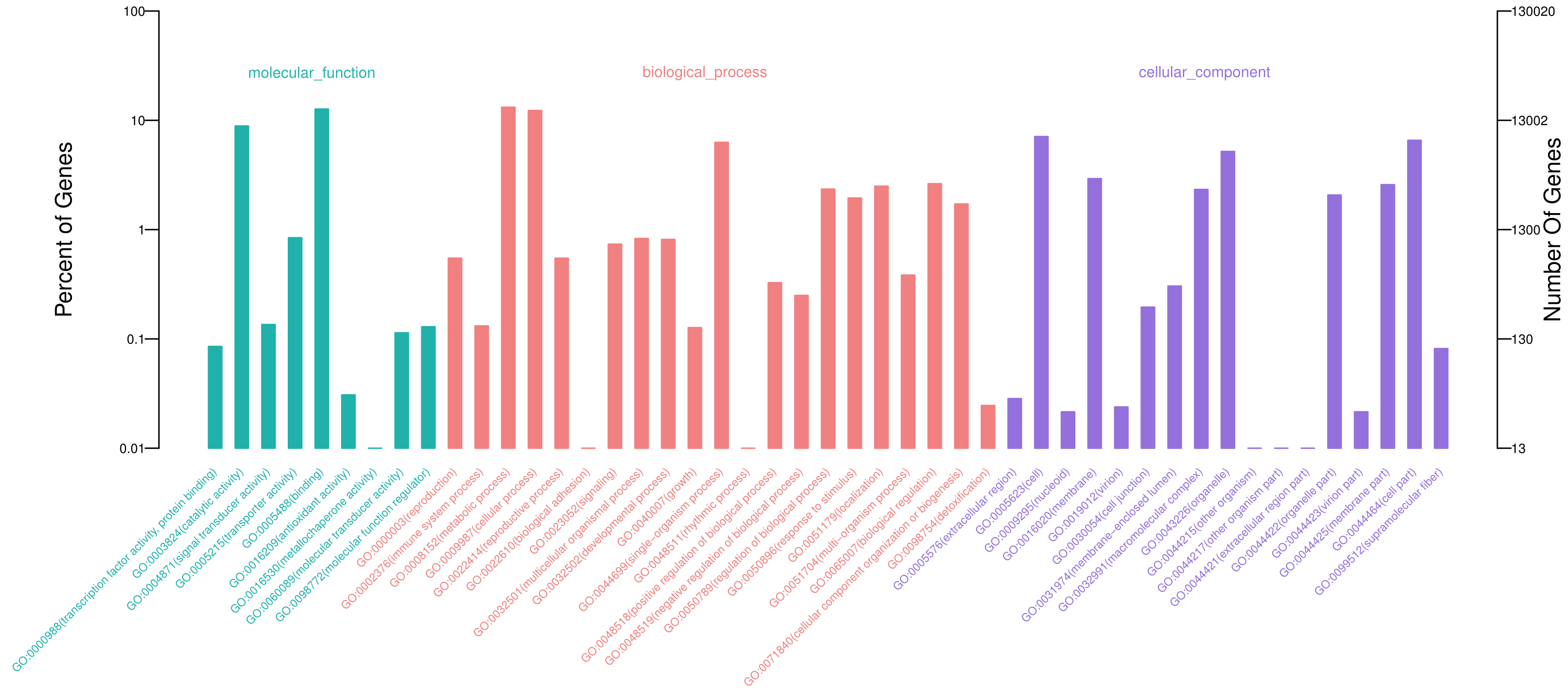
3.2. Functional Annotation
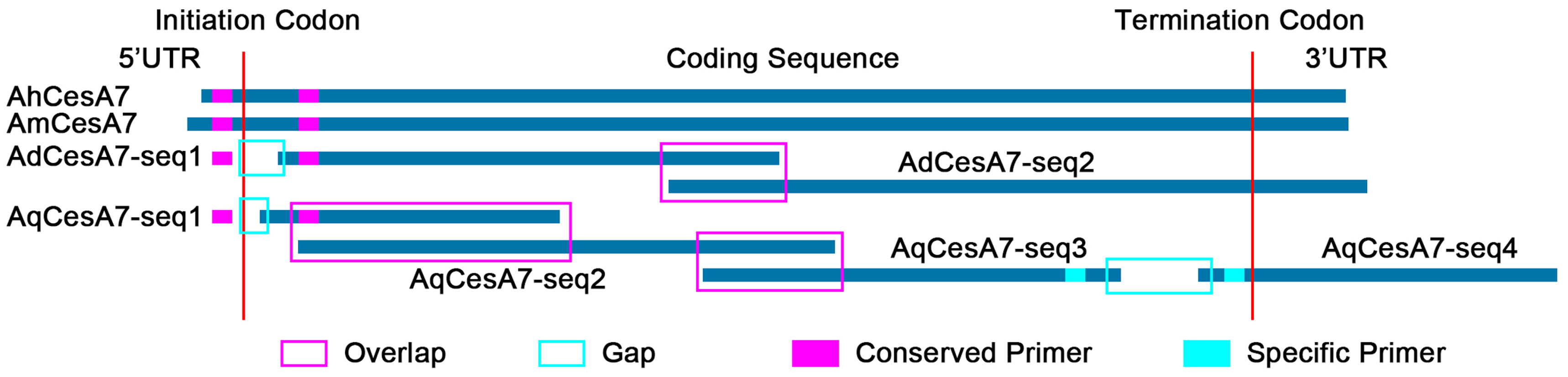
3.3. Identification and Cloning of CesA Genes
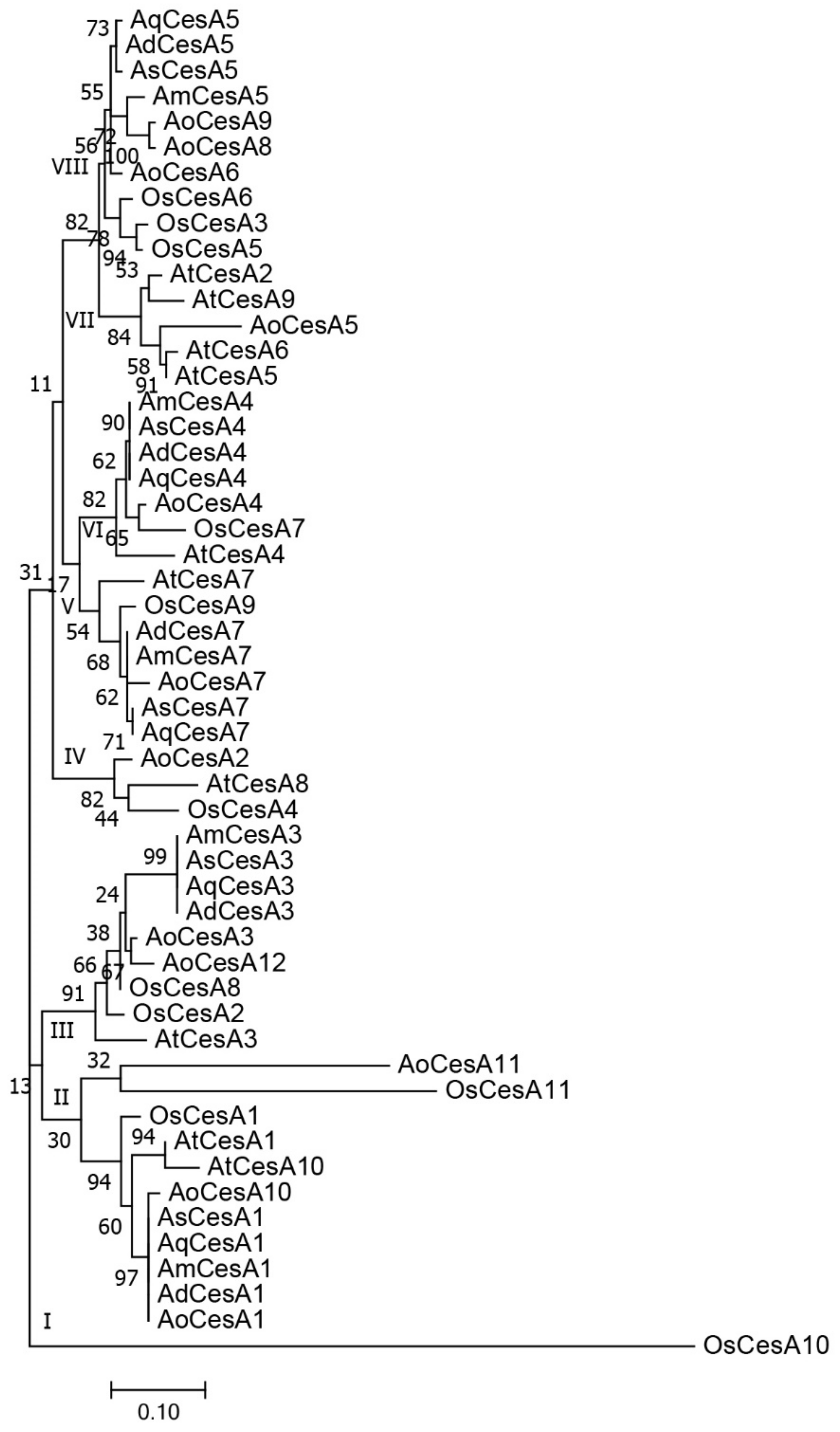
3.4. Phylogeny of CesA Genes
3.5. Expression Pattern of Agave CesA Genes
4. Discussion
4.1. Characterization of the A. H11648 Transcriptome
4.2. Potential Candidate CesA Genes Involved in Leaf Fiber Biosynthesis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Borland, A.M.; Griffiths, H.; Hartwell, J.; Smith, J.A. Exploiting the potential of plants with crassulacean acid metabolism for bioenergy production on marginal lands. J. Exp. Bot. 2009, 60, 2879–2896. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mai, Y.W.; Ye, L. Sisal fibre and its composites: A review of recent developments. Compos. Sci. Technol. 2000, 60, 2037–2055. [Google Scholar] [CrossRef]
- López-Romero, J.C.; Ayala-Zavala, J.F.; González-Aguilar, G.A.; Peña-Ramos, E.A.; González-Ríos, H. Biological activities of Agave by-products and their possible applications in food and pharmaceuticals. J. Sci. Food Agric. 2018, 98, 2461–2474. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cushman, J.C.; Borland, A.M.; Edwards, E.; Wullschleger, S.D.; Tuskan, G.A.; Owen, N.A.; Griffiths, H.; Smith, J.A.; De Paoli, H.C.; et al. A roadmap for research on crassulacean acid metabolism (CAM) to enhance sustainable food and bioenergy production in a hotter, drier world. New Phytol. 2015, 207, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.R. Agave as a model CAM crop system for a warming and drying world. Front. Plant Sci. 2015, 6, 684. [Google Scholar] [CrossRef] [PubMed]
- Robert, M.L.; Lim, K.Y.; Hanson, L.; Sanchez-Teyer, F.; Bennett, M.D.; Leitch, A.R.; Leitch, I.J. Wild and agronomically important Agave species (Asparagaceae) show proportional increases in chromosome number, genome size, and genetic markers with increasing ploidy. Bot. J. Linn. Soc. 2010, 158, 215–222. [Google Scholar] [CrossRef]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Nobel, P.S.; Hartsock, T.L. Temperature, water, and PAR influences on predicted and measured productivity of Agave deserti at various elevations. Oecologia 1986, 68, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.M.; Martin, J.A.; Simpson, J.; Abraham-Juarez, M.J.; Wang, Z.; Visel, A. De novo transcriptome assembly of drought tolerant CAM plants, Agave deserti and Agave tequilana. BMC Genom. 2013, 14, 563. [Google Scholar] [CrossRef]
- Abraham, P.E.; Yin, H.; Borland, A.M.; Weighill, D.; Lim, S.D.; De Paoli, H.C.; Engle, N.; Jones, P.C.; Agh, R.; Weston, D.J.; et al. Transcript, protein and metabolite temporal dynamics in the CAM plant Agave. Nat. Plants 2016, 2, 16178. [Google Scholar] [CrossRef]
- Yin, H.; Guo, H.B.; Weston, D.J.; Borland, A.M.; Ranjan, P.; Abraham, P.E.; Jawdy, S.S.; Wachira, J.; Tuskan, G.A.; Tschaplinski, T.J.; et al. Diel rewiring and positive selection of ancient plant proteins enabled evolution of CAM photosynthesis in Agave. BMC Genom. 2018, 19, 588. [Google Scholar] [CrossRef] [PubMed]
- Cedeño, M. Tequila production. Crit. Rev. Biotechnol. 1995, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Romero, C.; Martinez-Hernandez, A.; Mellado-Mojica, E.; López, M.G.; Simpson, J. Molecular and functional characterization of novel fructosyltransferases and invertases from Agave tequilana. PLoS ONE 2012, 7, e35878. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Gonzáleza, E.M.; López, M.G.; Délano-Frier, J.P.; Gómez-Leyva, J.F. Expression of the 1-SST and 1-FFT genes and consequent fructan accumulation in Agave tequilana and A. inaequidens is differentially induced by diverse (a)biotic-stress related elicitors. J. Plant Physiol. 2014, 171, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Pérez, S.A.; Espinal-Centeno, A.; Oropeza-Aburto, A.; Caballero-Pérez, J.; Falcon, F.; Aragón-Raygoza, A.; Sánchez-Segura, L.; Herrera-Estrella, L.; Cruz-Hernández, A.; Cruz-Ramírez, A. Transcriptional profiling of the CAM plant Agave salmiana reveals conservation of a genetic program for regeneration. Dev. Biol. 2018, 442, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Doblin, M.; Kurek, I.; Jacob-Wilk, D.; Delmer, D. Cellulose biosynthesis in plants: From genes to rosettes. Plant Cell Physiol. 2002, 43, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chen, J.; Bao, Y.; Liu, L.; Jiang, H.; An, X.; Dai, L.; Wang, B.; Peng, D. Transcript profiling reveals auxin and cytokinin signaling pathways and transcription regulation during in vitro organogenesis of ramie (Boehmeria nivea L. Gaud). PLoS ONE 2014, 9, e113768. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. Sequence Read Archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Betancur, L.; Singh, B.; Rapp, R.A.; Wendel, J.F.; Marks, M.D.; Roberts, A.W.; Haigler, C.H. Phylogenetically distinct cellulose synthase genes support secondary wall thickening in Arabidopsis shoot trichomes and cotton fiber. J. Integr. Plant Biol. 2010, 52, 205–220. [Google Scholar] [CrossRef]
- Wang, L.; Guo, K.; Li, Y.; Tu, Y.; Hu, H.; Wang, B.; Cui, X.; Peng, L. Expression profiling and integrative analysis of the CESA/CSL superfamily in rice. BMC Plant Biol. 2010, 10, 282. [Google Scholar] [CrossRef]
- Huang, X.; Bao, Y.N.; Wang, B.; Liu, L.J.; Chen, J.; Dai, L.J.; Peng, D.X. Identification and expression of Aux/IAA, ARF, and LBD family transcription factors in Boehmeria nivea. Biol. Plant. 2016, 60, 1–7. [Google Scholar] [CrossRef]
- Rychlik, W. OLIGO 7 primer analysis software. Methods Mol. Biol. 2007, 402, 35–60. [Google Scholar]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2017, 45, D37–D42. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- DNAMAN-Bioinformatics Solutions. Available online: www.lynnon.com (accessed on 30 January 2019).
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tan, Z.; Hu, B.; Yang, Z.; Xu, B.; Zhuang, L.; Huang, B. Selection and validation of reference genes for target gene analysis with quantitative RT-PCR in leaves and roots of bermudagrass under four different abiotic stresses. Physiol. Plant. 2015, 155, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Harkess, A.; Zhou, J.; Xu, C.; Bowers, J.E.; Van der Hulst, R.; Ayyampalayam, S.; Mercati, F.; Riccardi, P.; McKain, M.R.; Kakrana, A.; et al. The asparagus genome sheds light on the origin and evolution of a young Y chromosome. Nat. Commun. 2017, 8, 1279. [Google Scholar] [CrossRef]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef]
- Wei, C.; Yang, H.; Wang, S.; Zhao, J.; Liu, C.; Gao, L.; Xia, E.; Lu, Y.; Tai, Y.; She, G.; et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4151–E4158. [Google Scholar]
- Schmidt, M.H.; Vogel, A.; Denton, A.K.; Istace, B.; Wormit, A.; Van de Geest, H.; Bolger, M.E.; Alseekh, S.; Maß, J.; Pfaff, C.; et al. De Novo Assembly of a New Solanum pennellii Accession Using Nanopore Sequencing. Plant Cell 2017, 29, 2336–2348. [Google Scholar] [CrossRef]
- Twyford, A.D. The road to 10,000 plant genomes. Nat. Plants 2018, 4, 312–313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wan, X.; Xu, J.; Lin, L.; Qi, J. De novo assembly of kenaf (Hibiscus cannabinus) transcriptome using Illumina sequencing for gene discovery and marker identification. Mol. Breed. 2015, 35, 192. [Google Scholar] [CrossRef]
- Sadamoto, H.; Takahashi, H.; Okada, T.; Kenmoku, H.; Toyota, M.; Asakawa, Y. De novo sequencing and transcriptome analysis of the central nervous system of mollusc Lymnaea stagnalis by deep RNA sequencing. PLoS ONE 2012, 7, e42546. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Chase, M.W.; Reveal, J.L.; Fay, M.F. A subfamilial classification for the expanded asparagalean families Amaryllidaceae, Asparagaceae and Xanthorrhoeaceae. Bot. J. Linn. Soc. 2009, 161, 132–136. [Google Scholar] [CrossRef]
- Pear, J.; Kawagoe, Y.; Schreckengost, W.; Delmer, D.; Stalker, D. Higher plants contain homologs of the bacterial celA genes encoding the catalytic subunit of cellulose synthase. Proc. Natl. Acad. Sci. USA 1996, 93, 12637–12642. [Google Scholar] [CrossRef]
- Kim, H.; Triplett, B.; Zhang, H.; Lee, M.; Hinchliffe, D.; Li, P.; Fang, D. Cloning and characterization of homeologous cellulose synthase catalytic subunit 2 genes from allotetraploid cotton (Gossypium hirsutum L.). Gene 2012, 494, 181–189. [Google Scholar] [CrossRef]
- Liu, T.; Zhu, S.; Tang, Q.; Chen, P.; Yu, Y.; Tang, S. De novo assembly and characterization of transcriptome using Illumina paired-end sequencing and identification of CesA gene in ramie (Boehmeria nivea L. Gaud). BMC Genom. 2013, 14, 125. [Google Scholar] [CrossRef]
- Popper, Z.A. Evolution and diversity of green plant cell walls. Curr. Opin. Plant Biol. 2008, 11, 286–292. [Google Scholar] [CrossRef]
- Huang, X.; Wang, B.; Xi, J.; Zhang, Y.; He, C.; Zheng, J.; Gao, J.; Chen, H.; Zhang, S.; Wu, W.; et al. Transcriptome comparison reveals distinct selection patterns in domesticated and wild Agave species, the important CAM plants. Int. J. Genom. 2018, 2018, 5716518. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Xiao, M.; Xi, J.; He, C.; Zheng, J.; Chen, H.; Gao, J.; Zhang, S.; Wu, W.; Liang, Y.; et al. De Novo Transcriptome Assembly of Agave H11648 by Illumina Sequencing and Identification of Cellulose Synthase Genes in Agave Species. Genes 2019, 10, 103. https://doi.org/10.3390/genes10020103
Huang X, Xiao M, Xi J, He C, Zheng J, Chen H, Gao J, Zhang S, Wu W, Liang Y, et al. De Novo Transcriptome Assembly of Agave H11648 by Illumina Sequencing and Identification of Cellulose Synthase Genes in Agave Species. Genes. 2019; 10(2):103. https://doi.org/10.3390/genes10020103
Chicago/Turabian StyleHuang, Xing, Mei Xiao, Jingen Xi, Chunping He, Jinlong Zheng, Helong Chen, Jianming Gao, Shiqing Zhang, Weihuai Wu, Yanqiong Liang, and et al. 2019. "De Novo Transcriptome Assembly of Agave H11648 by Illumina Sequencing and Identification of Cellulose Synthase Genes in Agave Species" Genes 10, no. 2: 103. https://doi.org/10.3390/genes10020103
APA StyleHuang, X., Xiao, M., Xi, J., He, C., Zheng, J., Chen, H., Gao, J., Zhang, S., Wu, W., Liang, Y., Xie, L., & Yi, K. (2019). De Novo Transcriptome Assembly of Agave H11648 by Illumina Sequencing and Identification of Cellulose Synthase Genes in Agave Species. Genes, 10(2), 103. https://doi.org/10.3390/genes10020103