Severe Peripheral Joint Laxity is a Distinctive Clinical Feature of Spondylodysplastic-Ehlers-Danlos Syndrome (EDS)-B4GALT7 and Spondylodysplastic-EDS-B3GALT6

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
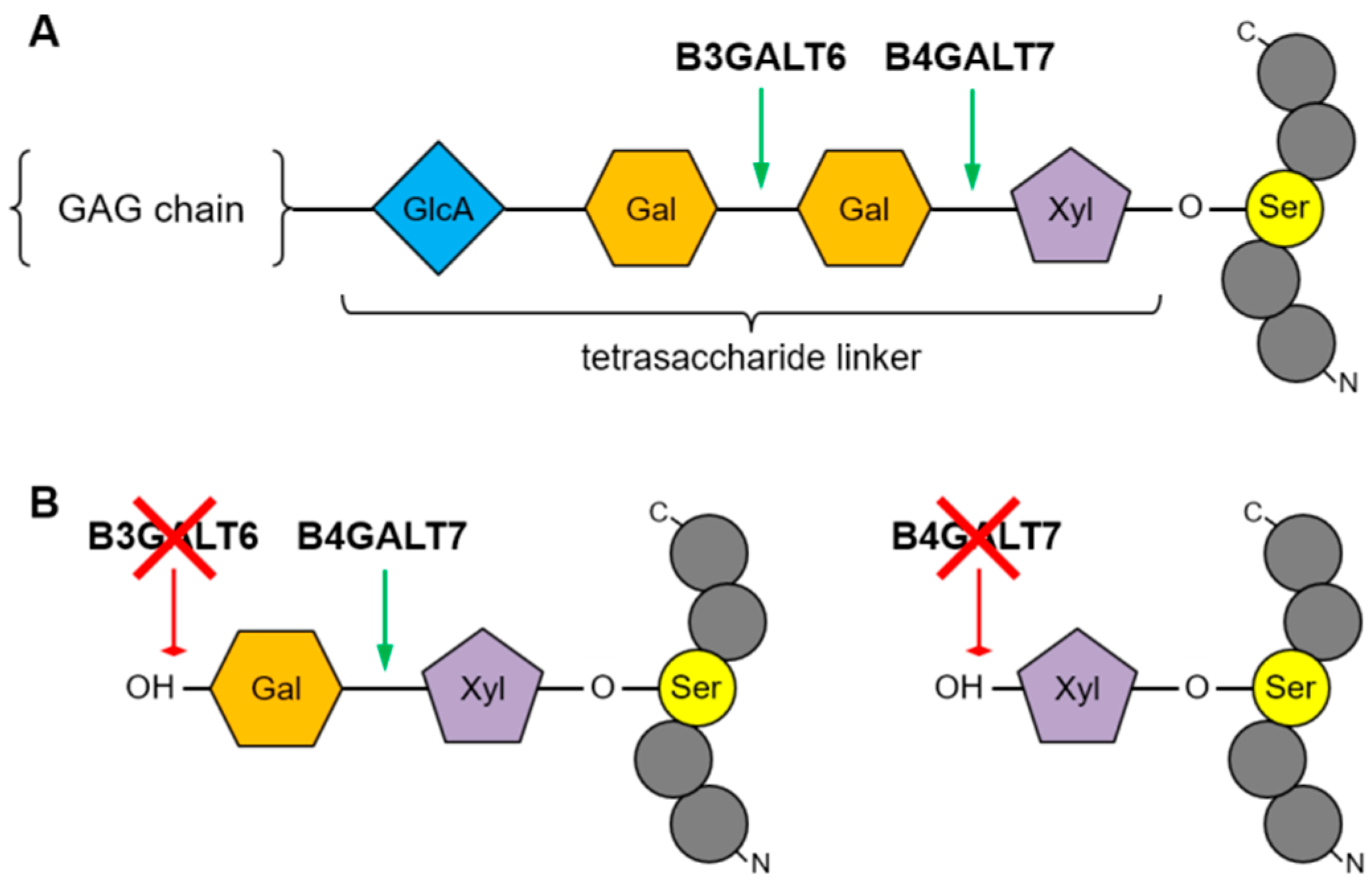
2.1. Metabolic Labeling of Fibroblast Cultures and PG Synthesis Analysis
2.2. Molecular Genetic Testing
3. Patients and Results
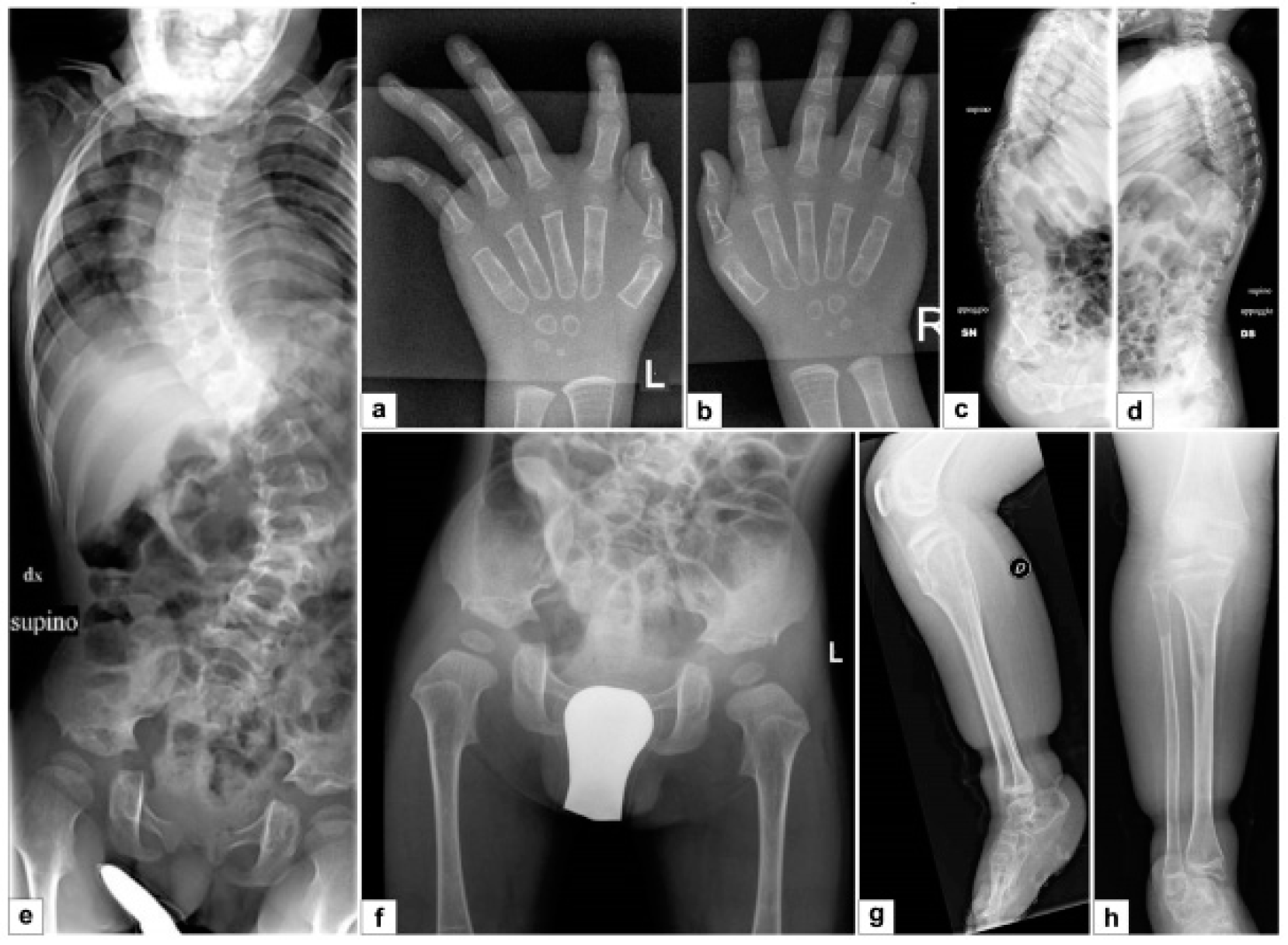
3.1. First Patient
3.1.1. Clinical Report
3.1.2. Molecular and Functional Analysis
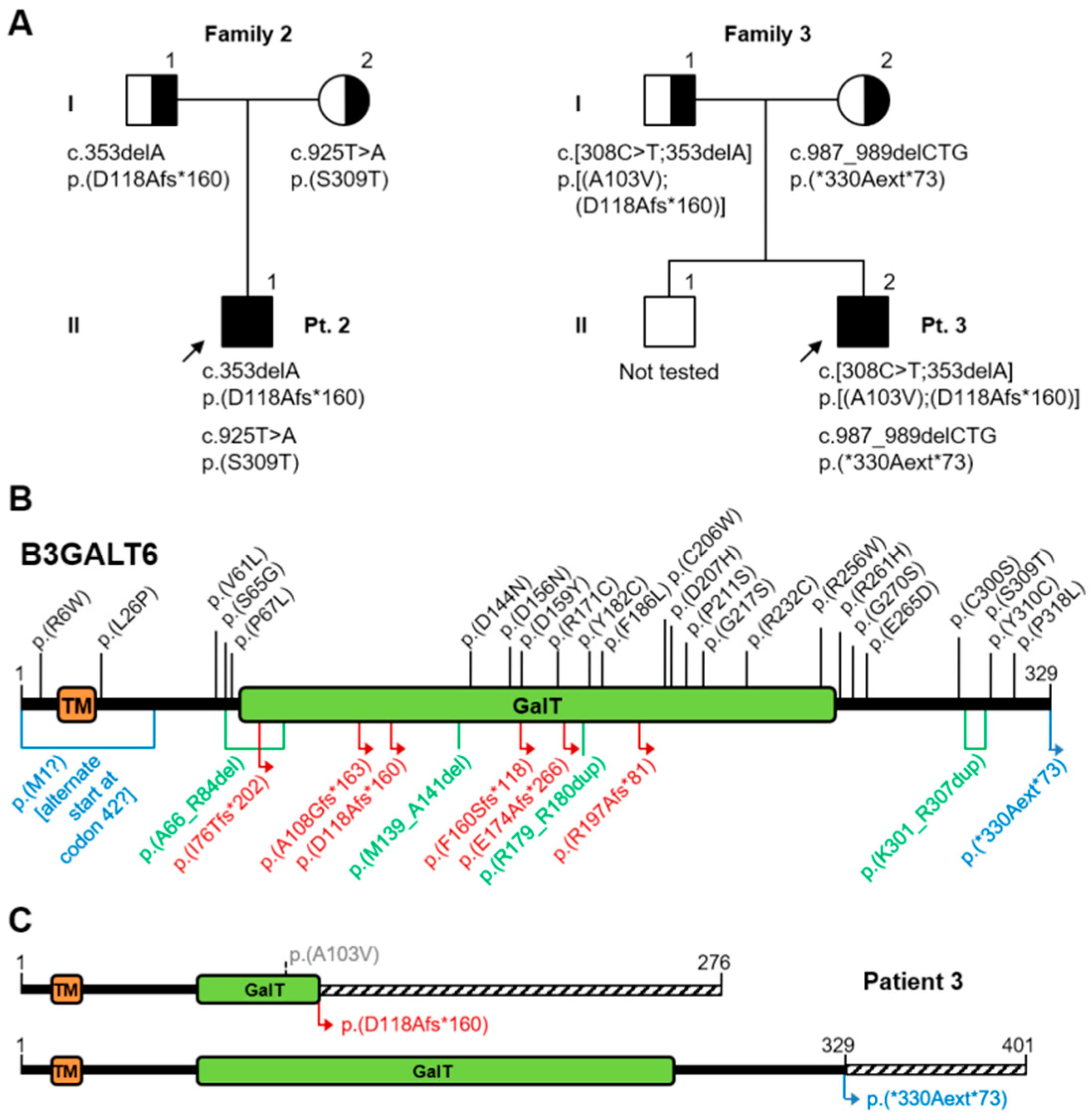
3.2. Second Patient
3.2.1. Clinical Report
3.2.2. Molecular Analysis
3.3. Third Patient
3.3.1. Clinical Report
3.3.2. Molecular Analysis
4. Discussion
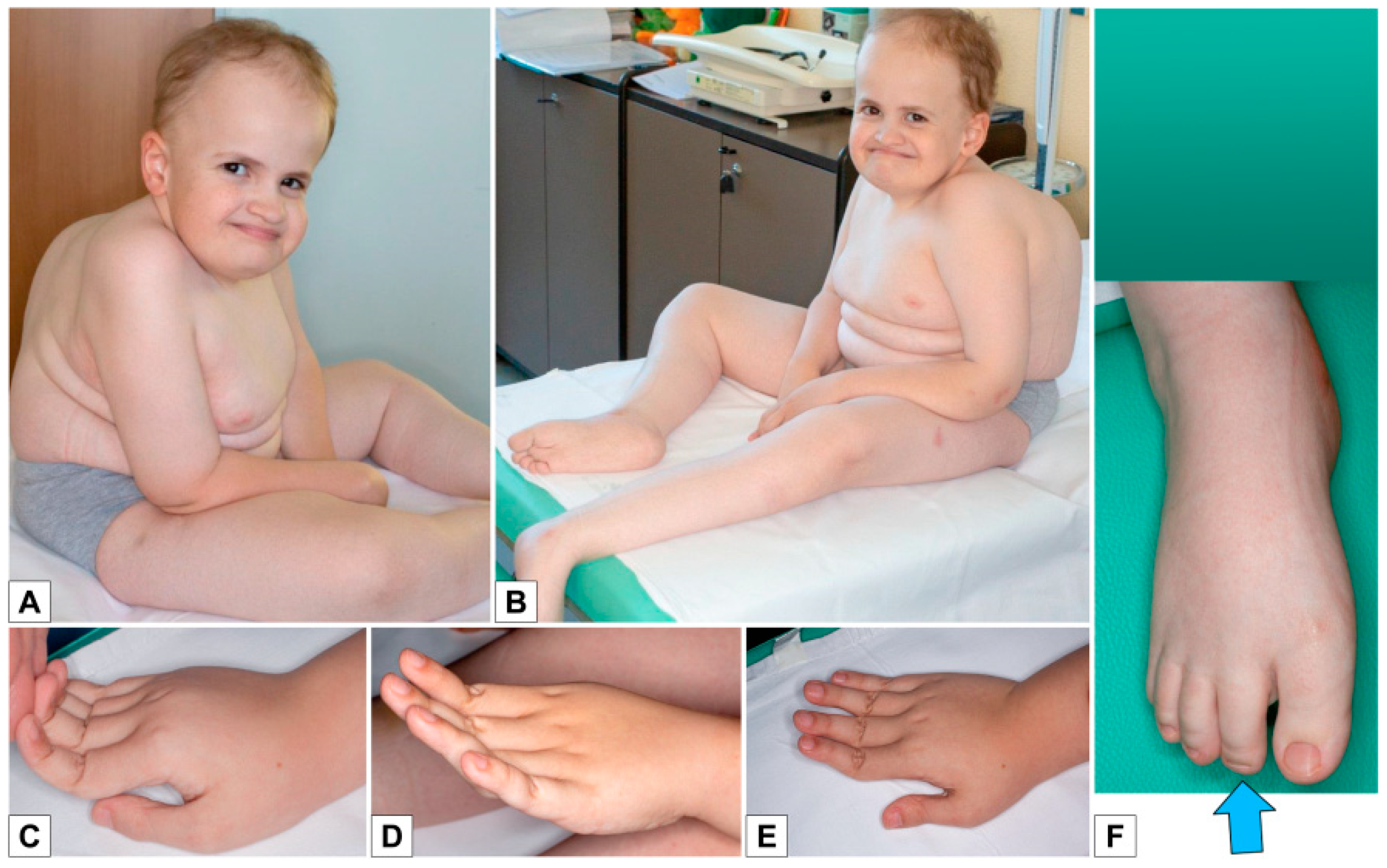
- the extreme distal joint hypermobility and soft, hyperextensible skin, particularly of the hands;
- the radiological signs, which are the main indicator for discriminating spEDS-B4GALT7, associated with radioulnar synostosis, and spEDS-B3GALT6, characterized by kyphoscoliosis (congenital or early onset and progressive) and by the skeletal signs of SEMDJL1 (platyspondyly, short iliac bones, elbow dislocation).
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kreuger, J.; Spillmann, D.; Li, J.P.; Lindahl, U. Interactions between heparan sulfate and proteins: The concept of specificity. J. Cell Biol. 2006, 174, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Couchman, J.R.; Pataki, C.A. An Introduction to Proteoglycans and Their Localization. J. Histochem. Cytochem. 2012, 60, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Bülow, H.E.; Hobert, O. The Molecular Diversity of Glycosaminoglycans Shapes Animal Development. Annu. Rev. Cell Dev. Biol. 2006, 22, 375–407. [Google Scholar] [CrossRef] [PubMed]
- Bui, C.; Huber, C.; Tuysuz, B.; Alanay, Y.; Bole-Feysot, C.; Leroy, J.G.; Mortier, G.; Nitschke, P.; Munnich, A.; Cormier-Daire, V. XYLT1 mutations in desbuquois dysplasia type 2. Am. J. Hum. Genet. 2014, 94, 405–414. [Google Scholar] [CrossRef] [PubMed]
- van Koningsbruggen, S.; Knoester, H.; Bakx, R.; Mook, O.; Knegt, L.; Cobben, J.M. Complete and partial XYLT1 deletion in a patient with neonatal short limb skeletal dysplasia. Am. J. Med. Genet. Part A 2016, 170, 510–514. [Google Scholar] [CrossRef]
- Jamsheer, A.; Olech, E.M.; Kozłowski, K.; Niedziela, M.; Sowińska-Seidler, A.; Obara-Moszyńska, M.; Latos-Bieleńska, A.; Karczewski, M.; Zemojtel, T. Exome sequencing reveals two novel compound heterozygous XYLT1 mutations in a Polish patient with Desbuquois dysplasia type 2 and growth hormone deficiency. J. Hum. Genet. 2016, 61, 577–583. [Google Scholar] [CrossRef]
- Guo, L.; Elcioglu, N.H.; Iida, A.; Demirkol, Y.K.; Aras, S.; Matsumoto, N.; Nishimura, G.; Miyake, N.; Ikegawa, S. Novel and recurrent XYLT1 mutations in two Turkish families with Desbuquois dysplasia, type 2. J. Hum. Genet. 2017, 62, 447–451. [Google Scholar] [CrossRef]
- Schreml, J.; Durmaz, B.; Cogulu, O.; Keupp, K.; Beleggia, F.; Pohl, E.; Milz, E.; Coker, M.; Ucar, S.K.; Nürnberg, G.; et al. The missing “link”: An autosomal recessive short stature syndrome caused by a hypofunctional XYLT1 mutation. Hum. Genet. 2014, 133, 29–39. [Google Scholar] [CrossRef]
- OMIM. Available online: https://www.omim.org/ (accessed on 1 August 2019).
- Munns, C.F.; Fahiminiya, S.; Poudel, N.; Munteanu, M.C.; Majewski, J.; Sillence, D.O.; Metcalf, J.P.; Biggin, A.; Glorieux, F.; Fassier, F.; et al. Homozygosity for frameshift mutations in XYLT2 result in a spondylo-ocular syndrome with bone fragility, cataracts, and hearing defects. Am. J. Hum. Genet. 2015, 96, 971–978. [Google Scholar] [CrossRef]
- Taylan, F.; Yavaş Abalı, Z.; Jäntti, N.; Güneş, N.; Darendeliler, F.; Baş, F.; Poyrazoğlu, Ş.; Tamçelik, N.; Tüysüz, B.; Mäkitie, O. Two novel mutations in XYLT2 cause spondyloocular syndrome. Am. J. Med. Genet. Part A 2017, 173, 3195–3200. [Google Scholar] [CrossRef] [PubMed]
- Taylan, F.; Costantini, A.; Coles, N.; Pekkinen, M.; Héon, E.; Şıklar, Z.; Berberoğlu, M.; Kämpe, A.; Kıykım, E.; Grigelioniene, G.; et al. Spondyloocular Syndrome: Novel Mutations in XYLT2 Gene and Expansion of the Phenotypic Spectrum. J. Bone Miner. Res. 2016, 31, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Baasanjav, S.; Al-Gazali, L.; Hashiguchi, T.; Mizumoto, S.; Fischer, B.; Horn, D.; Seelow, D.; Ali, B.R.; Aziz, S.A.A.; Langer, R.; et al. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am. J. Hum. Genet. 2011, 89, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Von Oettingen, J.E.; Tan, W.H.; Dauber, A. Skeletal dysplasia, global developmental delay, and multiple congenital anomalies in a 5-year-old boy-Report of the second family with B3GAT3 mutation and expansion of the phenotype. Am. J. Med. Genet. Part A 2014, 164, 1580–1586. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Schwarze, U.; Adam, M.P.; Byers, P.H.; Mefford, H.C. A homozygous B3GAT3 mutation causes a severe syndrome with multiple fractures, expanding the phenotype of linkeropathy syndromes. Am. J. Med. Genet. Part A 2015, 167, 2691–2696. [Google Scholar] [CrossRef]
- Budde, B.S.; Mizumoto, S.; Kogawa, R.; Becker, C.; Altmüller, J.; Thiele, H.; Rüschendorf, F.; Toliat, M.R.; Kaleschke, G.; Hämmerle, J.M.; et al. Skeletal dysplasia in a consanguineous clan from the island of Nias/Indonesia is caused by a novel mutation in B3GAT3. Hum. Genet. 2015, 134, 691–704. [Google Scholar] [CrossRef]
- Alazami, A.M.; Al-Qattan, S.M.; Faqeih, E.; Alhashem, A.; Alshammari, M.; Alzahrani, F.; Al-Dosari, M.S.; Patel, N.; Alsagheir, A.; Binabbas, B.; et al. Expanding the clinical and genetic heterogeneity of hereditary disorders of connective tissue. Hum. Genet. 2016, 135, 525–540. [Google Scholar] [CrossRef]
- Yauy, K.; Mau-Them, F.T.; Willems, M.; Coubes, C.; Blanchet, P.; Herlin, C.; Arrada, I.T.; Sanchez, E.; Faure, J.M.; Le Gac, M.P.; et al. B3GAT3-related disorder with craniosynostosis and bone fragility due to a unique mutation. Genet. Med. 2018, 20, 269–274. [Google Scholar] [CrossRef]
- Paganini, C.; Costantini, R.; Superti-Furga, A.; Rossi, A. Bone and connective tissue disorders caused by defects in glycosaminoglycan biosynthesis: A panoramic view. FEBS J. 2019, 286, 3008–3032. [Google Scholar] [CrossRef]
- Hernández, A.; Aguirre-Negrete, M.G.; Liparoli, J.C.; Cantú, J.M. Third case of a distinct variant of the Ehlers–Danlos Syndrome (EDS). Clin. Genet. 2008, 20, 222–224. [Google Scholar] [CrossRef]
- Hernández, A.; Aguirre-Negrete, M.G.; González-Flores, S.; Reynoso-Luna, M.C.; Fragoso, R.; Nazará, Z.; AND, G.T.; Cantú, J.M. Ehlers-Danlos features with progeroid facies and mild mental retardation: Further delineation of the syndrome. Clin. Genet. 1986, 30, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.; Aguirre-Negrete, M.G.; Ramírez-Soltero, S.; González-Mendoza, A.; Martínez, R.M.; Velázquez-Cabrera, A.; Cantú, J.M. A distinct variant of the Ehlers–Danlos syndrome.pdf. Clin. Genet. 1979, 16, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Kresse, H.; Rosthøj, S.; Quentin, E.; Hollmann, J.; Glössl, J.; Okada, S.; Tønnesen, T. Glycosaminoglycan-free small proteoglycan core protein is secreted by fibroblasts from a patient with a syndrome resembling progeroid. Am. J. Hum. Genet. 1987, 41, 436–453. [Google Scholar] [PubMed]
- Quentin, E.; Gladen, A.; Roden, L.; Kresse, H. A genetic defect in the biosynthesis of dermatan sulfate proteoglycan: Galactosyltransferase I deficiency in fibroblasts from a patient with a progeroid syndrome. Proc. Natl. Acad. Sci. USA 1990, 87, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.; Levery, S.B.; Mandel, U.; Kresse, H.; Schwientek, T.; Bennett, E.P.; Clausen, H. Cloning and expression of a proteoglycan UDP-galactose: β-xylose β1,4- galactosyltransferase I. A seventh member of the human β4- galactosyltransferase gene family. J. Biol. Chem. 1999, 274, 26165–26171. [Google Scholar] [CrossRef] [PubMed]
- Okajima, T.; Fukumoto, S.; Furukawat, K.; Urano, T.; Furukawa, K. Molecular basis for the progeroid variant of Ehlers–Danlos syndrome. Identification and characterization of two mutations in galactosyltransferase I gene. J. Biol. Chem. 1999, 274, 28841–28844. [Google Scholar] [CrossRef] [PubMed]
- Rahuel-Clermont, S.; Daligault, F.; Piet, M.-H.; Gulberti, S.; Netter, P.; Branlant, G.; Magdalou, J.; Lattard, V. Biochemical and thermodynamic characterization of mutated β1,4-galactosyltransferase 7 involved in the progeroid form of the Ehlers–Danlos syndrome. Biochem. J. 2010, 432, 303–311. [Google Scholar] [CrossRef]
- Faiyaz-Ul-Haque, M.; Zaidi, S.H.E.; Al-Ali, M.; Al-Mureikhi, M.S.; Kennedy, S.; Al-Thani, G.; Tsui, L.-C.; Teebi, A.S. A novel missense mutation in the galactosyltransferase-I (B4GALT7) gene in a family exhibiting facioskeletal anomalies and Ehlers–Danlos syndrome resembling the progeroid type. Am. J. Med. Genet. 2004, 128, 39–45. [Google Scholar] [CrossRef]
- Guo, M.H.; Stoler, J.; Lui, J.; Nilsson, O.; Bianchi, D.W.; Hirschhorn, J.N.; Dauber, A. Redefining the progeroid form of Ehlers–Danlos syndrome: Report of the fourth patient with B4GALT7 deficiency and review of the literature. Am. J. Med. Genet. Part A 2013, 161, 2519–2527. [Google Scholar]
- Cartault, F.; Munier, P.; Jacquemont, M.L.; Vellayoudom, J.; Doray, B.; Payet, C.; Randrianaivo, H.; Laville, J.M.; Munnich, A.; Cormier-Daire, V. Expanding the clinical spectrum of B4GALT7 deficiency: Homozygous p. R270C mutation with founder effect causes Larsen of Reunion Island syndrome. Eur. J. Hum. Genet. 2015, 23, 49–53. [Google Scholar] [CrossRef]
- Salter, C.G.; Davies, J.H.; Moon, R.J.; Fairhurst, J.; Bunyan, D.; Foulds, N. Further defining the phenotypic spectrum of B4GALT7 mutations. Am. J. Med. Genet. Part A 2016, 170, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Arunrut, T.; Sabbadini, M.; Jain, M.; Machol, K.; Scaglia, F.; Slavotinek, A. Corneal clouding, cataract, and colobomas with a novel missense mutation in B4GALT7—A review of eye anomalies in the linkeropathy syndromes. Am. J. Med. Genet. Part A 2016, 170, 2711–2718. [Google Scholar] [CrossRef] [PubMed]
- Beighton, P.; Kozlowski, K. Spondylo-Epi-Metaphyseal dysplasia with joint laxity and severe, progressive kyphoscoliosis. Skelet. Radiol. 1980, 5, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Beighton, P.; Kozlowski, K.; Gericke, G.; Wallis, G.; Grobler, L. Spondylo-epimetaphyseal dysplasia with joint laxity and severe, progressive kyphoscoliosis. A potentially lethal dwarfing disorder. S. Afr. Med. J. 1983, 64, 772–775. [Google Scholar] [PubMed]
- Kozlowski, K.; Beighton, P. Radiographic features of spondylo-epimetaphyseal dysplasia with joint laxity and progressive kyphoscoliosis. In RöFo-Fortschritte Auf Dem Gebiet der Röntgenstrahlen und der Bildgebenden Verfahren; Georg Thieme Verlag Stuttgart: New York, NY, USA, 1984; Volume 141, pp. 337–341. [Google Scholar]
- Beighton, P.; Gericke, G.; Kozlowski, K.; Grobler, L. The manifestations and natural history of spondylo-epi-metaphyseal dysplasia with joint laxity. Clin. Genet. 1984, 26, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Torrington, M.; Beighton, P. The ancestry of spondyloepimetaphyseal dysplasia with joint laxity (SEMDJL) in South Africa. Clin. Genet. 2008, 39, 210–213. [Google Scholar] [CrossRef]
- Beighton, P. Spondyloepimetaphyseal dysplasia with joint laxity (SEMDJL). J. Med. Genet. 1994, 31, 136–140. [Google Scholar] [CrossRef]
- Christianson, A.L.; Beighton, P. Spondyloepimetaphyseal dysplasia with joint laxity (SEMDJL) in three neonates. Genet. Couns. 1996, 7, 219–225. [Google Scholar]
- Hall, C.M.; Elçioglu, N.H.; Shaw, D.G. A distinct form of spondyloepimetaphyseal dysplasia with multiple dislocations. J. Med. Genet. 1998, 35, 566–572. [Google Scholar] [CrossRef]
- Vorster, A.A.; Beighton, P.; Ramesar, R.S. Spondyloepimetaphyseal dysplasia with joint laxity (Beighton type); mutation analysis in eight affected South African families. Clin. Genet. 2015, 87, 492–495. [Google Scholar] [CrossRef]
- Bonafe, L.; Cormier-Daire, V.; Hall, C.; Lachman, R.; Mortier, G.; Mundlos, S.; Nishimura, G.; Sangiorgi, L.; Savarirayan, R.; Sillence, D.; et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am. J. Med. Genet. Part A 2015, 167, 2869–2892. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Kariminejad, A.; Van Damme, T.; Gauche, C.; Syx, D.; Merhi-Soussi, F.; Gulberti, S.; Symoens, S.; Vanhauwaert, S.; Willaert, A.; et al. Defective initiation of glycosaminoglycan synthesis due to B3GALT6 mutations causes a pleiotropic Ehlers–Danlos-syndrome-like connective tissue disorder. Am. J. Hum. Genet. 2013, 92, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Mizumoto, S.; Miyake, N.; Kogawa, R.; Iida, A.; Ito, H.; Kitoh, H.; Hirayama, A.; Mitsubuchi, H.; Miyazaki, O.; et al. Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am. J. Hum. Genet. 2013, 92, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am. J. Med. Genet. Part C 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Brady, A.F.; Demirdas, S.; Fournel-Gigleux, S.; Ghali, N.; Giunta, C.; Kapferer-Seebacher, I.; Kosho, T.; Mendoza-Londono, R.; Pope, M.F.; Rohrbach, M.; et al. The Ehlers–Danlos syndromes, rare types. Am. J. Med. Genet. Part C 2017, 175, 70–115. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, T.; Pang, X.; Guillemyn, B.; Gulberti, S.; Syx, D.; De Rycke, R.; Kaye, O.; de Die-Smulders, C.E.M.; Pfundt, R.; Kariminejad, A.; et al. Biallelic B3GALT6 mutations cause spondylodysplastic Ehlers–Danlos syndrome. Hum. Mol. Genet. 2018, 27, 3475–3487. [Google Scholar] [CrossRef]
- Paganini, C.; Monti, L.; Costantini, R.; Besio, R.; Lecci, S.; Biggiogera, M.; Tian, K.; Schwartz, J.M.; Huber, C.; Cormier-Daire, V.; et al. Calcium activated nucleotidase 1 (CANT1) is critical for glycosaminoglycan biosynthesis in cartilage and endochondral ossification. Matrix Biol. 2019, 81, 70–90. [Google Scholar] [CrossRef]
- Nizon, M.; Huber, C.; De Leonardis, F.; Merrina, R.; Forlino, A.; Fradin, M.; Tuysuz, B.; Abu-Libdeh, B.Y.; Alanay, Y.; Albrecht, B.; et al. Further delineation of CANT1 phenotypic spectrum and demonstration of its role in proteoglycan synthesis. Hum. Mutat. 2012, 33, 1261–1266. [Google Scholar] [CrossRef]
- Ritelli, M.; Dordoni, C.; Cinquina, V.; Venturini, M.; Calzavara-Pinton, P.; Colombi, M. Expanding the clinical and mutational spectrum of B4GALT7-spondylodysplastic Ehlers–Danlos syndrome. Orphanet J. Rare Dis. 2017, 12, 153. [Google Scholar] [CrossRef]
- Sandler-Wilson, C.; Wambach, J.A.; Marshall, B.A.; Wegner, D.J.; McAlister, W.; Cole, F.S.; Shinawi, M. Phenotype and response to growth hormone therapy in siblings with B4GALT7 deficiency. Bone 2019, 124, 14–21. [Google Scholar] [CrossRef]
- Sellars, E.A.; Bosanko, K.A.; Lepard, T.; Garnica, A.; Schaefer, G.B. A newborn with complex skeletal abnormalities, joint contractures, and bilateral corneal clouding with sclerocornea. Semin. Pediatr. Neurol. 2014, 21, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Chiarelli, N.; Zoppi, N.; Dordoni, C.; Quinzani, S.; Traversa, M.; Venturini, M.; Calzavara-Pinton, P.; Colombi, M. Insights in the etiopathology of galactosyltransferase II (GalT-II) deficiency from transcriptome-wide expression profiling of skin fibroblasts of two sisters with compound heterozygosity for two novel B3GALT6 mutations. Mol. Genet. Metab. Rep. 2015, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Trejo, P.; Rauch, F.; Glorieux, F.H.; Ouellet, J.; Benaroch, T.; Campeau, P.M. Spondyloepimetaphysial Dysplasia with Joint Laxity in Three Siblings with B3GALT6 Mutations. Mol. Syndromol. 2017, 8, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Ben-Mahmoud, A.; Ben-Salem, S.; Al-Sorkhy, M.; John, A.; Ali, B.R.; Al-Gazali, L. A B3GALT6 variant in patient originally described as Al-Gazali syndrome and implicating the endoplasmic reticulum quality control in the mechanism of some β3GalT6-pathy mutations. Clin. Genet. 2018, 93, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Civic, N.; Furuichi, T.; Shimoda, S.; Mishima, K.; Higashiyama, H.; Idaira, Y.; Asada, Y.; Kitamura, H.; Yamasaki, S.; et al. The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-β signaling pathways. PLoS ONE 2008, 3, e3642. [Google Scholar] [CrossRef]
- Giunta, C.; Elçioglu, N.H.; Albrecht, B.; Eich, G.; Chambaz, C.; Janecke, A.R.; Yeowell, H.; Weis, M.; Eyre, D.R.; Kraenzlin, M.; et al. Spondylocheiro Dysplastic Form of the Ehlers–Danlos Syndrome—An Autosomal-Recessive Entity Caused by Mutations in the Zinc Transporter Gene SLC39A13. Am. J. Hum. Genet. 2008, 82, 1290–1305. [Google Scholar] [CrossRef] [PubMed]
- Dusanic, M.; Dekomien, G.; Lücke, T.; Vorgerd, M.; Weis, J.; Epplen, J.T.; Köhler, C.; Hoffjan, S. Novel Nonsense Mutation in SLC39A13 Initially Presenting as Myopathy: Case Report and Review of the Literature. Mol. Syndromol. 2018, 9, 100–109. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hernandez et al. [1979, 1981, 1986] [21,22,23] | Kresse et al. [1987][24] | Faiyaz-Ul-Haque et al. [2004][29] | Guo et al. [2013][30] | Cartault et al. [2015][31] | Arunrut et al. [2016][33] | Salter et al. [2016][32] | Ritelli et al. [2017][51] | Sandler-Wilson et al. [2019][52] | This study, Patient 1 | Total (patients with mutation) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genetics | Diagnosis (Various denominations) | A distinct variant of the EDS | EDS, progeroid type 1 | EDS, progeroid type 1 | EDS, progeroid type 1 | Larsen of Reunion Island syndrome | B4GALT7-linkeropathy phenotype | Phenotypic Spectrum of B4GALT7 | spEDS- B4GALT7 | spEDS- B4GALT7 | spEDS- B4GALT7 | spEDS- B4GALT7 |
| B4GALT7 variants | ? | c.[557C>A ];[617T>A] | c.[808C>T];[808C>T] | c.[122T>C];[808C>T] | c.[808C>T];[808C>T] | c.[970T>A];[970T>A] | c.[277dupC];[641G>A] c.[421C>T];[808C>T] | c.[829G>T];[829G>T] | c.[421C>T];[808C>T] | c.[277_278insC];[628C>T] | 26/33 Homozygous 7/33 Compound heterozygous | |
| Variants on protein | ? | p.[(A186D)];[(L206P)] | p.[(R270C)];[(R270C)] | p.[(L41P)];[(R270C)] | p.[(R270C)];[(R270C)] | p.[(C324S)];[(C324S)] | p.[(H93Pfs*73)];[(C214Y)] p.[(R141W)];[(R270C)] | p.[(E277*)];[(E277*)] | p.[(R141W)];[(R270C)] | p.[(H93Pfs*73)];[(H210Y)] | ||
| Gender | 5M | M | 1F 1M | M | 11M 11F | F | 1M 1F | F | 1M 1F | M | 17M 16F | |
| Age | 8y, 15y, 15y, 16y, 18y | 4y 9m | 2y, 33y | 10y | 4y, 46y | 5y | 3y 6m, 13y | 30y | 4y, 10y | 7y 8m | 2y → 46y | |
| Main features | Short staturea | 4/5 | + | 2/2 | + | 22/22 | + | 2/2 | + | 2/2 | + | 33/33 |
| Radiolunar synostosisc | n.a. | + | 2/2 | + | 10/21 | - | 2/2 | - | 2/2 | + | 19/32 | |
| Bowing of limbsa | n.a. | + | 2/2 | + | 21/21 | - | 2/2 | - | 2/2 | + | 30/32 | |
| Joint hypermobility especially of the handsc | 5/5 | + | 2/2 | + | 22/22 | + | 2/2 | + | 2/2 | + | 33/33 | |
| Skin hyperextensibility, soft, doughy skinb | 5/5 | + | 2/2 | + | 21/22 | + | 2/2 | + | 2/2 | + | 32/33 | |
| Facial dysmorphismsc | Progeroid facial appearance | 5/5 | mild | 0/2 | - | 0/22 | - | 0/2 | - | 0/2 | - | 1/33 |
| Short face | - | + | 2/2 | - | 22/22 | + | 2/2 | - | 2/2 | + | 31/33 | |
| Midface hypoplasia | - | - | 2/2 | - | + | 2/2 | - | 2/2 | + | 30/33 | ||
| Narrow mouth | - | + | 2/2 | - | + | 1/2 | + | 2/2 | + | 31/33 | ||
| Proptosis | - | + | 2/2 | - | + | 2/2 | + | 2/2 | + | 32/33 | ||
| Cleft palate | - | Bifid uvula | 0/2 | - | 1/22 | - | 1/2 | - | 1/2 | - | 4/33 | |
| Loose skin | - | + | 2/2 | + | n.a. | n.a. | 2/2 | + | n.a. | + | 8/8 | |
| Other clinical features | Delayed wound healing | 5/5 | + | 1/2 | + | n.a. | + | 2/2 | + | n.a | - | 7/8 |
| Cardiovascular abnormalities | 1/5 Aortic/Pulmonic Stenosis | n.a. | n.a. | - | n.a. | - | n.a. | - | n.a. | + :ASD | 1/4 | |
| Delayed motor developmentb | 5/5 | + | 2/2 | - | n.a. | + | 2/2 | + | 2/2 | + | 10/11 | |
| Delayed cognitive developmentb | 5/5 | n.a. | n.a. | Mild learning disabilities | 12/22 (learning disabilities) | n.a. | 1/2 Severe 1/2 n.a | - | 2/2 | - | 16/29 | |
| Muscle hypotoniaa | n.a. | + | 2/2 | mild | n.a. | + | 2/2 | + | 2/2 | + | 11/11 | |
| Ophthalmological abnormalitiesc | n.a. | - | 1/2 Mild esotropia and mild hypermetropia | Severe hyperopia, congenital ptosis, intermittent exotropia | 5/21 glaucoma 1/21 megalocornea | Nystagmus Iris and optic nerve colobomas Posterior subcapsular cataracts High hyperopia Right-sided ptosis | Severe hypermetropia Small optic nerves Hypermetropia Strabismus | - | 2/2 Blue sclerae 1/2 Severe hyperopia | Myopia | 13/32 | |
| Osteopeniab | n.a. | + | 2/2 | - | n.a. | n.a. | 2/2 | + | 1/2 | + | 8/10 | |
| Pes planusb | 5/5 | + | ½ | + | n.a. | + | 1/2 1/2 n.a. | + | 2/2 | + | 9/11 | |
| Bilateral elbow contractures or limited elbow movementc | n.a. | + | 2/2 | + | n.a. | n.a. | 2/2 | - | 1/2 | + | 8/10 | |
| Sensorineural hearing loss | n.a. | - | n.a. | n.a. | n.a. | n.a. | 1/2 Conductive hearing loss | + | n.a. | - | 2/7 | |
| Other less frequent features | Cryptorchidism 4/5 Inguinal hernia 1/5 Hypogonadism 1/5 Varicose veins 5/5 Multiple nevi 5/5 Dental anomalies 5/5 | Dental anomalies: defective and greyish enamel Clavicular exostoses | Yellow discoloration of teeth with defective enamel Mild eventration of the right hemidiaphragm Bilateral equinovarus deformity | Unilateral ptosis | Pectus carinatum 5/22 Bifid thumb 2/22 Scoliosis/ Kyphosis 6/22 | Pectus carinatum Scoliosis Broad fingertips Subluxation of the distal interphalangeal joints Absence of the pineal gland Prominent scalp veins | Irregular and fragile dentition Scoliosis Bilateral patellar dislocation | Bilateral hallux valgus Lymphedema Scoliosis Temporomandibular joint dislocation | 1/2 Chest wall deformity 2/2 Coronal cleft vertebrae 2/2 Sagittal craniosynostosis 1/2 Vesicoureteral reflux | Hip dysplasia Ulnar deviation of fingers | ||
| Malfait et al. 2013[44] | Nakajima et al. 2013[45] | Sellars et al. 2014[53] | Ritelli et al., 2015[54] | Alazami et al., 2016[18] | Trejo et al.2017[55] | Ben-Mahmoud et al. 2018[56] | Van Damme et al. 2018[48] | This study, Patient 2 | This study, Patient 3 | Total (patients with mutation) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genetics | Diagnosis (Various denominations) | EDS-like connective tissue disorder SEMDJL1 | B3GALT6 spectrum SEMDJL1 EDS-progeroid form | EDS progeroid type2 SEMDJL1 | EDS-like syndrome EDS progeroid type2 SEMDJL1 | B3GALT6- phenotype | SEMDJL1 | spEDS- B3GALT6 SEMDJL1 | spEDS- B3GALT6 | spEDS- B3GALT6 | spEDS- B3GALT6 | spEDS- B3GALT6 |
| B3GALT6 variants | c.[619G>C];[619G>C] c.[323_344del];[619G>C] c.[649G>A];[649G>A] | 9 Compound heterozygous, 1 Heterozygous; see Ref. (Table 3) | ? | c.[227delT];[766C>T] | c.[556T>C];[556T>C] c.[536_541dup];[536_541dup] | c.[511C>T];[901_921dup] | c.[618C>G];[618C>G] | 8 Compound heterozygous, 1 Homozygous; see Ref (Table 3) | c.[353delA];[925T>A] | c.[308C>T;353delA];[987_989delCTG] | 12/45 homozygous 32/45 compound heterozygous 1/45 heterozygous | |
| Variants on protein | p.[(D207H)];[(D207H)] p.[(A108Gfs*163)];[(D207H)] p.[(G217S)];[(G217S)] | See Figure 7 | p.[(D159Y)];[(E265D)] | p.[(I76Tfs*202)];[(R256W)] | p.[(F186L)];[(F186L)] p.[(R179_R180dup)];[(R179_R180dup)] | p.[(R171C)];[(K301_R307dup)] | p.[(C206W)];[(C206W)] | See Figure 7 | p.[(D118Afs*160)];[(S309T)] | p.[(A103V);(D118Afs*160)];[(*330Aext*73)] | ||
| Gender | 2M 3F | 6M 6F | M | 2F | 1M 4F | 3F | 1M 2F | 7M 5F | M | M | 20M 25F | |
| Age | 1y8m →27y | 1m →34y | 6m | 21y, 25y | 6w → 6y | 12y, 15y, 15y | 4d →2m | 8m →37y | 12y 7m | 13y 3m | 4d →37y | |
| Main features | Short stature a | 2/3 | 12/12 | + | 2/2 | 3/3 | 3/3 | n.a. | 10/10 | + | + | 35/36 |
| Kyphoscoliosis(congenital or early onset, progressive) c | 3/4 | 12/12 | - | 2/2 | 4/5 | 3/3 | n.a. | 10/10 | + | + | 36/39 | |
| Bowing of limbs a | 2/4 | n.a. | + | n.a. | n.a. | 3/3 | n.a. | 10/10 | - | - | 16/20 | |
| Joint hypermobility especially of the hands c | 4/4 | 7/10 | n.a. | 2/2 | 5/5 | 3/3 | 2/2 | 10/10 | + | + | 33/36 | |
| Skin hyperextensibility, soft, doughy skin b | 4/4 | 6/10 | n.a. | 2/2 | 4/5 | 3/3 | 2/2 | 9/10 | + | + | 33/38 | |
| Skeletal changes SEMDJL1 d | 3/4 | 12/12 | n.a. | 2/2 | 2/2 | 0/1 | 3/3 | 10/10 | + | + | 34/36 | |
| Facial dysmorphismsc | Prominent forehead | 4/4 | 9/10 | + | 2/2 | 4/5 | 0/3 | 1/1 | 8/10 | + | + | 31/38 |
| Sparse hair | 2/4 | 3/10 | - | 0/2 | n.a | 2/3 | 0/1 | 3/3 | + | + | 12/26 | |
| Midface hypoplasia | 2/4 | n.a. | + | 1/2 | 4/5 | 2/3 | 1/1 | 8/10 | + | + | 21/28 | |
| Blue sclerae | 3/4 | 7/10 | n.a. | 2/2 | 4/5 | 1/3 | n.a. | 6/10 | + | + | 25/36 | |
| Proptosis | 2/4 | 7/10 | + | 0/2 | n.a. | 3/3 | n.a. | 7/10 | - | - | 20/32 | |
| Cleft palate | 0/4 | 1/10 | - | 0/2 | n.a. | 0/3 | - | - | - | - | 2/25 | |
| Other clinical features | Joint hand contractures c | 2/3 | 3/12 | + | 2/2 | 1/5 | 2/2 | 3/3 | 10/10 | - | - | 24/40 |
| Cardiovascular anomalies | n.a. | Mitral regurgitation 1/? | n.a. | 2 Mitral valve prolapse | 1 Aortic valve stenosis 1 Mitral valve prolapse/? | n.a. | n.a. | Aortic root aneurysm 3/8 Cardiac valve anomalies 2/8 | Aortic root aneurysm Mitral valve prolapse | Mitral valve prolapse | 12/? | |
| Delayed motor development b | 4/4 | 2/? | + | n.a. | 4/5 | 2/2 | n.a. | 5/9 | + | + | 17/24 | |
| Delayed cognitive development b | 2/2 | n.a. | n.a. | 0/2 | 4/5 | n.a. | n.a. | 3/8 | - | - | 10/20 | |
| Muscle hypotonia a | 4/4 | 5/12 | + | 1/1 | 2/? | 3/3 (1 mild) | 1/1 | 5/9 | + | + | 25/37 | |
| Ophthalmological anomalies | Myopia 2/4 Retinal detachment1/4 | n.a. | Corneal opacity Sclerocornea | 0/2 | n.a. | n.a | Corneal opacity 3/3 | Glaucoma and optic nerve atrophy 1/10 Microcornea 1/10 | - | - | 7/19 | |
| Osteopenia b | 4/4 | n.a. | + | 2/2 | 3/3 | 0/3 | 2/2 | 8/8 | + | + | 22/26 | |
| Pes planus b | 2/2 | n.a. | n.a. | 2/2 | n.a. | n.a | 0/2 | n.a. | + | + | 6/8 | |
| Talipes equinovarus c | 3/4 | 4/12 | + | n.a | 3/5 | n.a | 2/2 | 10/10 | + | + | 23/37 | |
| Peculiar fingers c | 3/4 | 7/11 | + | 2/2 | n.a. | n.a | 2/2 | n.a. | + | + | 17/22 | |
| Anomalies of dentition, discoloration of teeth c | 3/4 | n.a. | n.a. | 2/2 | n.a. | n.a | n.a. | 8/9 | + | + | 13/17 | |
| Less frequent features | Excessive wrinkling of palmar skin (hands and feet) 2/4 Pectus deformity 3/4 | Elbow dislocation 9/10 Limited elbow movement 9/11 Carpal synostosis 1/10 Short metacarpals 6/10 Hip dislocation 5/12 Epiphyseal dysplasia of femoral head 4/12 | Radioulnar synostosis Early death | 2/2 Genu valgus 2/2 Hallux valgus | 4/5 Multiple fractures Bilateral disclocated radial head 1/5 Pectus carinatum | 3/3 Bilateral radioulnar dislocation 3/3 Hip dysplasia 2/3 Hearing loss 1/3 Pectus carinatum 1/3 Ulnar deviation of the fingers | Contractures of the large joints 2/2 Radioulnar synostosis 2/2 Oligodactyly of the right 3rd finger 1/2 Spontaneous fractures 2/2 Early death 3/3 | Sensorineural and conductive hearing loss 1/10 Cervical spine instability 3/7 Laryngeal cleft 1/10 Tracheomalacia 2/10 Spontaneous repeated pneumothoraces 1/10 Chronic respiratory insufficiency 2/10 Pectus carinatum 1/10 excavatum 1/10 Wilms tumor 1/10 Joint dislocations 10/10 Hip dysplasia 4/6 Fractures 8/9 Hallux valgus 3/10 | Prominent superficial veins Limited elbow extension Ptosic kidney, Bilateral caliceal and ureteral dilatation Fractures Right hip dysplasia Recurrent luxation of the toes, Hypoplastic nails Hallux valgus | Prominent superficial veins Barrett’s oesophagus Limited elbow extension Bilateral cryptorchidism Hypoplastic nails | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caraffi, S.G.; Maini, I.; Ivanovski, I.; Pollazzon, M.; Giangiobbe, S.; Valli, M.; Rossi, A.; Sassi, S.; Faccioli, S.; Di Rocco, M.; et al. Severe Peripheral Joint Laxity is a Distinctive Clinical Feature of Spondylodysplastic-Ehlers-Danlos Syndrome (EDS)-B4GALT7 and Spondylodysplastic-EDS-B3GALT6. Genes 2019, 10, 799. https://doi.org/10.3390/genes10100799
Caraffi SG, Maini I, Ivanovski I, Pollazzon M, Giangiobbe S, Valli M, Rossi A, Sassi S, Faccioli S, Di Rocco M, et al. Severe Peripheral Joint Laxity is a Distinctive Clinical Feature of Spondylodysplastic-Ehlers-Danlos Syndrome (EDS)-B4GALT7 and Spondylodysplastic-EDS-B3GALT6. Genes. 2019; 10(10):799. https://doi.org/10.3390/genes10100799
Chicago/Turabian StyleCaraffi, Stefano Giuseppe, Ilenia Maini, Ivan Ivanovski, Marzia Pollazzon, Sara Giangiobbe, Maurizia Valli, Antonio Rossi, Silvia Sassi, Silvia Faccioli, Maja Di Rocco, and et al. 2019. "Severe Peripheral Joint Laxity is a Distinctive Clinical Feature of Spondylodysplastic-Ehlers-Danlos Syndrome (EDS)-B4GALT7 and Spondylodysplastic-EDS-B3GALT6" Genes 10, no. 10: 799. https://doi.org/10.3390/genes10100799
APA StyleCaraffi, S. G., Maini, I., Ivanovski, I., Pollazzon, M., Giangiobbe, S., Valli, M., Rossi, A., Sassi, S., Faccioli, S., Di Rocco, M., Magnani, C., Campos-Xavier, B., Unger, S., Superti-Furga, A., & Garavelli, L. (2019). Severe Peripheral Joint Laxity is a Distinctive Clinical Feature of Spondylodysplastic-Ehlers-Danlos Syndrome (EDS)-B4GALT7 and Spondylodysplastic-EDS-B3GALT6. Genes, 10(10), 799. https://doi.org/10.3390/genes10100799