Role of Mitochondrial Glycerol-3-Phosphate Dehydrogenase in Metabolic Adaptations of Prostate Cancer

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Models of mGPDH Overexpression
2.3. Electrophoretic Analyses
2.4. In-gel Activity Staining of mGPDH
2.5. Western Blot Analysis
2.6. Proteomic Analysis
2.7. Enzyme Activity Assay
2.8. Oxygen Consumption and Extracellular Acidification
2.9. Reactive Oxygen Species Production
2.10. Cellular Fractionation
2.11. Scratch Assay
2.12. Statistic Analysis
3. Results
3.1. Energetics of Prostate Cancer Cell Lines
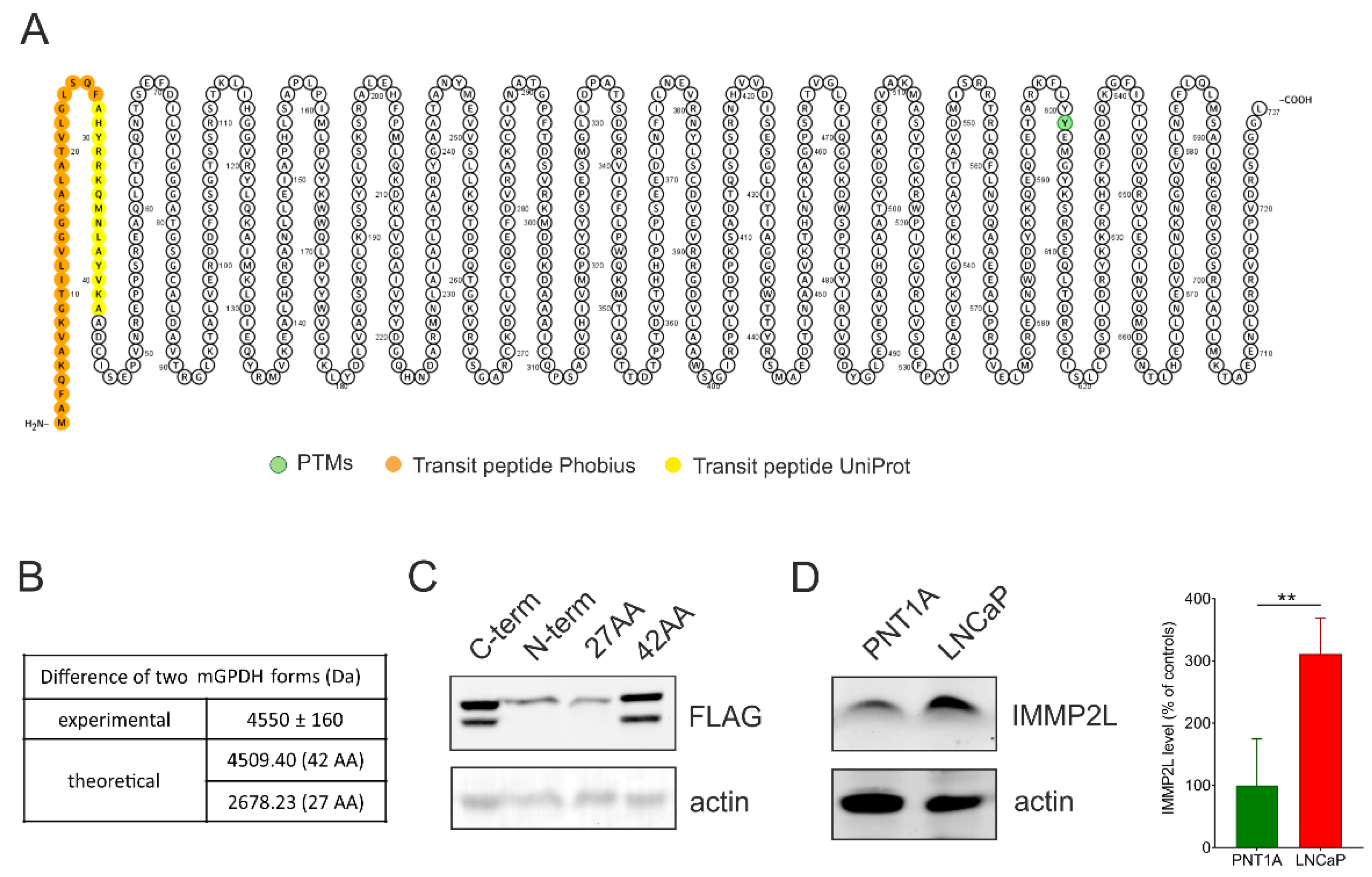
3.2. mGPDH Processing
3.3. Role of IMMP2L
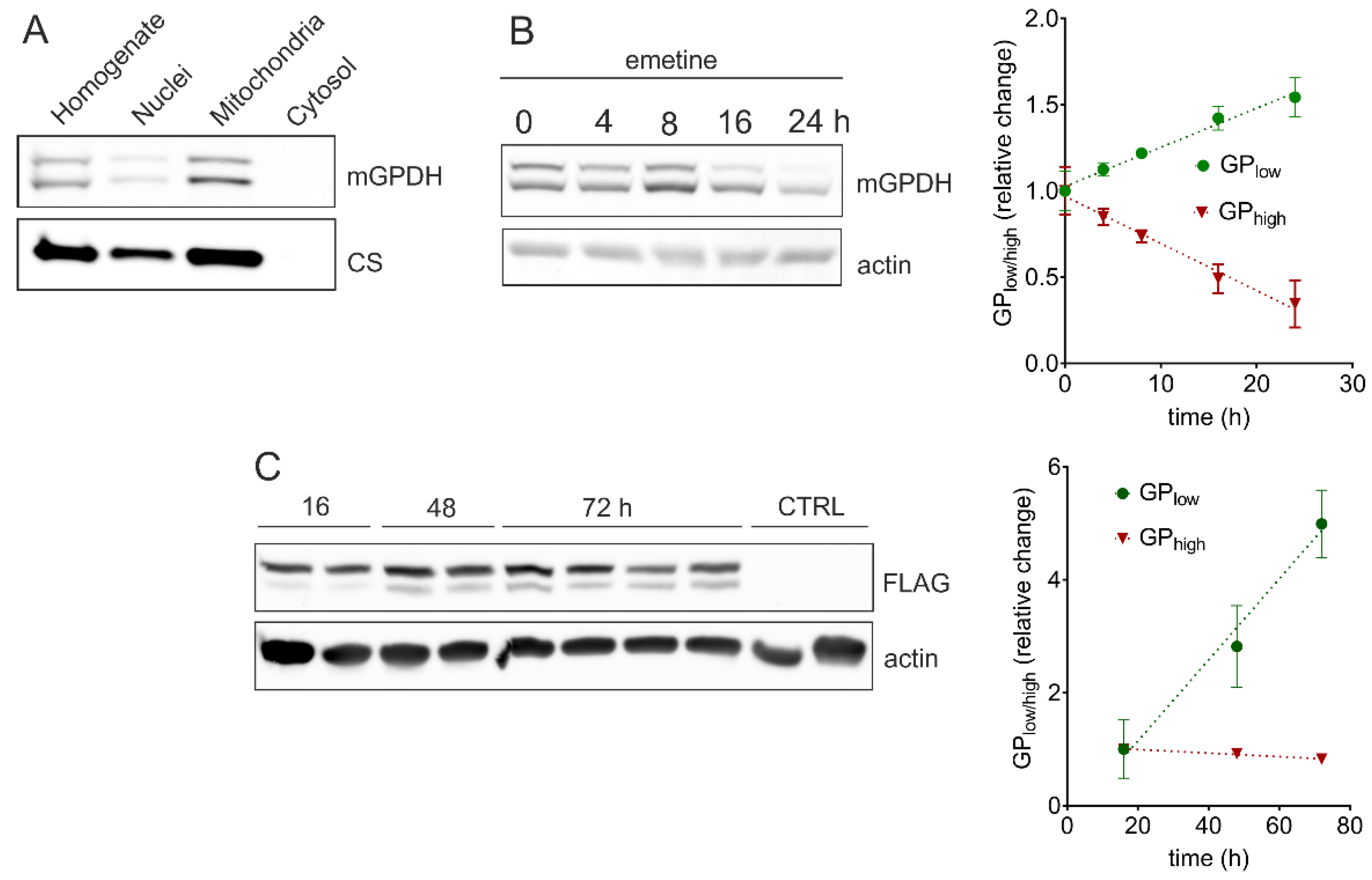
3.4. Kinetics of mGPDH Processing
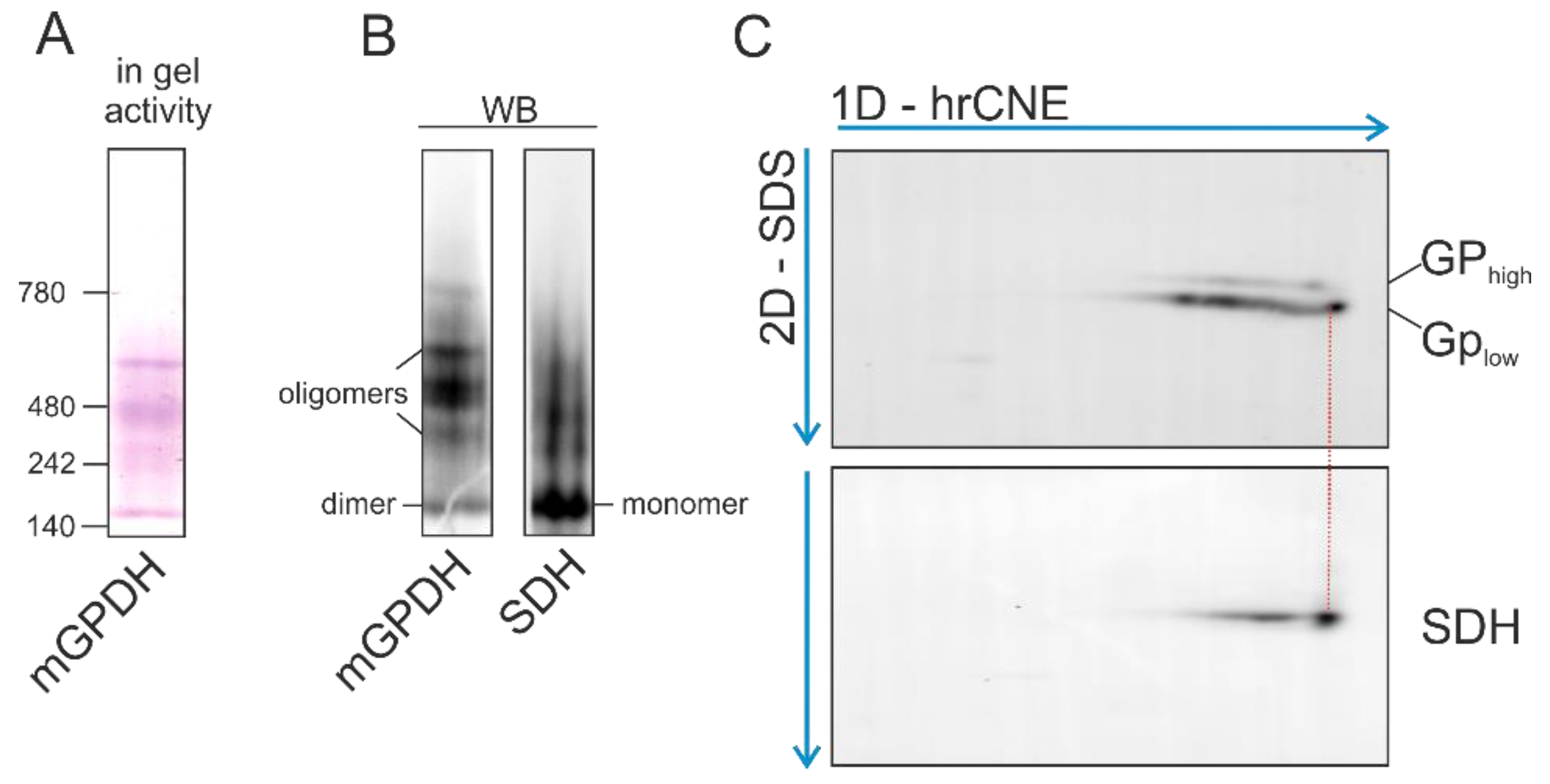
3.5. Native Forms of mGPDH
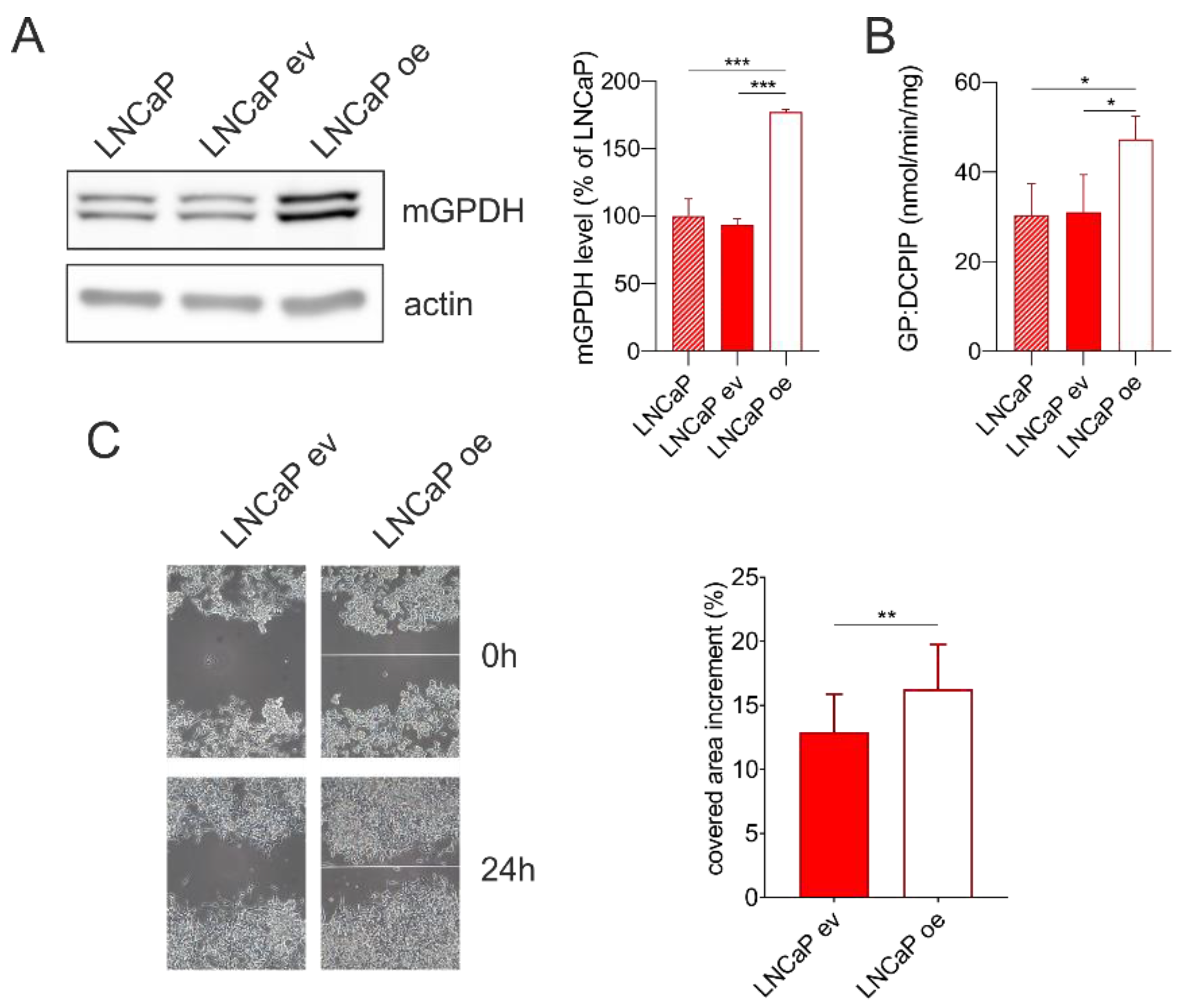
3.6. mGPDH Content Modulates Cell Migration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Culp, M.B.; Soerjomataram, I.; Efstathiou, J.A.; Bray, F.; Jemal, A. Recent Global Patterns in Prostate Cancer Incidence and Mortality Rates. Eur. Urol. 2020, 77, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Cutruzzola, F.; Giardina, G.; Marani, M.; Macone, A.; Paiardini, A.; Rinaldo, S.; Paone, A. Glucose Metabolism in the Progression of Prostate Cancer. Front. Physiol. 2017, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Costello, L.C.; Franklin, R.B.; Feng, P. Mitochondrial function, zinc, and intermediary metabolism relationships in normal prostate and prostate cancer. Mitochondrion 2005, 5, 143–153. [Google Scholar] [CrossRef]
- Singh, K.K.; Desouki, M.M.; Franklin, R.B.; Costello, L.C. Mitochondrial aconitase and citrate metabolism in malignant and nonmalignant human prostate tissues. Mol. Cancer 2006, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Gonthier, K.; Poluri, R.T.K.; Audet-Walsh, E. Functional genomic studies reveal the androgen receptor as a master regulator of cellular energy metabolism in prostate cancer. J. Steroid Biochem. Mol. Biol. 2019, 191, 105367. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7. [Google Scholar] [CrossRef]
- Stoykova, G.E.; Schlaepfer, I.R. Lipid Metabolism and Endocrine Resistance in Prostate Cancer, and New Opportunities for Therapy. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Bader, D.A.; Hartig, S.M.; Putluri, V.; Foley, C.; Hamilton, M.P.; Smith, E.A.; Saha, P.K.; Panigrahi, A.; Walker, C.; Zong, L.; et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 2019, 1, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, P.; Koc, E.; Sonpavde, G.; Singh, R.; Singh, K.K. Mitochondrial localization, import, and mitochondrial function of the androgen receptor. J. Biol. Chem. 2019, 294, 6621–6634. [Google Scholar] [CrossRef] [PubMed]
- Dueregger, A.; Schopf, B.; Eder, T.; Hofer, J.; Gnaiger, E.; Aufinger, A.; Kenner, L.; Perktold, B.; Ramoner, R.; Klocker, H.; et al. Differential Utilization of Dietary Fatty Acids in Benign and Malignant Cells of the Prostate. PLoS ONE 2015, 10, e0135704. [Google Scholar] [CrossRef] [PubMed]
- Pertega-Gomes, N.; Felisbino, S.; Massie, C.E.; Vizcaino, J.R.; Coelho, R.; Sandi, C.; Simoes-Sousa, S.; Jurmeister, S.; Ramos-Montoya, A.; Asim, M.; et al. A glycolytic phenotype is associated with prostate cancer progression and aggressiveness: A role for monocarboxylate transporters as metabolic targets for therapy. J. Pathol. 2015, 236, 517–530. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijon, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glode, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef]
- Schopf, B.; Weissensteiner, H.; Schafer, G.; Fazzini, F.; Charoentong, P.; Naschberger, A.; Rupp, B.; Fendt, L.; Bukur, V.; Giese, I.; et al. OXPHOS remodeling in high-grade prostate cancer involves mtDNA mutations and increased succinate oxidation. Nat. Commun. 2020, 11, 1487. [Google Scholar] [CrossRef]
- Chowdhury, S.K.; Gemin, A.; Singh, G. High activity of mitochondrial glycerophosphate dehydrogenase and glycerophosphate-dependent ROS production in prostate cancer cell lines. Biochem. Biophys. Res. Commun. 2005, 333, 1139–1145. [Google Scholar] [CrossRef]
- Chowdhury, S.K.; Raha, S.; Tarnopolsky, M.A.; Singh, G. Increased expression of mitochondrial glycerophosphate dehydrogenase and antioxidant enzymes in prostate cancer cell lines/cancer. Free Radic. Res. 2007, 41, 1116–1124. [Google Scholar] [CrossRef]
- Mracek, T.; Drahota, Z.; Houstek, J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta 2013, 1827, 401–410. [Google Scholar] [CrossRef]
- Drahota, Z.; Chowdhury, S.K.; Floryk, D.; Mracek, T.; Wilhelm, J.; Rauchova, H.; Lenaz, G.; Houstek, J. Glycerophosphate-dependent hydrogen peroxide production by brown adipose tissue mitochondria and its activation by ferricyanide. J. Bioenerg. Biomembr. 2002, 34, 105–113. [Google Scholar] [CrossRef]
- Mracek, T.; Pecinova, A.; Vrbacky, M.; Drahota, Z.; Houstek, J. High efficiency of ROS production by glycerophosphate dehydrogenase in mammalian mitochondria. Arch. Biochem. Biophys. 2009, 481, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.M.; Coleman, R.A. Enzymes of glycerolipid synthesis in eukaryotes. Annu. Rev. Biochem. 1980, 49, 459–487. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.M.; Osnos, M.; Rivlin, R.S. Thyroid hormone regulation of mitochondrial alpha-glycerophosphate dehydrogenase in liver and hepatoma. Cancer Res. 1970, 30, 1764–1768. [Google Scholar] [PubMed]
- Pedersen, P.L. Tumor mitochondria and the bioenergetics of cancer cells. Prog. Exp. Tumor. Res. 1978, 22, 190–274. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H. Tricine-SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Karas, M.; Schagger, H. High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol. Cell. Proteom. 2007, 6, 1215–1225. [Google Scholar] [CrossRef]
- Frederiks, W.M.; Marx, F.; Myagkaya, G.L. A histochemical study of changes in mitochondrial enzyme activities of rat liver after ischemia in vitro. Virchows Arch. B 1986, 51, 321–329. [Google Scholar] [CrossRef]
- Mracek, T.; Jesina, P.; Krivakova, P.; Bolehovska, R.; Cervinkova, Z.; Drahota, Z.; Houstek, J. Time-course of hormonal induction of mitochondrial glycerophosphate dehydrogenase biogenesis in rat liver. Biochim. Biophys. Acta 2005, 1726, 217–223. [Google Scholar] [CrossRef]
- Hartmannova, H.; Piherova, L.; Tauchmannova, K.; Kidd, K.; Acott, P.D.; Crocker, J.F.; Oussedik, Y.; Mallet, M.; Hodanova, K.; Stranecky, V.; et al. Acadian variant of Fanconi syndrome is caused by mitochondrial respiratory chain complex I deficiency due to a non-coding mutation in complex I assembly factor NDUFAF6. Hum. Mol. Genet. 2016, 25, 4062–4079. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Pajuelo Reguera, D.; Cunatova, K.; Vrbacky, M.; Pecinova, A.; Houstek, J.; Mracek, T.; Pecina, P. Cytochrome c Oxidase Subunit 4 Isoform Exchange Results in Modulation of Oxygen Affinity. Cells 2020, 9. [Google Scholar] [CrossRef]
- Mracek, T.; Pecina, P.; Vojtiskova, A.; Kalous, M.; Sebesta, O.; Houstek, J. Two components in pathogenic mechanism of mitochondrial ATPase deficiency: Energy deprivation and ROS production. Exp. Gerontol. 2006, 41, 683–687. [Google Scholar] [CrossRef]
- Bentlage, H.A.; Wendel, U.; Schagger, H.; ter Laak, H.J.; Janssen, A.J.; Trijbels, J.M. Lethal infantile mitochondrial disease with isolated complex I deficiency in fibroblasts but with combined complex I and IV deficiencies in muscle. Neurology 1996, 47, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Wee, W.K.; Chen, H.; Ong, L.T.; Qu, J.; Tan, H.F.; Tan, S.M. Expression of kindlin-3 in melanoma cells impedes cell migration and metastasis. Cell Adhes. Migr. 2017, 11, 419–433. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Kall, L.; Krogh, A.; Sonnhammer, E.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef]
- Compute pI/MW Tool. Available online: https://web.expasy.org/compute_pi/ (accessed on 8 June 2020).
- Lu, B.; Poirier, C.; Gaspar, T.; Gratzke, C.; Harrison, W.; Busija, D.; Matzuk, M.M.; Andersson, K.E.; Overbeek, P.A.; Bishop, C.E. A mutation in the inner mitochondrial membrane peptidase 2-like gene (Immp2l) affects mitochondrial function and impairs fertility in mice. Biol. Reprod. 2008, 78, 601–610. [Google Scholar] [CrossRef]
- Yuan, L.; Zhai, L.; Qian, L.; Huang, D.; Ding, Y.; Xiang, H.; Liu, X.; Thompson, J.W.; Liu, J.; He, Y.H.; et al. Switching off IMMP2L signaling drives senescence via simultaneous metabolic alteration and blockage of cell death. Cell Res. 2018, 28, 625–643. [Google Scholar] [CrossRef]
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667. [Google Scholar] [CrossRef]
- Jadvar, H.; Desai, B.; Ji, L.; Conti, P.S.; Dorff, T.B.; Groshen, S.G.; Pinski, J.K.; Quinn, D.I. Baseline 18F-FDG PET/CT parameters as imaging biomarkers of overall survival in castrate-resistant metastatic prostate cancer. J. Nucl. Med. 2013, 54, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Elia, I.; Schmieder, R.; Christen, S.; Fendt, S.M. Organ-Specific Cancer Metabolism and Its Potential for Therapy. Handb. Exp. Pharmacol. 2016, 233, 321–353. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Koza, R.A.; Kozak, U.C.; Brown, L.J.; Leiter, E.H.; MacDonald, M.J.; Kozak, L.P. Sequence and tissue-dependent RNA expression of mouse FAD-linked glycerol-3-phosphate dehydrogenase. Arch. Biochem. Biophys. 1996, 336, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Daley, B.; Gaskins, K.; Vasko, V.V.; Boufraqech, M.; Patel, D.; Sourbier, C.; Reece, J.; Cheng, S.Y.; Kebebew, E.; et al. Metformin Targets Mitochondrial Glycerophosphate Dehydrogenase to Control Rate of Oxidative Phosphorylation and Growth of Thyroid Cancer In Vitro and In Vivo. Clin. Cancer Res. 2018, 24, 4030–4043. [Google Scholar] [CrossRef]
- Houstek, J.; Drahota, Z. The regulation of glycerol 3-phosphate oxidase of rate brownadipose tissue mitochondria by long-chain free fatty acids. Mol. Cell. Biochem. 1975, 7, 45–50. [Google Scholar] [CrossRef]
- Ballif, B.A.; Carey, G.R.; Sunyaev, S.R.; Gygi, S.P. Large-scale identification and evolution indexing of tyrosine phosphorylation sites from murine brain. J. Proteome Res. 2008, 7, 311–318. [Google Scholar] [CrossRef]
- DeNardo, B.D.; Holloway, M.P.; Ji, Q.; Nguyen, K.T.; Cheng, Y.; Valentine, M.B.; Salomon, A.; Altura, R.A. Quantitative phosphoproteomic analysis identifies activation of the RET and IGF-1R/IR signaling pathways in neuroblastoma. PLoS ONE 2013, 8, e82513. [Google Scholar] [CrossRef]
- MacPherson, L.; Tokatlidis, K. Protein trafficking in the mitochondrial intermembrane space: Mechanisms and links to human disease. Biochem. J. 2017, 474, 2533–2545. [Google Scholar] [CrossRef]
- Yeh, J.I.; Chinte, U.; Du, S. Structure of glycerol-3-phosphate dehydrogenase, an essential monotopic membrane enzyme involved in respiration and metabolism. Proc. Natl. Acad. Sci. USA 2008, 105, 3280–3285. [Google Scholar] [CrossRef]
- Allen, K.N.; Entova, S.; Ray, L.C.; Imperiali, B. Monotopic Membrane Proteins Join the Fold. Trends Biochem. Sci. 2019, 44, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int Conf. Intell Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar] [PubMed]
- TMpred. Available online: https://embnet.vital-it.ch/software/TMPRED_form.html (accessed on 8 June 2020).
- Bharadwaj, M.S.; Zhou, Y.; Molina, A.J.; Criswell, T.; Lu, B. Examination of bioenergetic function in the inner mitochondrial membrane peptidase 2-like (Immp2l) mutant mice. Redox Biol. 2014, 2, 1008–1015. [Google Scholar] [CrossRef]
- Mracek, T.; Holzerova, E.; Drahota, Z.; Kovarova, N.; Vrbacky, M.; Jesina, P.; Houstek, J. ROS generation and multiple forms of mammalian mitochondrial glycerol-3-phosphate dehydrogenase. Biochim. Biophys. Acta 2014, 1837, 98–111. [Google Scholar] [CrossRef]
- Denisenko, T.V.; Gorbunova, A.S.; Zhivotovsky, B. Mitochondrial Involvement in Migration, Invasion and Metastasis. Front. Cell Dev. Biol. 2019, 7, 355. [Google Scholar] [CrossRef]
- Lehuede, C.; Dupuy, F.; Rabinovitch, R.; Jones, R.G.; Siegel, P.M. Metabolic Plasticity as a Determinant of Tumor Growth and Metastasis. Cancer Res. 2016, 76, 5201–5208. [Google Scholar] [CrossRef]
- Orr, A.L.; Ashok, D.; Sarantos, M.R.; Ng, R.; Shi, T.; Gerencser, A.A.; Hughes, R.E.; Brand, M.D. Novel inhibitors of mitochondrial sn-glycerol 3-phosphate dehydrogenase. PLoS ONE 2014, 9, e89938. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecinová, A.; Alán, L.; Brázdová, A.; Vrbacký, M.; Pecina, P.; Drahota, Z.; Houštěk, J.; Mráček, T. Role of Mitochondrial Glycerol-3-Phosphate Dehydrogenase in Metabolic Adaptations of Prostate Cancer. Cells 2020, 9, 1764. https://doi.org/10.3390/cells9081764
Pecinová A, Alán L, Brázdová A, Vrbacký M, Pecina P, Drahota Z, Houštěk J, Mráček T. Role of Mitochondrial Glycerol-3-Phosphate Dehydrogenase in Metabolic Adaptations of Prostate Cancer. Cells. 2020; 9(8):1764. https://doi.org/10.3390/cells9081764
Chicago/Turabian StylePecinová, Alena, Lukáš Alán, Andrea Brázdová, Marek Vrbacký, Petr Pecina, Zdeněk Drahota, Josef Houštěk, and Tomáš Mráček. 2020. "Role of Mitochondrial Glycerol-3-Phosphate Dehydrogenase in Metabolic Adaptations of Prostate Cancer" Cells 9, no. 8: 1764. https://doi.org/10.3390/cells9081764
APA StylePecinová, A., Alán, L., Brázdová, A., Vrbacký, M., Pecina, P., Drahota, Z., Houštěk, J., & Mráček, T. (2020). Role of Mitochondrial Glycerol-3-Phosphate Dehydrogenase in Metabolic Adaptations of Prostate Cancer. Cells, 9(8), 1764. https://doi.org/10.3390/cells9081764