Cholesterol as an Endogenous Ligand of ERRα Promotes ERRα-Mediated Cellular Proliferation and Metabolic Target Gene Expression in Breast Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Extraction and Identification of ERRα Endogenous Ligands
2.2. GST-ERRα Pull Down Assay
2.3. Tryptophan Fluorescence Quenching Assay
2.4. Cell Culture
2.5. Antibodies
2.6. Luciferase Reporter Assay to Determine Cholesterol’s Effect on ERRα’s Transcriptional Activity
2.7. Immunoblotting and Immunoprecipitation
2.8. siRNA Transfection
2.9. RNA Preparation and Analysis
2.10. Immunocytochemistry (ICC)
2.11. Cell Proliferation Assay
2.12. Migration Assay
2.13. Statistical Analysis
3. Results
3.1. Identification of Cholesterol as a Candidate Endogenous Ligand of ERRα
3.2. Cholesterol Directly Binds to ERRα and Increases Its Transcriptional Activity
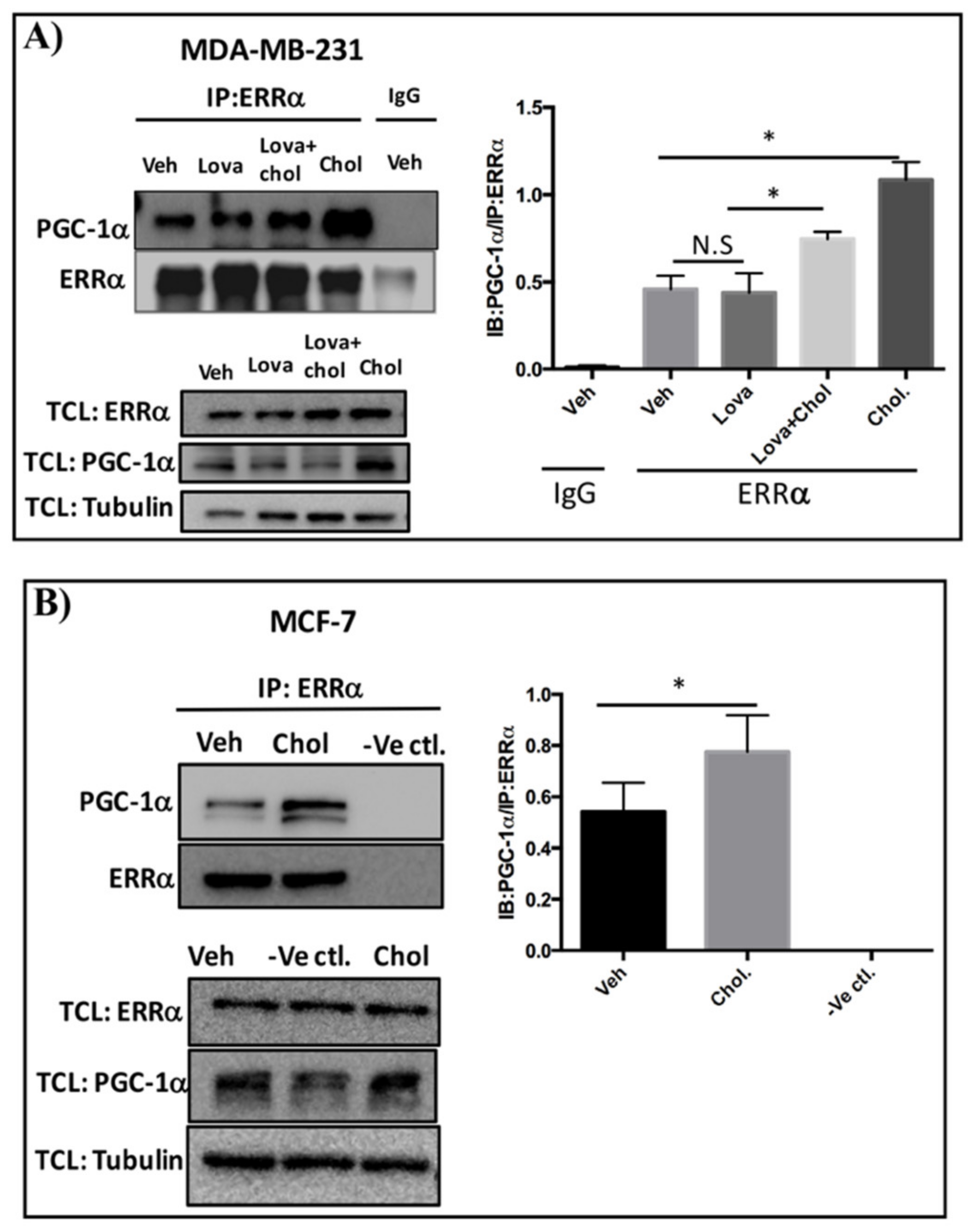
3.3. Cholesterol Enhances ERRα-PGC-1α Interaction in Breast Cancer Cells
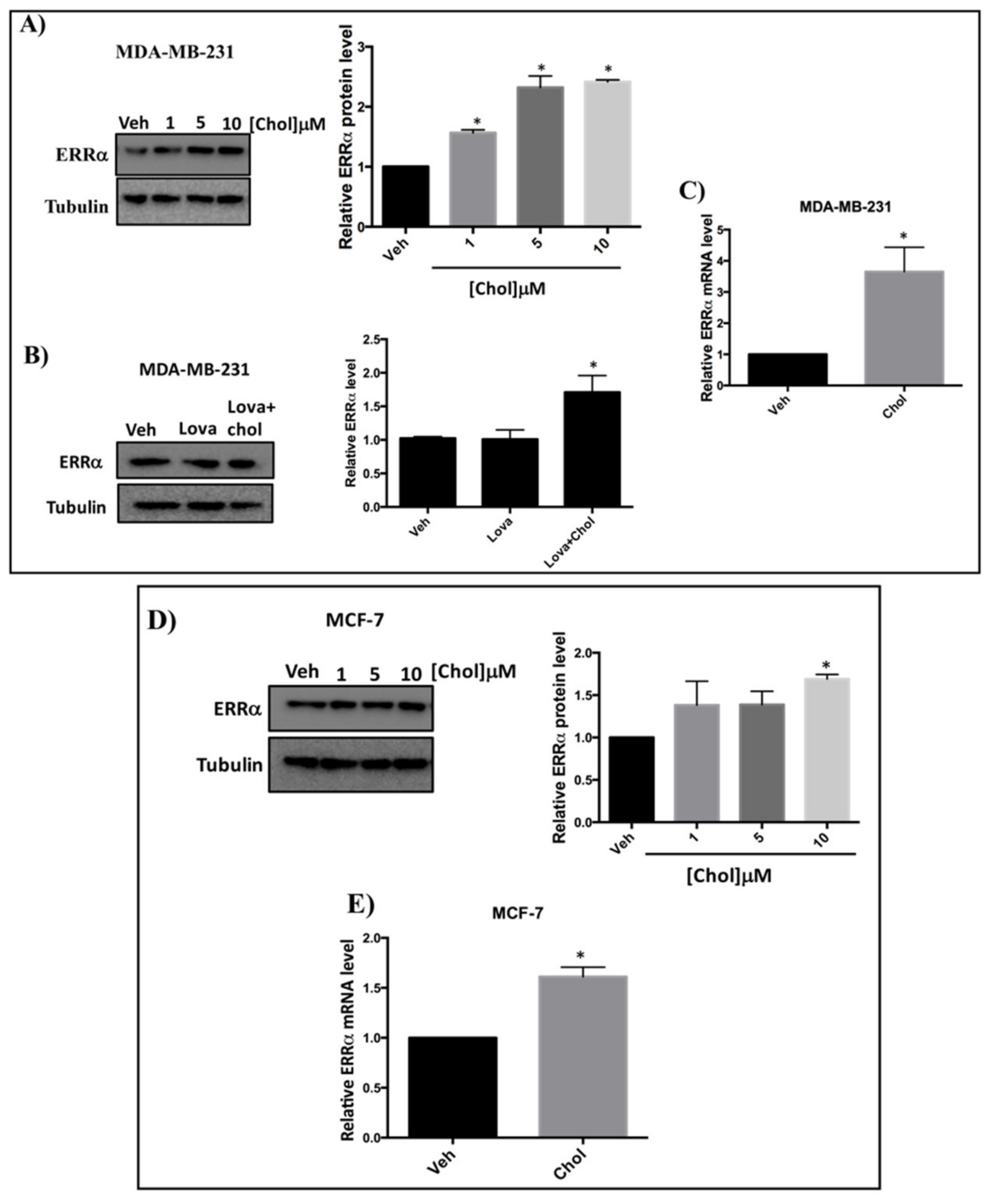
3.4. Cholesterol Increases ERRα Levels in Breast Cancer Cells
3.5. Cholesterol Enhances ERRα-Induced Metabolic Target Genes Through ERRα Pathway
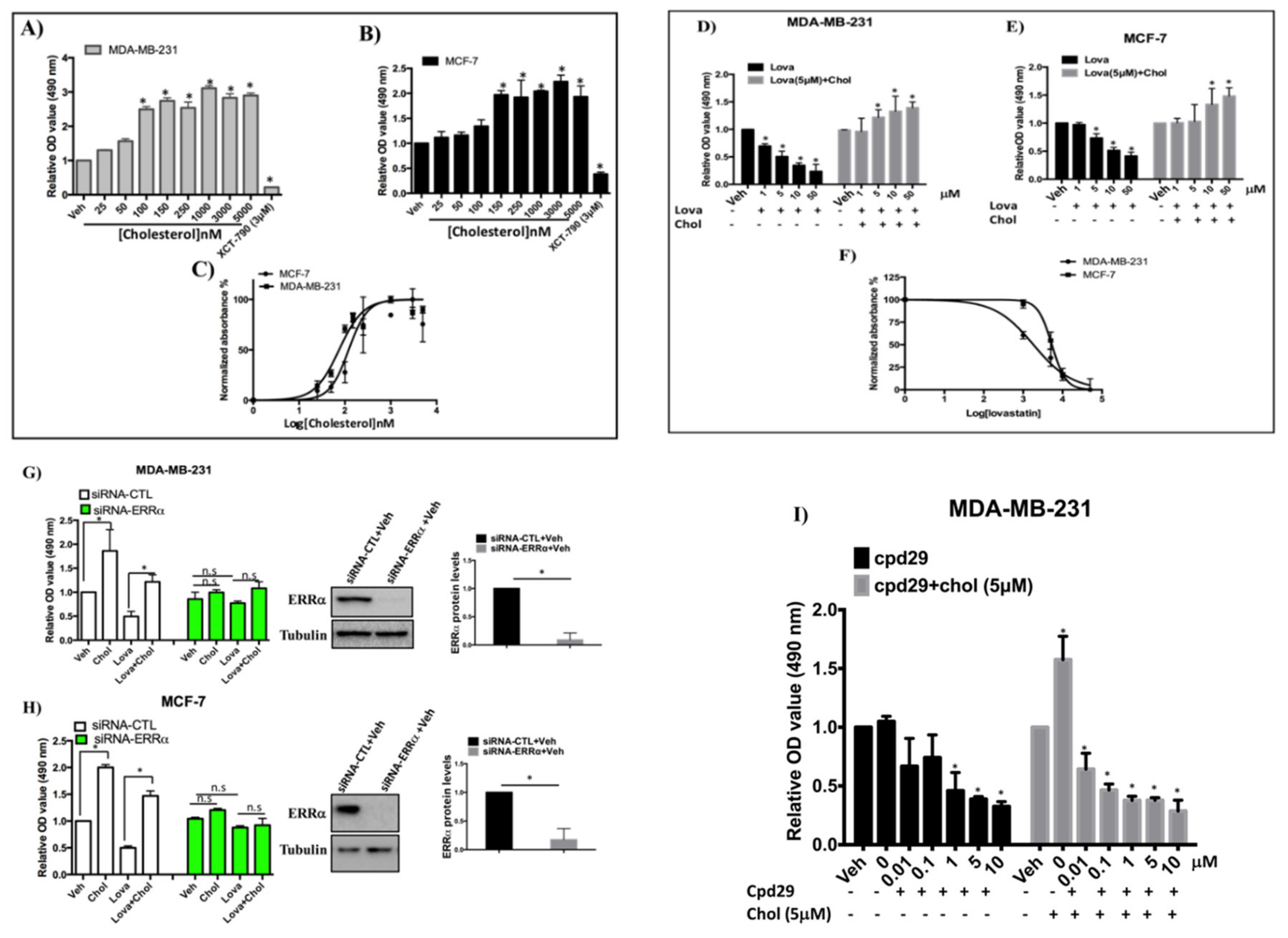
3.6. Cholesterol Enhances Cellular Proliferation of Breast Cancer Cells via the ERRα Pathway
3.7. Cholesterol Rescues the Inhibitory Effect of Lovastatin on Cellular Migration, but not that of XCT-790 in MDA-MB-231 Cells
4. Discussion
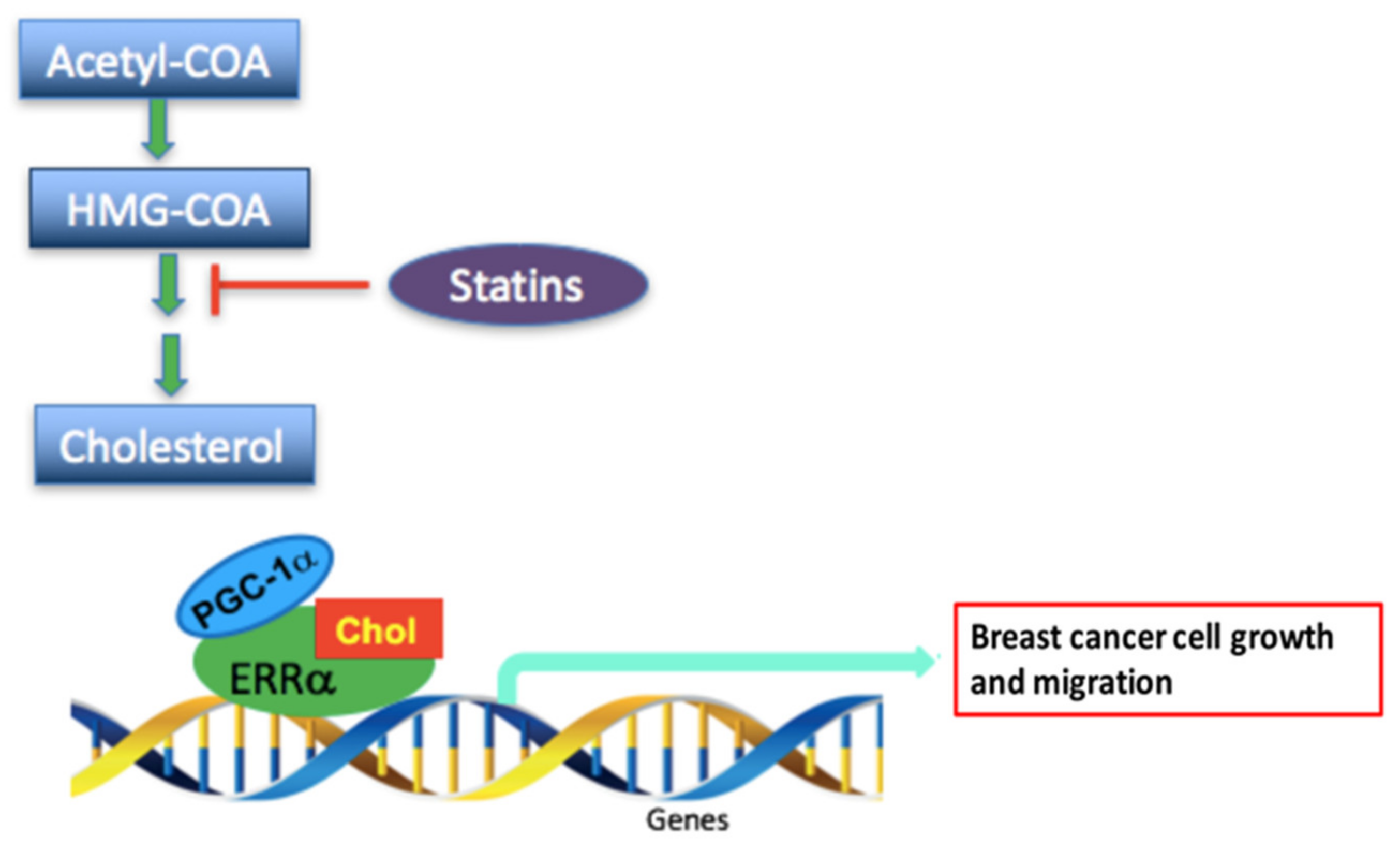
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- McDonnell, D.P.; Park, S.; Goulet, M.T.; Jasper, J.; Wardell, S.E.; Chang, C.Y.; Norris, J.D.; Guyton, J.R.; Nelson, E.R. Obesity, cholesterol metabolism, and breast cancer pathogenesis. Cancer Res. 2014, 74, 4976–4982. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.R.; Chang-Claude, J.; Goode, E.L.; Couch, F.J.; Nevanlinna, H.; Milne, R.L.; Gaudet, M.; Schmidt, M.K.; Broeks, A.; Cox, A.; et al. Associations of breast cancer risk factors with tumor subtypes: A pooled analysis from the Breast Cancer Association Consortium studies. J. Natl. Cancer Inst. 2011, 103, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Pierobon, M.; Frankenfeld, C.L. Obesity as a risk factor for triple-negative breast cancers: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2013, 137, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, F.; Kaaks, R.; Vainio, H. Overweight, obesity, and cancer risk. Lancet Oncol. 2002, 3, 565–574. [Google Scholar] [CrossRef]
- Garcia-Estevez, L.; Moreno-Bueno, G. Updating the role of obesity and cholesterol in breast cancer. BCR Breast Cancer Res. 2019, 21, 35. [Google Scholar] [CrossRef]
- Kotepui, M. Diet and risk of breast cancer. Contemp. Oncol. 2016, 20, 13–19. [Google Scholar] [CrossRef]
- Danilo, C.; Frank, P.G. Cholesterol and breast cancer development. Curr. Opin. Pharmacol. 2012, 12, 677–682. [Google Scholar] [CrossRef]
- Alikhani, N.; Ferguson, R.D.; Novosyadlyy, R.; Gallagher, E.J.; Scheinman, E.J.; Yakar, S.; LeRoith, D. Mammary tumor growth and pulmonary metastasis are enhanced in a hyperlipidemic mouse model. Oncogene 2013, 32, 961–967. [Google Scholar] [CrossRef]
- Ferguson, R.D.; Gallagher, E.J.; Cohen, D.; Tobin-Hess, A.; Alikhani, N.; Novosyadlyy, R.; Haddad, N.; Yakar, S.; LeRoith, D. Hyperinsulinemia promotes metastasis to the lung in a mouse model of Her2-mediated breast cancer. Endocr. Relat. Cancer 2013, 20, 391–401. [Google Scholar] [CrossRef]
- Nelson, E.R.; Wardell, S.E.; Jasper, J.S.; Park, S.; Suchindran, S.; Howe, M.K.; Carver, N.J.; Pillai, R.V.; Sullivan, P.M.; Sondhi, V.; et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 2013, 342, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Pelton, K.; Coticchia, C.M.; Curatolo, A.S.; Schaffner, C.P.; Zurakowski, D.; Solomon, K.R.; Moses, M.A. Hypercholesterolemia induces angiogenesis and accelerates growth of breast tumors in vivo. Am. J. Pathol. 2014, 184, 2099–2110. [Google Scholar] [CrossRef] [PubMed]
- Laisupasin, P.; Thompat, W.; Sukarayodhin, S.; Sornprom, A.; Sudjaroen, Y. Comparison of Serum Lipid Profiles between Normal Controls and Breast Cancer Patients. J. Lab. Physicians 2013, 5, 38–41. [Google Scholar] [CrossRef]
- Nelson, E.R.; Chang, C.-y.; McDonnell, D.P. Cholesterol and breast cancer pathophysiology. TEM Trends Endocrinol. Metab. 2014, 25, 649–655. [Google Scholar] [CrossRef]
- Baek, A.E.; Yu, Y.-R.A.; He, S.; Wardell, S.E.; Chang, C.-Y.; Kwon, S.; Pillai, R.V.; McDowell, H.B.; Thompson, J.W.; Dubois, L.G.; et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat. Commun. 2017, 8, 864. [Google Scholar] [CrossRef] [PubMed]
- Munsell, M.F.; Sprague, B.L.; Berry, D.A.; Chisholm, G.; Trentham-Dietz, A. Body mass index and breast cancer risk according to postmenopausal estrogen-progestin use and hormone receptor status. Epidemiol. Rev. 2014, 36, 114–136. [Google Scholar] [CrossRef]
- Chang, C.-y.; Kazmin, D.; Jasper, J.S.; Kunder, R.; Zuercher, W.J.; McDonnell, D.P. The metabolic regulator ERRα, a downstream target of HER2/IGF-1R, as a therapeutic target in breast cancer. Cancer Cell 2011, 20, 500–510. [Google Scholar] [CrossRef][Green Version]
- Stein, R.A.; Chang, C.Y.; Kazmin, D.A.; Way, J.; Schroeder, T.; Wergin, M.; Dewhirst, M.W.; McDonnell, D.P. Estrogen-related receptor alpha is critical for the growth of estrogen receptor-negative breast cancer. Cancer Res. 2008, 68, 8805–8812. [Google Scholar] [CrossRef]
- Deblois, G.; Giguère, V. Functional and physiological genomics of estrogen-related receptors (ERRs) in health and disease. Biochim. Biophys. Acta 2011, 1812, 1032–1040. [Google Scholar] [CrossRef]
- Chen, E.I.; Hewel, J.; Krueger, J.S.; Tiraby, C.; Weber, M.R.; Kralli, A.; Becker, K.; Yates, J.R., 3rd; Felding-Habermann, B. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007, 67, 1472–1486. [Google Scholar] [CrossRef]
- Deblois, G.; Hall, J.A.; Perry, M.C.; Laganiere, J.; Ghahremani, M.; Park, M.; Hallett, M.; Giguere, V. Genome-wide identification of direct target genes implicates estrogen-related receptor alpha as a determinant of breast cancer heterogeneity. Cancer Res. 2009, 69, 6149–6157. [Google Scholar] [CrossRef] [PubMed]
- Giguere, V.; Yang, N.; Segui, P.; Evans, R.M. Identification of a new class of steroid hormone receptors. Nature 1988, 331, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.; Bostner, J.; Sun, Y.; Miller, L.D.; Alayev, A.; Schwartz, N.S.; Lager, E.; Fornander, T.; Nordenskjöld, B.; Yu, J.J.; et al. ERRα Is a Marker of Tamoxifen Response and Survival in Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Berman, A.Y.; Manna, S.; Schwartz, N.S.; Katz, Y.E.; Sun, Y.; Behrmann, C.A.; Yu, J.J.; Plas, D.R.; Alayev, A.; Holz, M.K. ERRα regulates the growth of triple-negative breast cancer cells via S6K1-dependent mechanism. Signal. Transduct. Target. Ther. 2017, 2, 17035. [Google Scholar] [CrossRef]
- Kraus, R.J.; Ariazi, E.A.; Farrell, M.L.; Mertz, J.E. Estrogen-related receptor alpha 1 actively antagonizes estrogen receptor-regulated transcription in MCF-7 mammary cells. J. Biol. Chem. 2002, 277, 24826–24834. [Google Scholar] [CrossRef] [PubMed]
- Bianco, S.; Lanvin, O.; Tribollet, V.; Macari, C.; North, S.; Vanacker, J.M. Modulating estrogen receptor-related receptor-alpha activity inhibits cell proliferation. J. Biol. Chem. 2009, 284, 23286–23292. [Google Scholar] [CrossRef]
- Chisamore, M.J.; Wilkinson, H.A.; Flores, O.; Chen, J.D. Estrogen-related receptor-alpha antagonist inhibits both estrogen receptor-positive and estrogen receptor-negative breast tumor growth in mouse xenografts. Mol. Cancer Ther. 2009, 8, 672–681. [Google Scholar] [CrossRef]
- Ghanbari, F.; Hebert-Losier, A.; Barry, J.; Poirier, D.; Giguere, V.; Mader, S.; Philip, A. Isolation and functional characterization of a novel endogenous inverse agonist of estrogen related receptors (ERRs) from human pregnancy urine. J. Steroid Biochem. Mol. Biol. 2019, 191, 105352. [Google Scholar] [CrossRef]
- Wei, W.; Schwaid, A.G.; Wang, X.; Wang, X.; Chen, S.; Chu, Q.; Saghatelian, A.; Wan, Y. Ligand Activation of ERRalpha by Cholesterol Mediates Statin and Bisphosphonate Effects. Cell Metab. 2016, 23, 479–491. [Google Scholar] [CrossRef]
- Traboulsi, T.; El Ezzy, M.; Dumeaux, V.; Audemard, E.; Mader, S. Role of SUMOylation in differential ERα transcriptional repression by tamoxifen and fulvestrant in breast cancer cells. Oncogene 2019, 38, 1019–1037. [Google Scholar] [CrossRef]
- Tremblay, G.B.; Bergeron, D.; Giguere, V. 4-Hydroxytamoxifen is an isoform-specific inhibitor of orphan estrogen-receptor-related (ERR) nuclear receptors beta and gamma. Endocrinology 2001, 142, 4572–4575. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Paudel, H.K. 14-3-3zeta Mediates Tau Aggregation in Human Neuroblastoma M17 Cells. PLoS ONE 2016, 11, e0160635. [Google Scholar] [CrossRef]
- Anderson, G.L.; Manson, J.; Wallace, R.; Lund, B.; Hall, D.; Davis, S.; Shumaker, S.; Wang, C.Y.; Stein, E.; Prentice, R.L. Implementation of the Women’s Health Initiative study design. Ann. Epidemiol. 2003, 13, S5–S17. [Google Scholar] [CrossRef]
- Bahl, M.; Ennis, M.; Tannock, I.F.; Hux, J.E.; Pritchard, K.I.; Koo, J.; Goodwin, P.J. Serum lipids and outcome of early-stage breast cancer: Results of a prospective cohort study. Breast Cancer Res. Treat. 2005, 94, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Jiralerspong, S.; Kim, E.S.; Dong, W.; Feng, L.; Hortobagyi, G.N.; Giordano, S.H. Obesity, diabetes, and survival outcomes in a large cohort of early-stage breast cancer patients. Ann. Oncol. 2013, 24, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Kwan, M.L.; Habel, L.A.; Flick, E.D.; Quesenberry, C.P.; Caan, B. Post-diagnosis statin use and breast cancer recurrence in a prospective cohort study of early stage breast cancer survivors. Breast Cancer Res. Treat. 2008, 109, 573–579. [Google Scholar] [CrossRef]
- Ahern, T.P.; Pedersen, L.; Tarp, M.; Cronin-Fenton, D.P.; Garne, J.P.; Silliman, R.A.; Sorensen, H.T.; Lash, T.L. Statin prescriptions and breast cancer recurrence risk: A Danish nationwide prospective cohort study. J. Natl. Cancer Inst. 2011, 103, 1461–1468. [Google Scholar] [CrossRef]
- Beckwitt, C.H.; Brufsky, A.; Oltvai, Z.N.; Wells, A. Statin drugs to reduce breast cancer recurrence and mortality. BCR Breast Cancer Res. 2018, 20, 144. [Google Scholar] [CrossRef]
- Michalik, M.; Soczek, E.; Kosinska, M.; Rak, M.; Wojcik, K.A.; Lasota, S.; Pierzchalska, M.; Czyz, J.; Madeja, Z. Lovastatin-induced decrease of intracellular cholesterol level attenuates fibroblast-to-myofibroblast transition in bronchial fibroblasts derived from asthmatic patients. Eur J. Pharm. 2013, 704, 23–32. [Google Scholar] [CrossRef]
- Matusewicz, L.; Meissner, J.; Toporkiewicz, M.; Sikorski, A.F. The effect of statins on cancer cells—Review. Tumor Biol. 2015, 36, 4889–4904. [Google Scholar] [CrossRef]
- Zaleska, M.; Mozenska, O.; Bil, J. Statins use and cancer: An update. Future Oncol. 2018, 14, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Safi, R.; Liu, X.; Baldi, R.; Liu, W.; Liu, J.; Locasale, J.W.; Chang, C.Y.; McDonnell, D.P. Inhibition of ERRalpha Prevents Mitochondrial Pyruvate Uptake Exposing NADPH-Generating Pathways as Targetable Vulnerabilities in Breast Cancer. Cell Rep. 2019, 27, 3587–3601. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Chang, C.Y.; Safi, R.; Liu, X.; Baldi, R.; Jasper, J.S.; Anderson, G.R.; Liu, T.; Rathmell, J.C.; Dewhirst, M.W.; et al. ERRα-Regulated Lactate Metabolism Contributes to Resistance to Targeted Therapies in Breast Cancer. Cell Rep. 2016, 15, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Deblois, G.; Smith, H.W.; Tam, I.S.; Gravel, S.P.; Caron, M.; Savage, P.; Labbé, D.P.; Bégin, L.R.; Tremblay, M.L.; Park, M.; et al. ERRα mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nat. Commun. 2016, 7, 12156. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Morinibu, A.; Kobayashi, M.; Zhu, Y.; Wang, X.; Goto, Y.; Yeom, C.J.; Zhao, T.; Hirota, K.; Shinomiya, K.; et al. Aberrant IDH3α expression promotes malignant tumor growth by inducing HIF-1-mediated metabolic reprogramming and angiogenesis. Oncogene 2014, 34, 4758. [Google Scholar] [CrossRef]
- Guda, M.R.; Asuthkar, S.; Labak, C.M.; Tsung, A.J.; Alexandrov, I.; Mackenzie, M.J.; Prasad, D.V.; Velpula, K.K. Targeting PDK4 inhibits breast cancer metabolism. Am. J. Cancer Res. 2018, 8, 1725–1738. [Google Scholar] [PubMed]
- Hervouet, E.; Simonnet, H.; Godinot, C. Mitochondria and reactive oxygen species in renal cancer. Biochimie 2007, 89, 1080–1088. [Google Scholar] [CrossRef]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef]
- Stein, R.A.; Gaillard, S.; McDonnell, D.P. Estrogen-related receptor alpha induces the expression of vascular endothelial growth factor in breast cancer cells. J. Steroid Biochem. Mol. Biol. 2009, 114, 106–112. [Google Scholar] [CrossRef]
- Fradet, A.; Sorel, H.; Bouazza, L.; Goehrig, D.; Depalle, B.; Bellahcene, A.; Castronovo, V.; Follet, H.; Descotes, F.; Aubin, J.E.; et al. Dual function of ERRalpha in breast cancer and bone metastasis formation: Implication of VEGF and osteoprotegerin. Cancer Res. 2011, 71, 5728–5738. [Google Scholar] [CrossRef]
- Chang, C.Y.; McDonnell, D.P. Molecular pathways: The metabolic regulator estrogen-related receptor alpha as a therapeutic target in cancer. Clin. Cancer Res. 2012, 18, 6089–6095. [Google Scholar] [CrossRef]
- Lanvin, O.; Bianco, S.; Kersual, N.; Chalbos, D.; Vanacker, J.M. Potentiation of ICI182,780 (Fulvestrant)-induced estrogen receptor-alpha degradation by the estrogen receptor-related receptor-alpha inverse agonist XCT790. J. Biol. Chem. 2007, 282, 28328–28334. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.M.; Wilson, B.J.; Yang, X.-J.; Giguère, V. Phosphorylation-dependent sumoylation regulates estrogen-related receptor-alpha and -gamma transcriptional activity through a synergy control motif. Mol. Endocrinol. 2008, 22, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Casaburi, I.; Chimento, A.; De Luca, A.; Nocito, M.; Sculco, S.; Avena, P.; Trotta, F.; Rago, V.; Sirianni, R.; Pezzi, V. Cholesterol as an Endogenous ERRα Agonist: A New Perspective to Cancer Treatment. Front. Endocrinol. 2018, 9, 525. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Miki, Y.; Moriya, T.; Shimada, N.; Ishida, T.; Hirakawa, H.; Ohuchi, N.; Sasano, H. Estrogen-related receptor alpha in human breast carcinoma as a potent prognostic factor. Cancer Res. 2004, 64, 4670–4676. [Google Scholar] [CrossRef]
- Jarzabek, K.; Koda, M.; Kozlowski, L.; Sulkowski, S.; Kottler, M.-L.; Wolczynski, S. The significance of the expression of ERRalpha as a potential biomarker in breast cancer. J. Steroid Biochem. Mol. Biol. 2009, 113, 127–133. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Lunt, S.Y.; Dayton, T.L.; Fiske, B.P.; Israelsen, W.J.; Mattaini, K.R.; Vokes, N.I.; Stephanopoulos, G.; Cantley, L.C.; Metallo, C.M.; et al. Metabolic pathway alterations that support cell proliferation. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 325–334. [Google Scholar] [CrossRef]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghanbari, F.; Mader, S.; Philip, A. Cholesterol as an Endogenous Ligand of ERRα Promotes ERRα-Mediated Cellular Proliferation and Metabolic Target Gene Expression in Breast Cancer Cells. Cells 2020, 9, 1765. https://doi.org/10.3390/cells9081765
Ghanbari F, Mader S, Philip A. Cholesterol as an Endogenous Ligand of ERRα Promotes ERRα-Mediated Cellular Proliferation and Metabolic Target Gene Expression in Breast Cancer Cells. Cells. 2020; 9(8):1765. https://doi.org/10.3390/cells9081765
Chicago/Turabian StyleGhanbari, Faegheh, Sylvie Mader, and Anie Philip. 2020. "Cholesterol as an Endogenous Ligand of ERRα Promotes ERRα-Mediated Cellular Proliferation and Metabolic Target Gene Expression in Breast Cancer Cells" Cells 9, no. 8: 1765. https://doi.org/10.3390/cells9081765
APA StyleGhanbari, F., Mader, S., & Philip, A. (2020). Cholesterol as an Endogenous Ligand of ERRα Promotes ERRα-Mediated Cellular Proliferation and Metabolic Target Gene Expression in Breast Cancer Cells. Cells, 9(8), 1765. https://doi.org/10.3390/cells9081765