Role of Elevated Intracellular S-Adenosylhomocysteine in the Pathogenesis of Alcohol-Related Liver Disease

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Isolation of Hepatocytes and Experimental Design
2.3. Estimation of TGs
2.4. Messenger RNA Quantification
2.5. Western Blotting
2.6. Fluorescence Microscopy
2.7. Statistical Analyses
3. Results
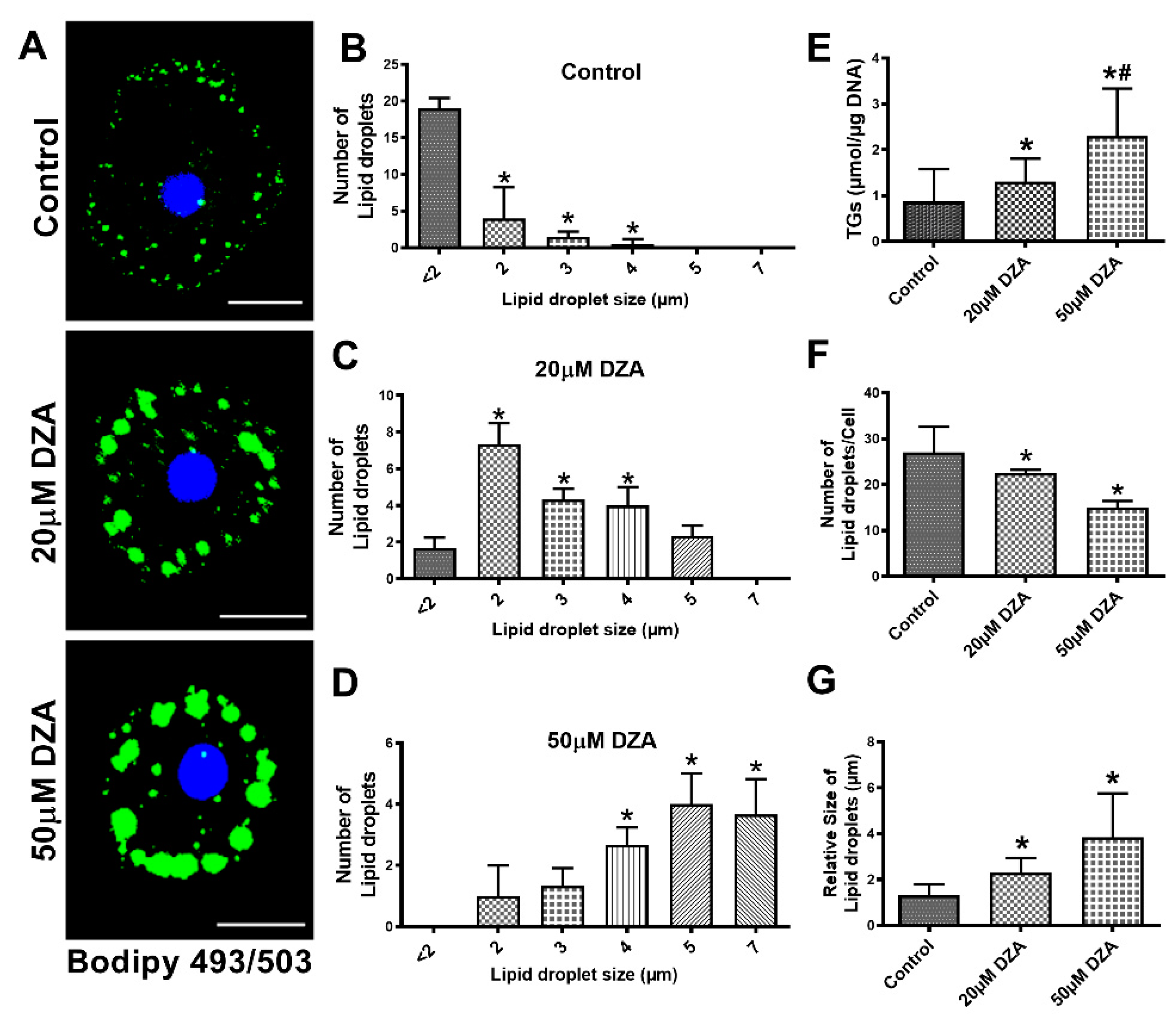
3.1. DZA Treatment Causes TG Level Increase and Accumulation of Larger-Sized LDs in Hepatocytes
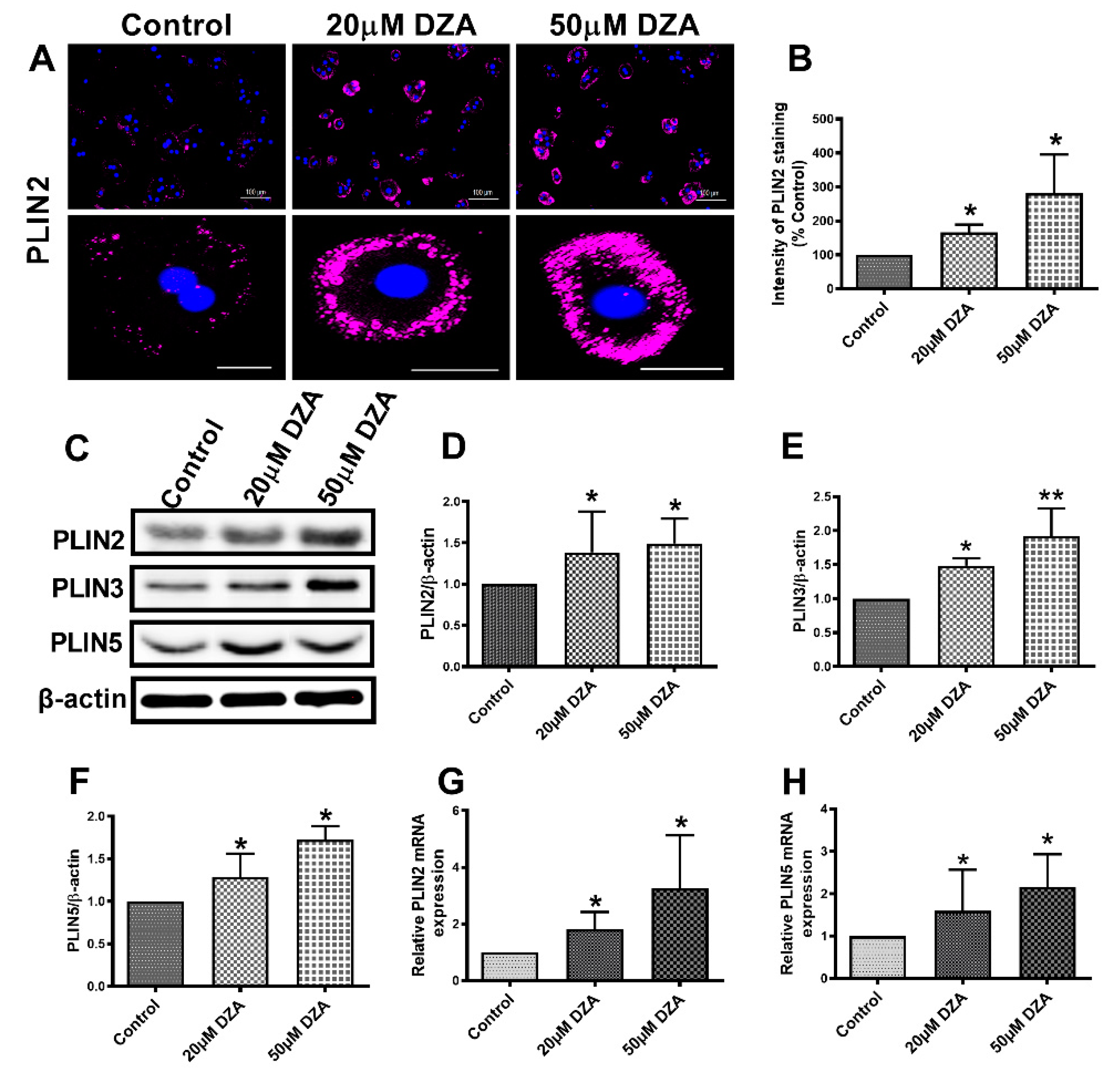
3.2. DZA Treatment Increase PLIN2, PLIN3 and PLIN5
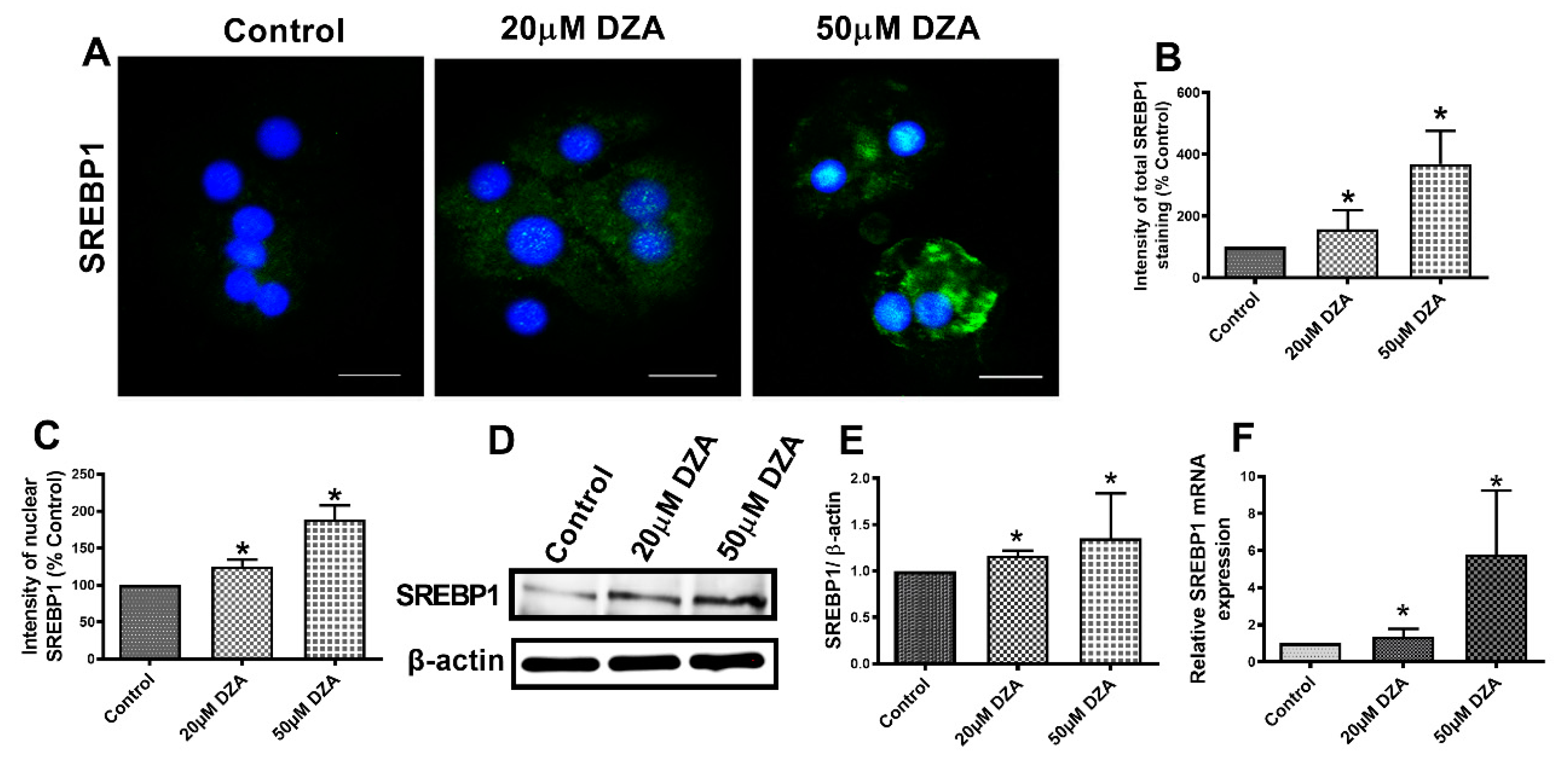
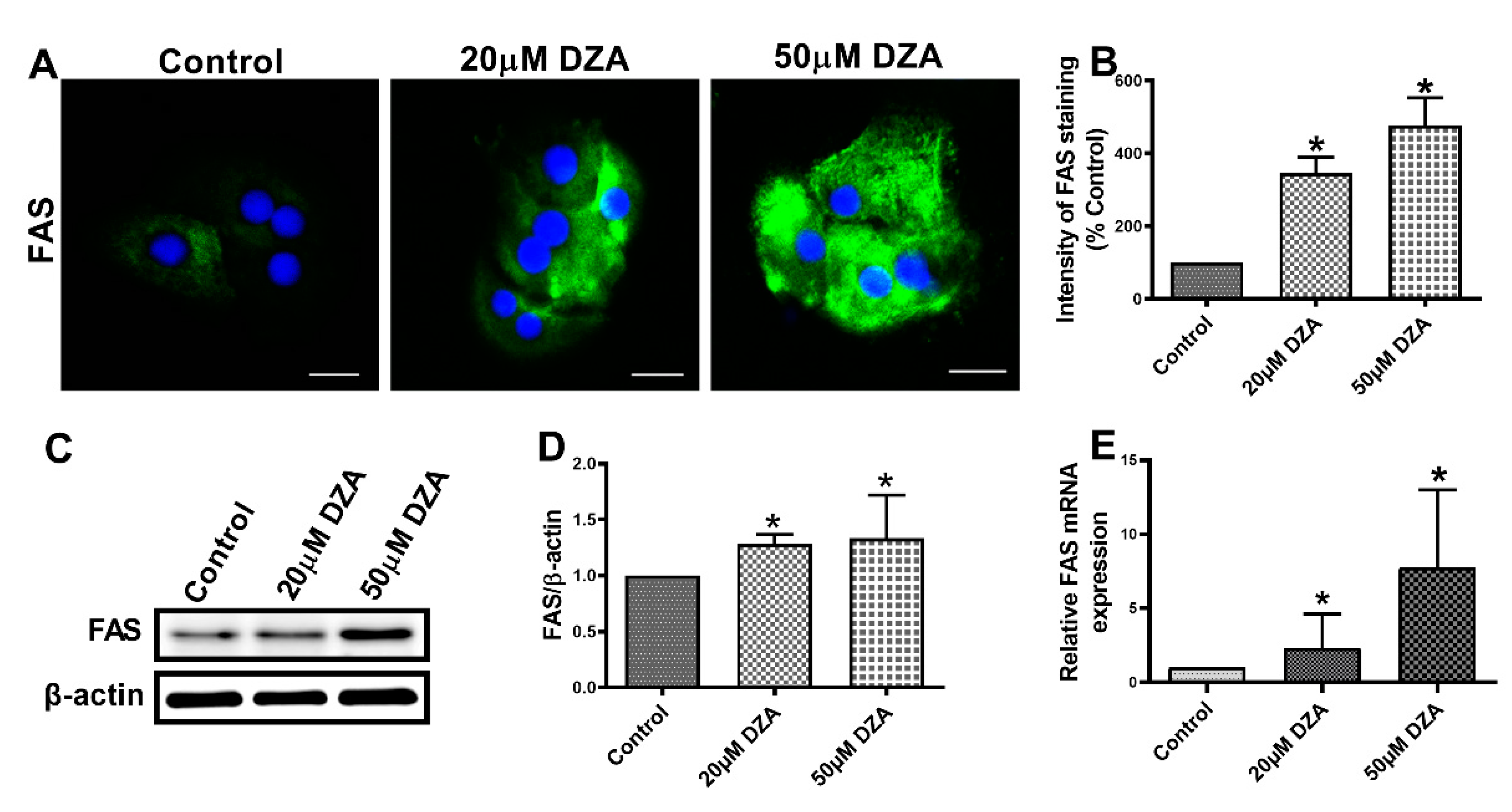
3.3. DZA Treatment Increases Sterol Regulatory Element-Binding Protein 1 (SREBP1) and Fatty Acid Synthase (FAS) Levels
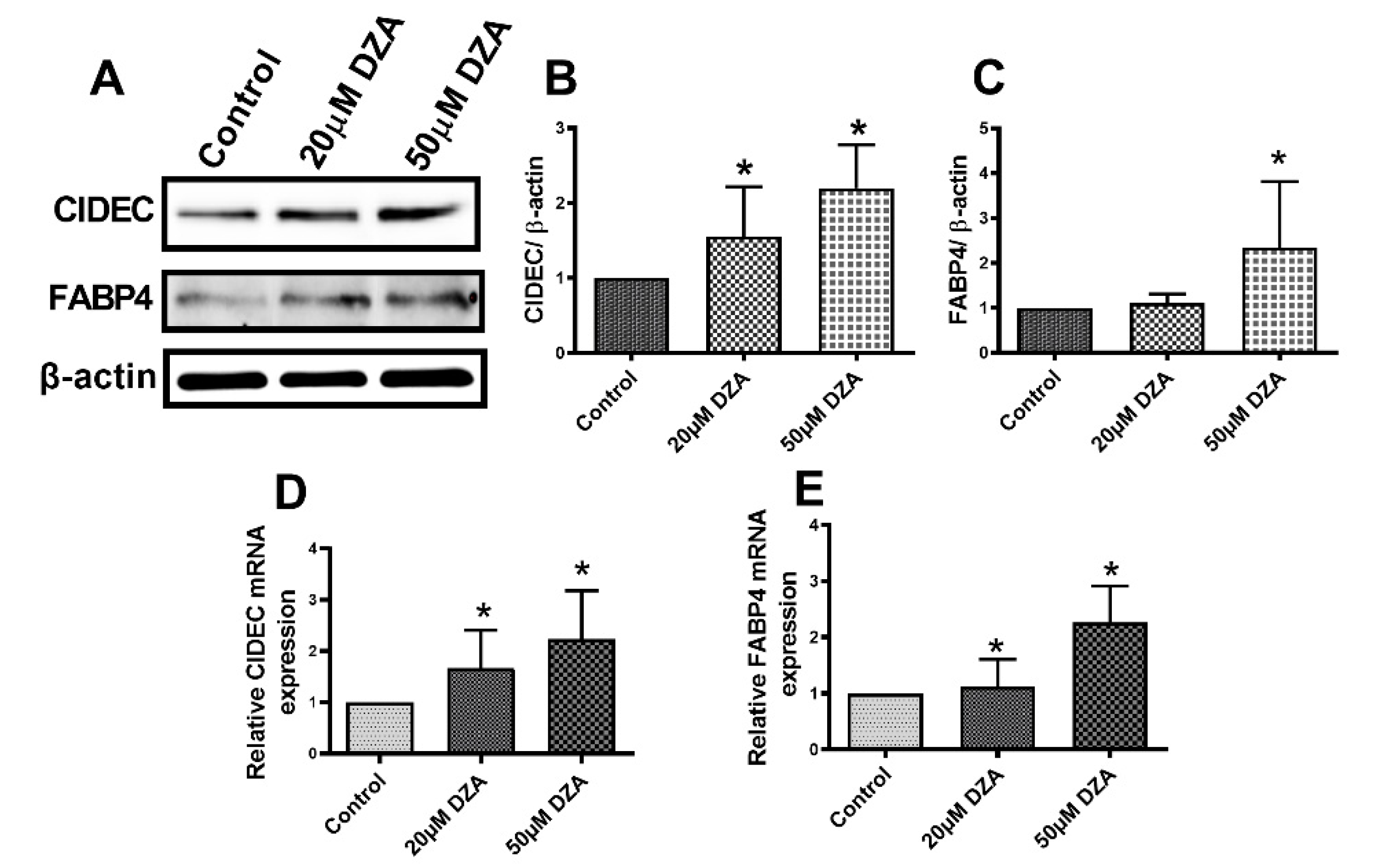
3.4. DZA Treatment Increase Cell Death-Inducing DNA Fragmentation Factor Alpha-like Effector C (CIDEC) and Fatty Acid Binding Protein 4 (FABP4) Levels
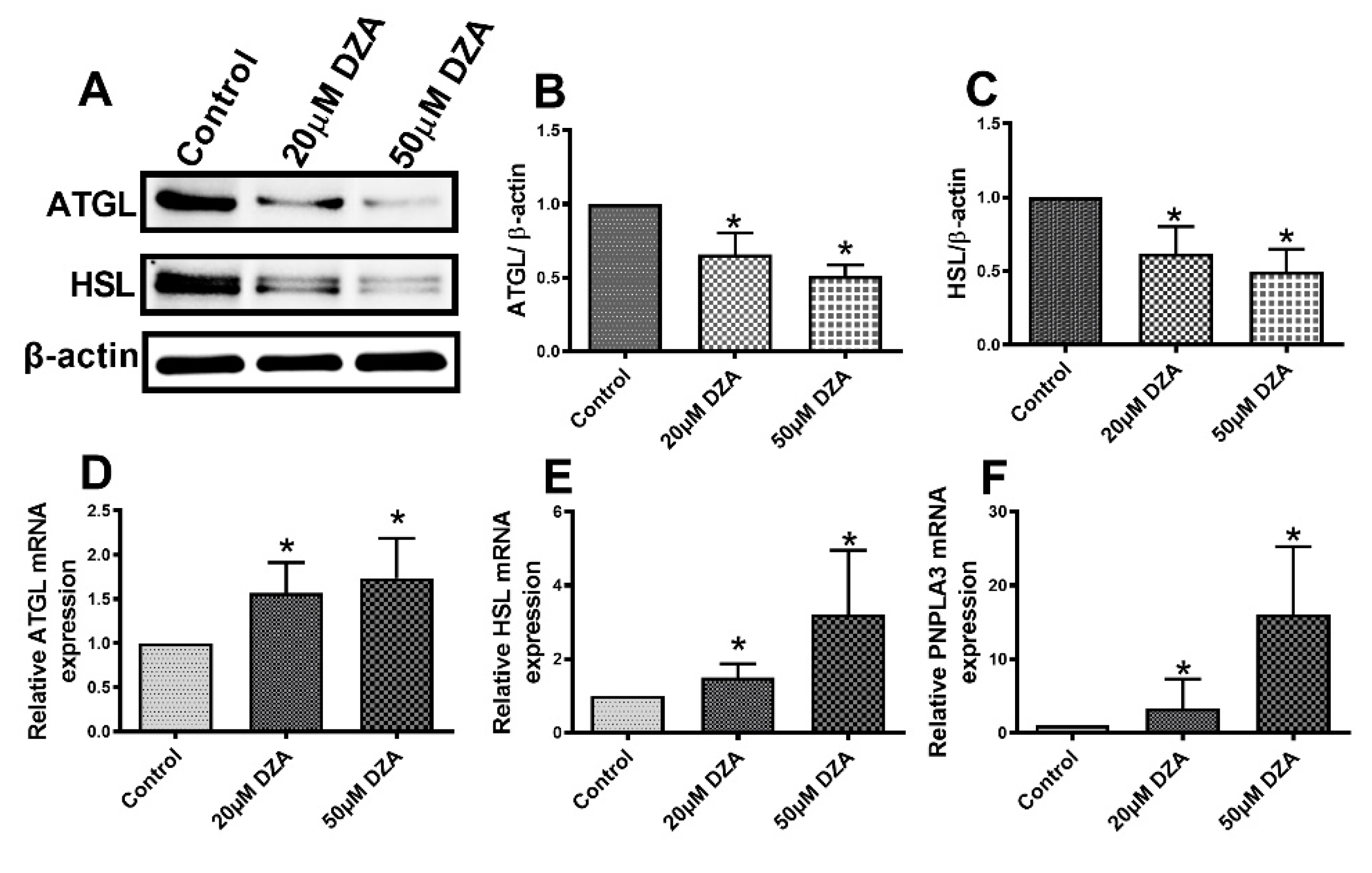
3.5. Lipases are Downregulated in Hepatocytes after DZA Treatment
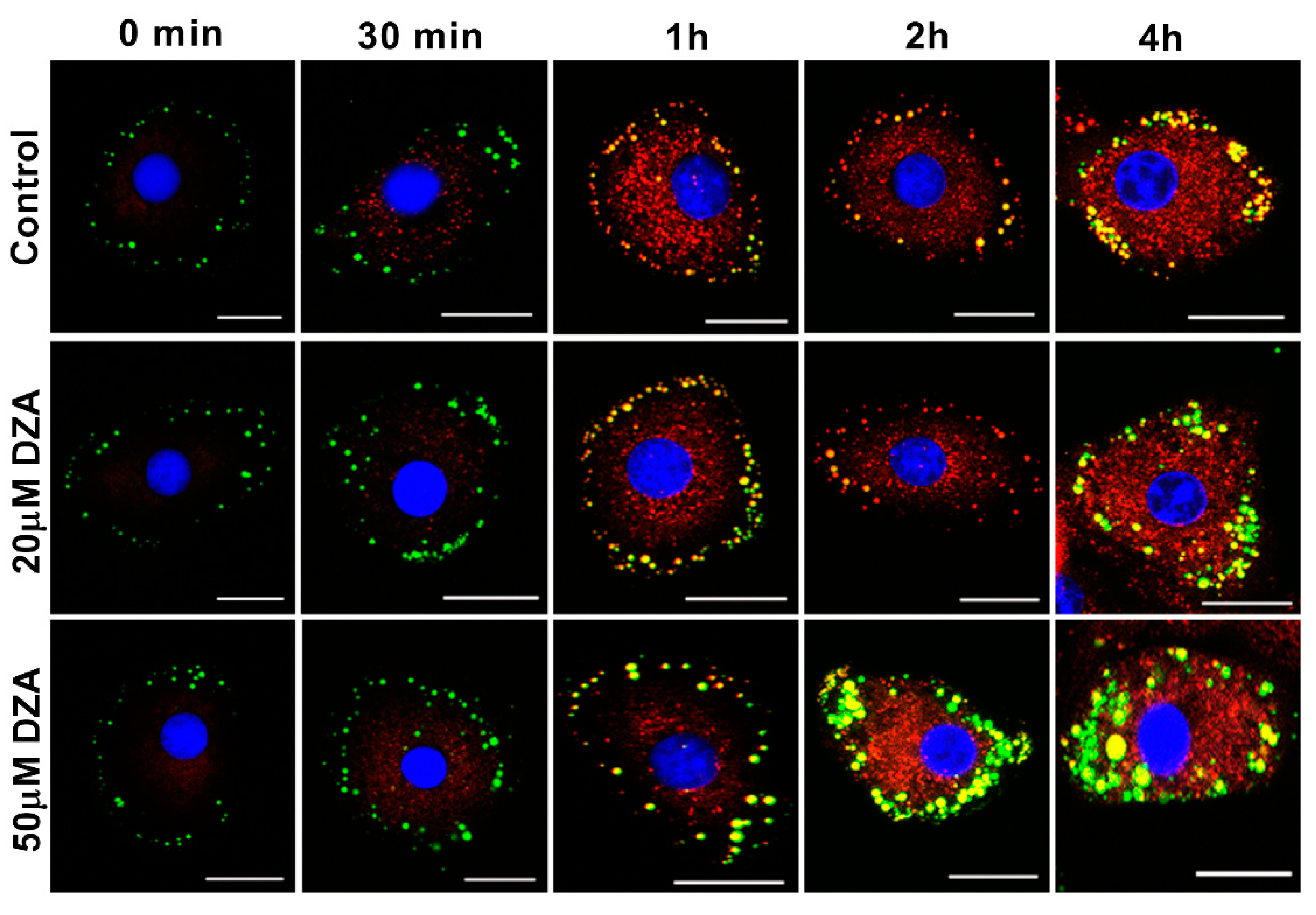
3.6. DZA Affects Nascent and Preformed LDs
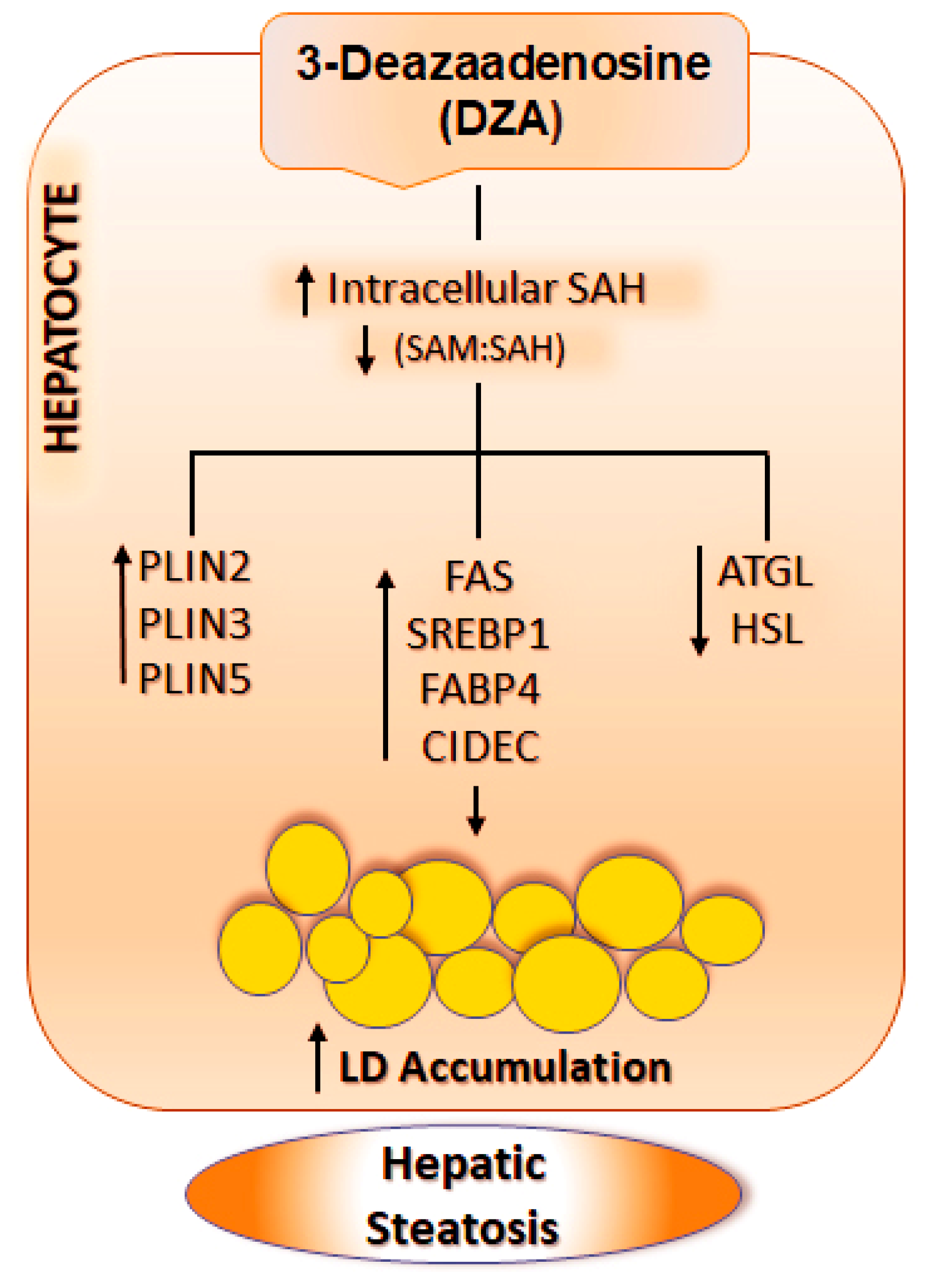
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kharbanda, K.K.; Todero, S.L.; King, A.L.; Osna, N.A.; McVicker, B.L.; Tuma, D.J.; Wisecarver, J.L.; Bailey, S.M. Betaine treatment attenuates chronic ethanol-induced hepatic steatosis and alterations to the mitochondrial respiratory chain proteome. Int. J. Hepatol. 2012, 2012, 962183. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.; Townsend, E.; Rickertsen, C.; Hains, A.; Brown, E.; Inwards, E.G.; Stoeckman, A.K.; Matis, M.P.; Sampathkumar, R.S.; Osna, N.; et al. Decreasing Phosphatidylcholine on the Surface of the Lipid Droplet Correlates with Altered Protein Binding and Steatosis. Cells 2018, 7, 230. [Google Scholar] [CrossRef]
- Fujimoto, T.; Parton, R.G. Not Just Fat: The Structure and Function of the Lipid Droplet. Cold Spring Harb. Perspect. Boil. 2011, 3, a004838. [Google Scholar] [CrossRef]
- Welte, M.A.; Gould, A.P. Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1260–1272. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Rasineni, K.; Ganesan, M.; Feng, D.; McVicker, B.L.; McNiven, M.A.; Osna, N.A.; Mott, J.L.; Casey, C.A.; Kharbanda, K.K. Structure, function and metabolism of hepatic and adipose tissue lipid droplets: Implications in alcoholic liver disease. Curr. Mol. Pharmacol. 2017, 10, 237–248. [Google Scholar] [CrossRef]
- Carr, R.M.; Ahima, R.S. Pathophysiology of lipid droplet proteins in liver diseases. Exp. Cell Res. 2016, 340, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Farese, R.V., Jr. The life of lipid droplets. Biochim. Biophys. Acta 2009, 1791, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Bartz, R.; Li, W.H.; Venables, B.; Zehmer, J.K.; Roth, M.R.; Welti, R.; Anderson, R.G.; Liu, P.; Chapman, K.D. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J. Lipid Res. 2007, 48, 837–847. [Google Scholar] [CrossRef]
- Brasaemle, D.L. Thematic review series: Adipocyte biology. The perilipin family of structural lipid droplet proteins: Stabilization of lipid droplets and control of lipolysis. J. Lipid Res. 2007, 48, 2547–2559. [Google Scholar] [CrossRef]
- Bickel, P.E.; Tansey, J.T.; Welte, M.A. Pat proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim. Biophys. Acta 2009, 1791, 419–440. [Google Scholar] [CrossRef]
- Ahmadian, M.; Wang, Y.; Sul, H.S. Lipolysis in adipocytes. Int. J. Biochem. Cell Biol. 2010, 42, 555–559. [Google Scholar] [CrossRef]
- Kharbanda, K.K.; Mailliard, M.E.; Baldwin, C.R.; Beckenhauer, H.C.; Sorrell, M.F.; Tuma, D.J. Betaine attenuates alcoholic steatosis by restoring phosphatidylcholine generation via the phosphatidylethanolamine methyltransferase pathway. J. Hepatol. 2007, 46, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K. Alcoholic liver disease and methionine metabolism. Sem. Liver Dis. 2009, 29, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K. Methionine metabolic pathway in alcoholic liver injury. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Brasaemle, D.L.; Subramanian, V.; Garcia, A.; Marcinkiewicz, A.; Rothenberg, A. Perilipin a and the control of triacylglycerol metabolism. Mol. Cell Biochem. 2009, 326, 15–21. [Google Scholar] [CrossRef]
- Hall, A.M.; Brunt, E.M.; Chen, Z.; Viswakarma, N.; Reddy, J.K.; Wolins, N.E.; Finck, B.N. Dynamic and differential regulation of proteins that coat lipid droplets in fatty liver dystrophic mice. J. Lipid Res. 2010, 51, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; Ahn, S.G.; Lee, J.H.; Kim, H.S.; Kim, J.W.; Rhim, H.; Jeong, S.W.; Kim, I.K. 3-deazaadenosine, a s-adenosylhomocysteine hydrolase inhibitor, has dual effects on nf-kappab regulation. Inhibition of nf-kappab transcriptional activity and promotion of ikappabalpha degradation. J. Biol. Chem. 1999, 274, 18981–18988. [Google Scholar] [CrossRef]
- Kharbanda, K.K.; Rogers, D.D., 2nd; Mailliard, M.E.; Siford, G.L.; Barak, A.J.; Beckenhauer, H.C.; Sorrell, M.F.; Tuma, D.J. Role of elevated s-adenosylhomocysteine in rat hepatocyte apoptosis: Protection by betaine. Biochem. Pharmacol. 2005, 70, 1883–1890. [Google Scholar] [CrossRef]
- Barak, A.J.; Beckenhauer, H.C.; Mailliard, M.E.; Kharbanda, K.K.; Tuma, D.J. Betaine lowers elevated s-adenosylhomocysteine levels in hepatocytes from ethanol-fed rats. J. Nutr. 2003, 133, 2845–2848. [Google Scholar] [CrossRef]
- Song, Z.; Zhou, Z.; Uriarte, S.; Wang, L.; Kang, Y.J.; Chen, T.; Barve, S.; McClain, C.J. S-adenosylhomocysteine sensitizes to tnf-alpha hepatotoxicity in mice and liver cells: A possible etiological factor in alcoholic liver disease. Hepatology 2004, 40, 989–997. [Google Scholar] [CrossRef]
- Song, Z.; Zhou, Z.; Song, M.; Uriarte, S.; Chen, T.; Deaciuc, I.; McClain, C.J. Alcohol-induced s-adenosylhomocysteine accumulation in the liver sensitizes to tnf hepatotoxicity: Possible involvement of mitochondrial s-adenosylmethionine transport. Biochem. Pharmacol. 2007, 74, 521–531. [Google Scholar] [CrossRef]
- Xiaoli, A.M.; Song, Z.; Yang, F. Lipogenic srebp-1a/c transcription factors activate expression of the iron regulator hepcidin, revealing cross-talk between lipid and iron metabolisms. J. Biol. Chem. 2019, 294, 12743–12753. [Google Scholar] [CrossRef]
- Asturias, F.J.; Chadick, J.Z.; Cheung, I.K.; Stark, H.; Witkowski, A.; Joshi, A.K.; Smith, S. Structure and molecular organization of mammalian fatty acid synthase. Nat. Struct. Mol. Biol. 2005, 12, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhou, L.; Li, P. Cide proteins and lipid metabolism. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Storch, J.; Thumser, A.E. The fatty acid transport function of fatty acid-binding proteins. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2000, 1486, 28–44. [Google Scholar] [CrossRef]
- Sahini, N.; Borlak, J. Recent insights into the molecular pathophysiology of lipid droplet formation in hepatocytes. Prog. Lipid Res. 2014, 54, 86–112. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, X.; Liu, P. Lipid droplet proteins and metabolic diseases. Biochim. Biophys. Acta Mol. Basis. Dis. 2018, 1864, 1968–1983. [Google Scholar] [CrossRef]
- Brasaemle, D.L.; Barber, T.; Wolins, N.E.; Serrero, G.; Blanchette-Mackie, E.J.; Londos, C. Adipose differentiation-related protein is an ubiquitously expressed lipid storage droplet-associated protein. J. Lipid Res. 1997, 38, 2249–2263. [Google Scholar] [PubMed]
- Straub, B.K.; Stoeffel, P.; Heid, H.; Zimbelmann, R.; Schirmacher, P. Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology 2008, 47, 1936–1946. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Coleman, R.A.; Kraemer, F.B.; McManaman, J.L.; Obin, M.S.; Puri, V.; Yan, Q.W.; Miyoshi, H.; Mashek, D.G. The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Investig. 2011, 121, 2102–2110. [Google Scholar] [CrossRef]
- Mak, K.M.; Ren, C.; Ponomarenko, A.; Cao, Q.; Lieber, C.S. Adipose differentiation-related protein is a reliable lipid droplet marker in alcoholic fatty liver of rats. Alcohol. Clin. Exp. Res. 2008, 32, 683–689. [Google Scholar] [CrossRef]
- Orlicky, D.J.; Roede, J.R.; Bales, E.; Greenwood, C.; Greenberg, A.; Petersen, D.; McManaman, J.L. Chronic ethanol consumption in mice alters hepatocyte lipid droplet properties. Alcohol. Clin. Exp. Res. 2011, 35, 1020–1033. [Google Scholar] [CrossRef]
- Chang, B.H.; Li, L.; Saha, P.; Chan, L. Absence of adipose differentiation related protein upregulates hepatic vldl secretion, relieves hepatosteatosis, and improves whole body insulin resistance in leptin-deficient mice. J. Lipid Res. 2010, 51, 2132–2142. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.M.; Peralta, G.; Yin, X.; Ahima, R.S. Absence of perilipin 2 prevents hepatic steatosis, glucose intolerance and ceramide accumulation in alcohol-fed mice. PLoS ONE 2014, 9, e97118. [Google Scholar] [CrossRef]
- Carr, R.M.; Patel, R.T.; Rao, V.; Dhir, R.; Graham, M.J.; Crooke, R.M.; Ahima, R.S. Reduction of tip47 improves hepatic steatosis and glucose homeostasis in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R996–R1003. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.F.; Wu, C.P.; Tang, C.K.; Tsai, C.W.; Rouhova, L.; Wu, Y.L. Identification of regulatory host genes involved in sigma virus replication using rnai knockdown in drosophila. Insects 2019, 10, 339. [Google Scholar] [CrossRef] [PubMed]
- Trevino, M.B.; Mazur-Hart, D.; Machida, Y.; King, T.; Nadler, J.; Galkina, E.V.; Poddar, A.; Dutta, S.; Imai, Y. Liver perilipin 5 expression worsens hepatosteatosis but not insulin resistance in high fat-fed mice. Mol. Endocrinol. 2015, 29, 1414–1425. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, Y.; Gao, X.; Li, L.; Yuan, Y.; Liu, F.; Zhang, L.; Wu, J.; Hu, P.; Zhang, X.; et al. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 2015, 61, 870–882. [Google Scholar] [CrossRef]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. Fat signals--lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef]
- Quiroga, A.D.; Lehner, R. Pharmacological intervention of liver triacylglycerol lipolysis: The good, the bad and the ugly. Biochem. Pharmacol. 2018, 155, 233–241. [Google Scholar] [CrossRef]
- Reid, B.N.; Ables, G.P.; Otlivanchik, O.A.; Schoiswohl, G.; Zechner, R.; Blaner, W.S.; Goldberg, I.J.; Schwabe, R.F.; Chua, S.C., Jr.; Huang, L.S. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J. Biol. Chem. 2008, 283, 13087–13099. [Google Scholar] [CrossRef]
- Wu, J.W.; Wang, S.P.; Alvarez, F.; Casavant, S.; Gauthier, N.; Abed, L.; Soni, K.G.; Yang, G.; Mitchell, G.A. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology 2011, 54, 122–132. [Google Scholar] [CrossRef]
- Fuchs, C.D.; Claudel, T.; Trauner, M. Role of metabolic lipases and lipolytic metabolites in the pathogenesis of nafld. Trends Endocrinol. Metab. 2014, 25, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Warner, D.R.; Feng, W.; Joshi-Barve, S.; McClain, C.J.; Seth, D.; Zhong, W.; Zhou, Z.; Osna, N.A.; Kharbanda, K.K. Mechanisms, biomarkers and targets for therapy in alcohol-associated liver injury: From genetics to nutrition: Summary of the isbra 2018 symposium. Alcohol 2020, 83, 105–114. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Daly, A.K.; Day, C.P. Genetics of alcoholic liver disease. Semin. Liver Dis. 2015, 35, 361–374. [Google Scholar] [PubMed]
- Restrepo, R.J.; Lim, R.W.; Korthuis, R.J.; Shukla, S.D. Binge alcohol alters pnpla3 levels in liver through epigenetic mechanism involving histone h3 acetylation. Alcohol 2017, 60, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. Srebps: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Duband-Goulet, I.; Woerner, S.; Gasparini, S.; Attanda, W.; Konde, E.; Tellier-Lebegue, C.; Craescu, C.T.; Gombault, A.; Roussel, P.; Vadrot, N.; et al. Subcellular localization of srebp1 depends on its interaction with the c-terminal region of wild-type and disease related a-type lamins. Exp. Cell Res. 2011, 317, 2800–2813. [Google Scholar] [CrossRef]
- Shimano, H.; Horton, J.D.; Hammer, R.E.; Shimomura, I.; Brown, M.S.; Goldstein, J.L. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated srebp-1a. J. Clin. Investig. 1996, 98, 1575–1584. [Google Scholar] [CrossRef]
- Shearn, C.T.; Smathers, R.L.; Backos, D.S.; Reigan, P.; Orlicky, D.J.; Petersen, D.R. Increased carbonylation of the lipid phosphatase pten contributes to akt2 activation in a murine model of early alcohol-induced steatosis. Free Radic. Biol. Med. 2013, 65, 680–692. [Google Scholar] [CrossRef]
- You, M.; Fischer, M.; Deeg, M.A.; Crabb, D.W. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (srebp). J. Biol. Chem. 2002, 277, 29342–29347. [Google Scholar] [CrossRef]
- Danesch, U.; Hoeck, W.; Ringold, G.M. Cloning and transcriptional regulation of a novel adipocyte-specific gene, fsp27. Caat-enhancer-binding protein (c/ebp) and c/ebp-like proteins interact with sequences required for differentiation-dependent expression. J. Biol. Chem. 1992, 267, 7185–7193. [Google Scholar] [PubMed]
- Puri, V.; Konda, S.; Ranjit, S.; Aouadi, M.; Chawla, A.; Chouinard, M.; Chakladar, A.; Czech, M.P. Fat-specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. J. Biol. Chem. 2007, 282, 34213–34218. [Google Scholar] [CrossRef] [PubMed]
- Slayton, M.; Gupta, A.; Balakrishnan, B.; Puri, V. Cide proteins in human health and disease. Cells 2019, 8, 238. [Google Scholar] [CrossRef] [PubMed]
- Langhi, C.; Baldan, A. Cidec/fsp27 is regulated by peroxisome proliferator-activated receptor alpha and plays a critical role in fasting- and diet-induced hepatosteatosis. Hepatology 2015, 61, 1227–1238. [Google Scholar] [CrossRef]
- Xu, X.; Park, J.G.; So, J.S.; Lee, A.H. Transcriptional activation of fsp27 by the liver-enriched transcription factor crebh promotes lipid droplet growth and hepatic steatosis. Hepatology 2015, 61, 857–869. [Google Scholar] [CrossRef]
- Hall, A.M.; Brunt, E.M.; Klein, S.; Finck, B.N. Hepatic expression of cell death-inducing dffa-like effector c in obese subjects is reduced by marked weight loss. Obesity 2010, 18, 417–419. [Google Scholar] [CrossRef]
- Thomes, P.G.; Rasineni, K.; Yang, L.; Donohue, T.M., Jr.; Kubik, J.L.; McNiven, M.A.; Casey, C.A. Ethanol withdrawal mitigates fatty liver by normalizing lipid catabolism. Am. J. Physiol.-Gastrointest. Liver Physiol. 2019, 316, G509–G518. [Google Scholar] [CrossRef]
- Schott, M.B.; Rasineni, K.; Weller, S.G.; Schulze, R.J.; Sletten, A.C.; Casey, C.A.; McNiven, M.A. Beta-adrenergic induction of lipolysis in hepatocytes is inhibited by ethanol exposure. J. Biol. Chem. 2017, 292, 11815–11828. [Google Scholar] [CrossRef]
- Leung, T.M.; Lu, Y.; Yan, W.; Moron-Concepcion, J.A.; Ward, S.C.; Ge, X.; Conde de la Rosa, L.; Nieto, N. Argininosuccinate synthase conditions the response to acute and chronic ethanol-induced liver injury in mice. Hepatology 2012, 55, 1596–1609. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arumugam, M.K.; Talawar, S.; Listenberger, L.; Donohue, T.M., Jr.; Osna, N.A.; Kharbanda, K.K. Role of Elevated Intracellular S-Adenosylhomocysteine in the Pathogenesis of Alcohol-Related Liver Disease. Cells 2020, 9, 1526. https://doi.org/10.3390/cells9061526
Arumugam MK, Talawar S, Listenberger L, Donohue TM Jr., Osna NA, Kharbanda KK. Role of Elevated Intracellular S-Adenosylhomocysteine in the Pathogenesis of Alcohol-Related Liver Disease. Cells. 2020; 9(6):1526. https://doi.org/10.3390/cells9061526
Chicago/Turabian StyleArumugam, Madan Kumar, Sharanappa Talawar, Laura Listenberger, Terrence M. Donohue, Jr., Natalia A. Osna, and Kusum K. Kharbanda. 2020. "Role of Elevated Intracellular S-Adenosylhomocysteine in the Pathogenesis of Alcohol-Related Liver Disease" Cells 9, no. 6: 1526. https://doi.org/10.3390/cells9061526
APA StyleArumugam, M. K., Talawar, S., Listenberger, L., Donohue, T. M., Jr., Osna, N. A., & Kharbanda, K. K. (2020). Role of Elevated Intracellular S-Adenosylhomocysteine in the Pathogenesis of Alcohol-Related Liver Disease. Cells, 9(6), 1526. https://doi.org/10.3390/cells9061526