Heterogeneity of Metabolic Vulnerability in Imatinib-Resistant Gastrointestinal Stromal Tumor


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Human GIST Cell Lines and Imatinib-Resistant Derivatives
2.3. GEO Dataset Analysis
2.4. Flow Cytometry for Mitochondrial Mass Measurement and Reactive Oxygen Species (ROS)
2.5. Glucose Uptake Assay
2.6. Immunofluoresence Analysis
2.7. Real-Time Glycolytic and Mitochondrial Respiration Rate Measurement
2.8. Cell Proliferation Assay
2.9. Cell Viability Assay
2.10. Giemsa Staining of Adherent Cells
2.11. Annexin V Apoptosis Assay
2.12. Western Blot Analysis
2.13. Statistical Analysis
3. Results
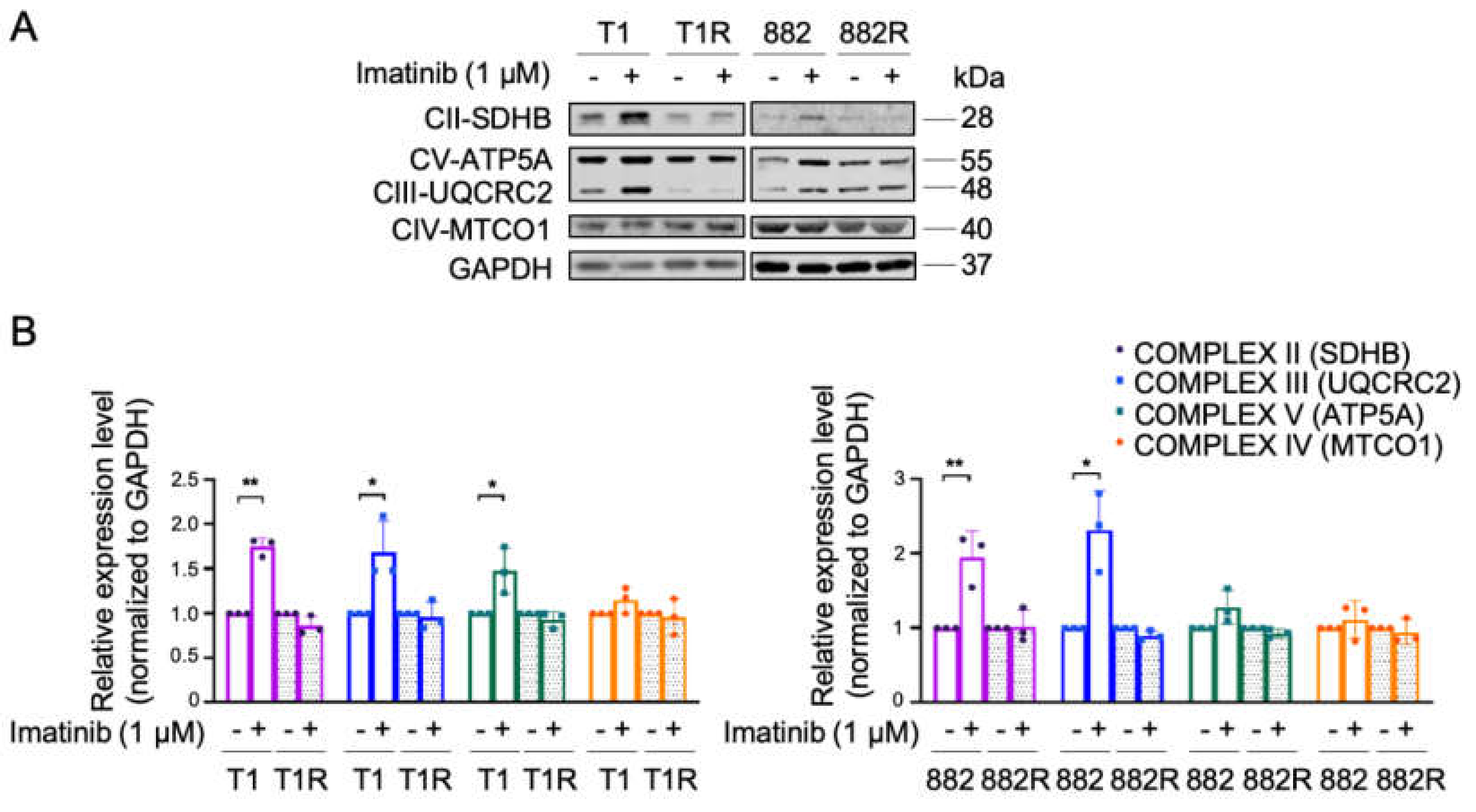
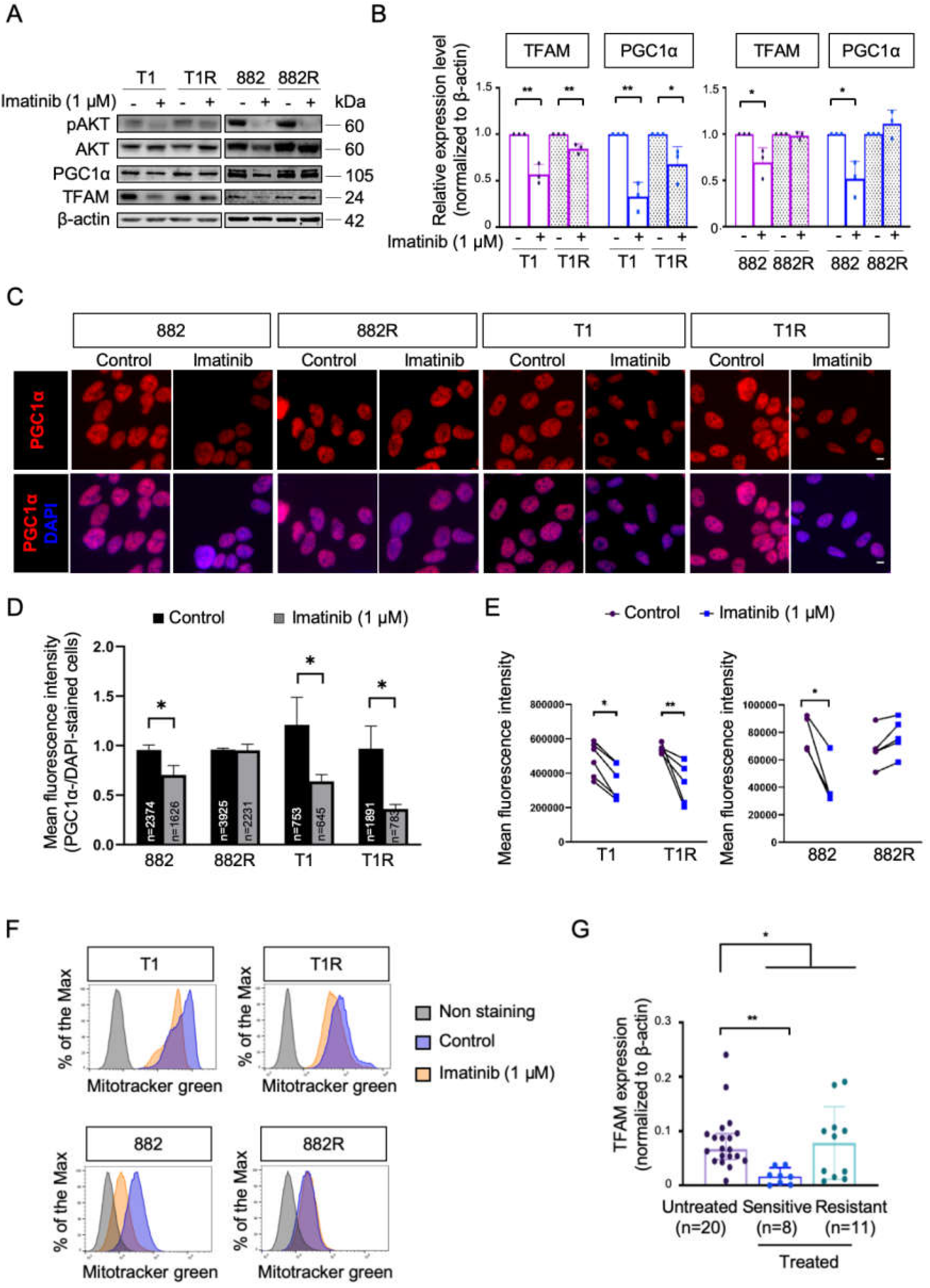
3.1. Characterization of OXPHOS and Mitochondrial Biogenesis upon Imatinib Treatment
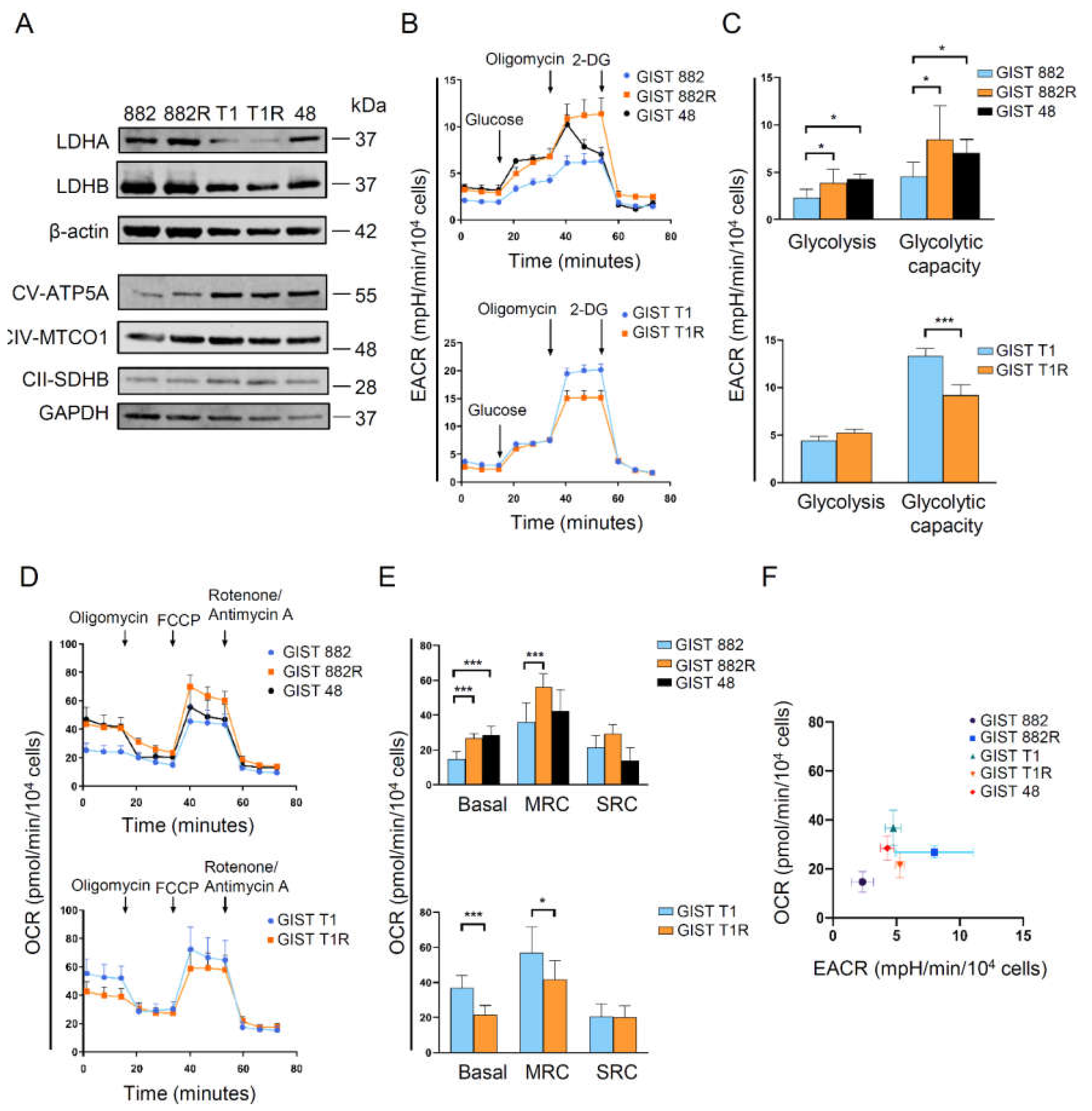
3.2. Heterogeneity of Energy Metabolism among Imatinib-Resistant GIST Cells
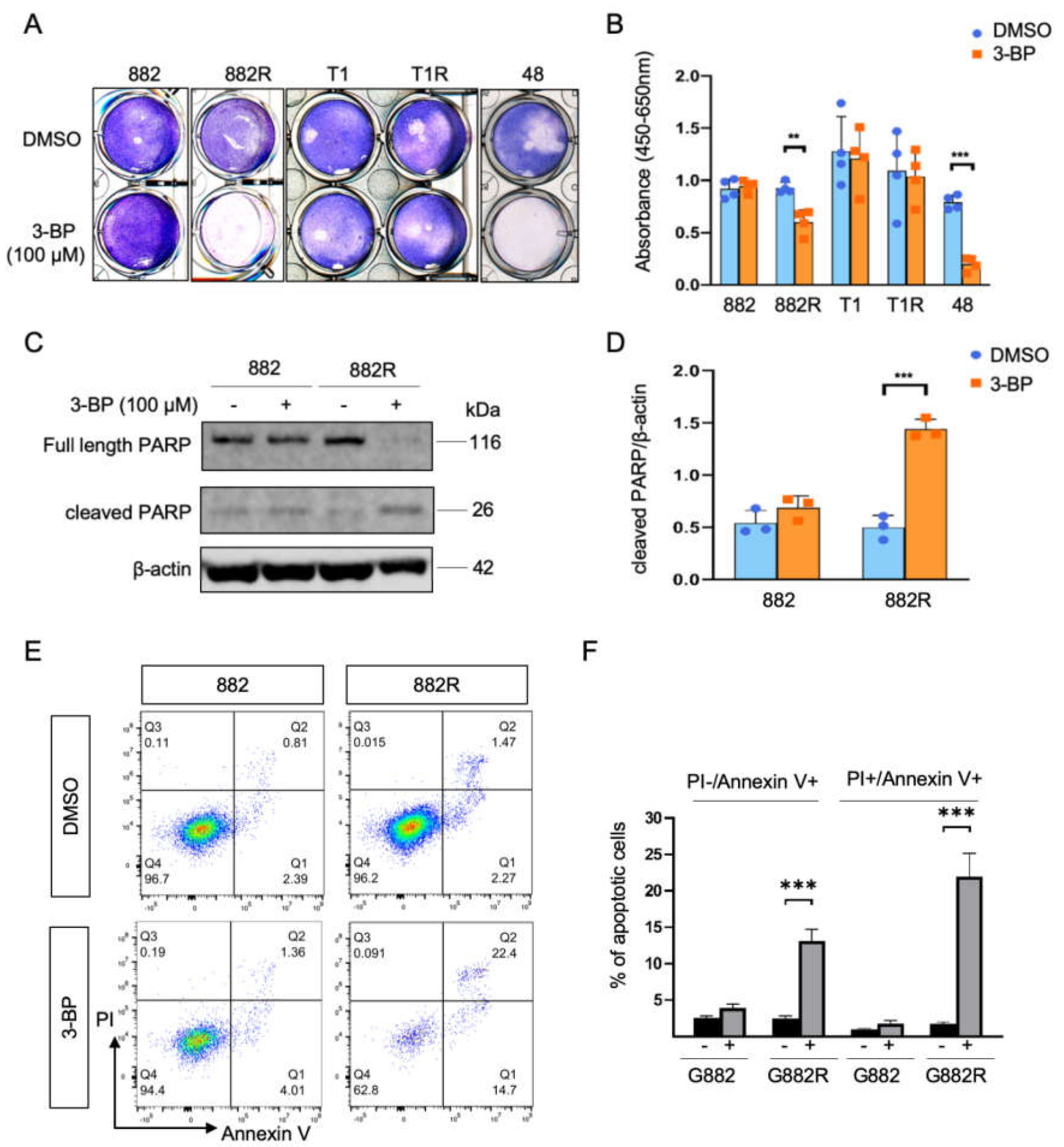
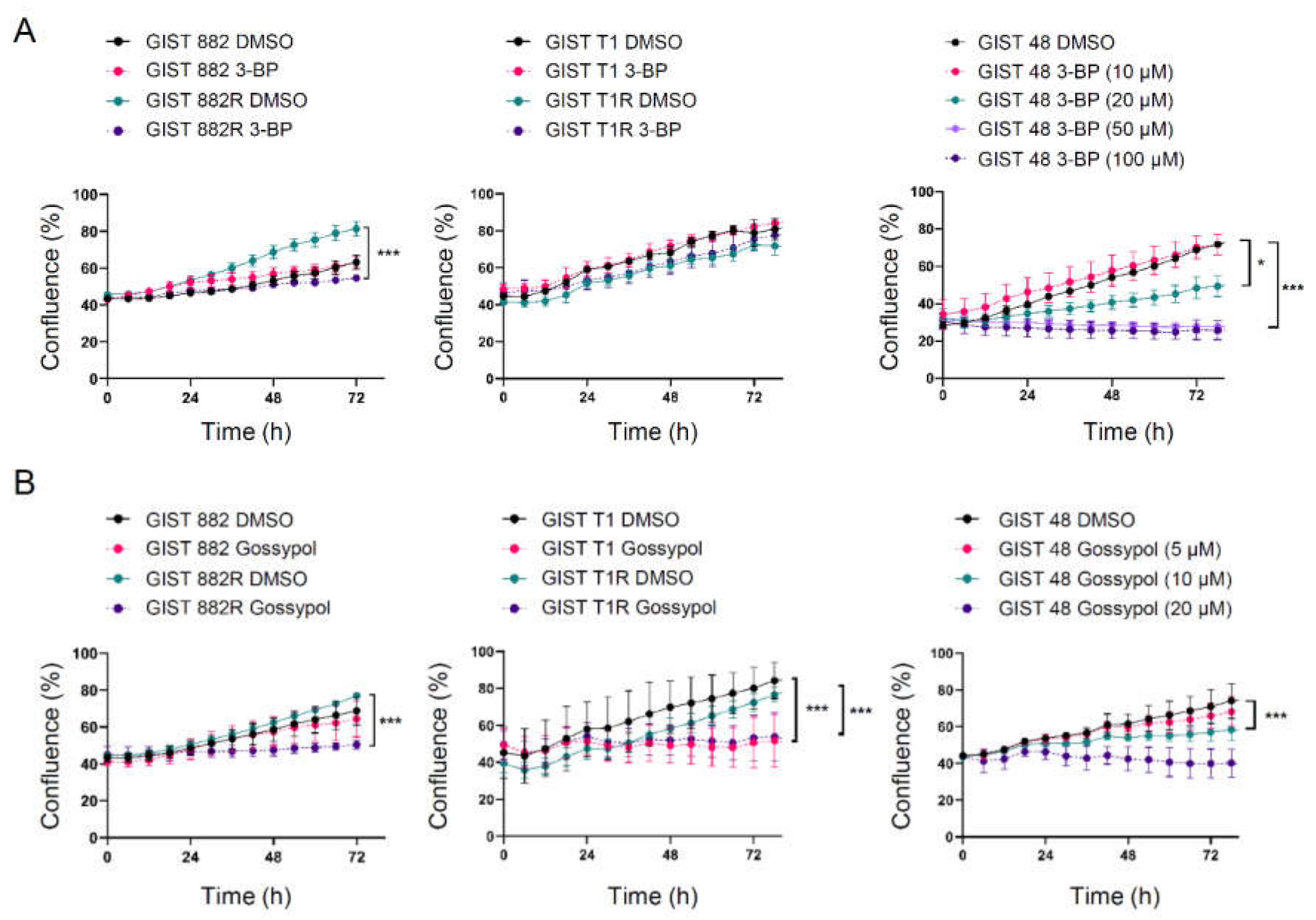
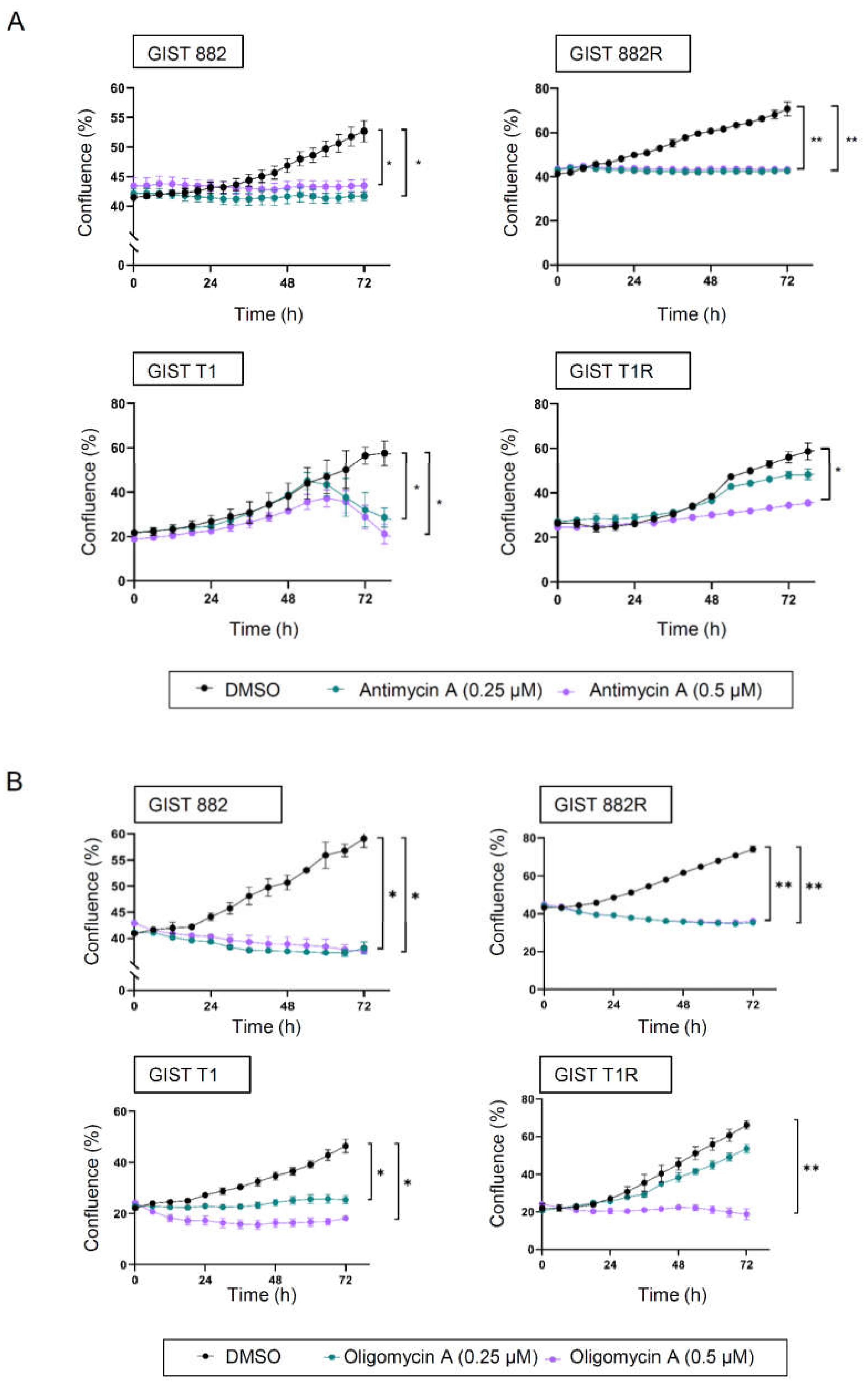
3.3. Cytotoxic Responses to Inhibition of Glycolysis and Oxidative Phosphorylation
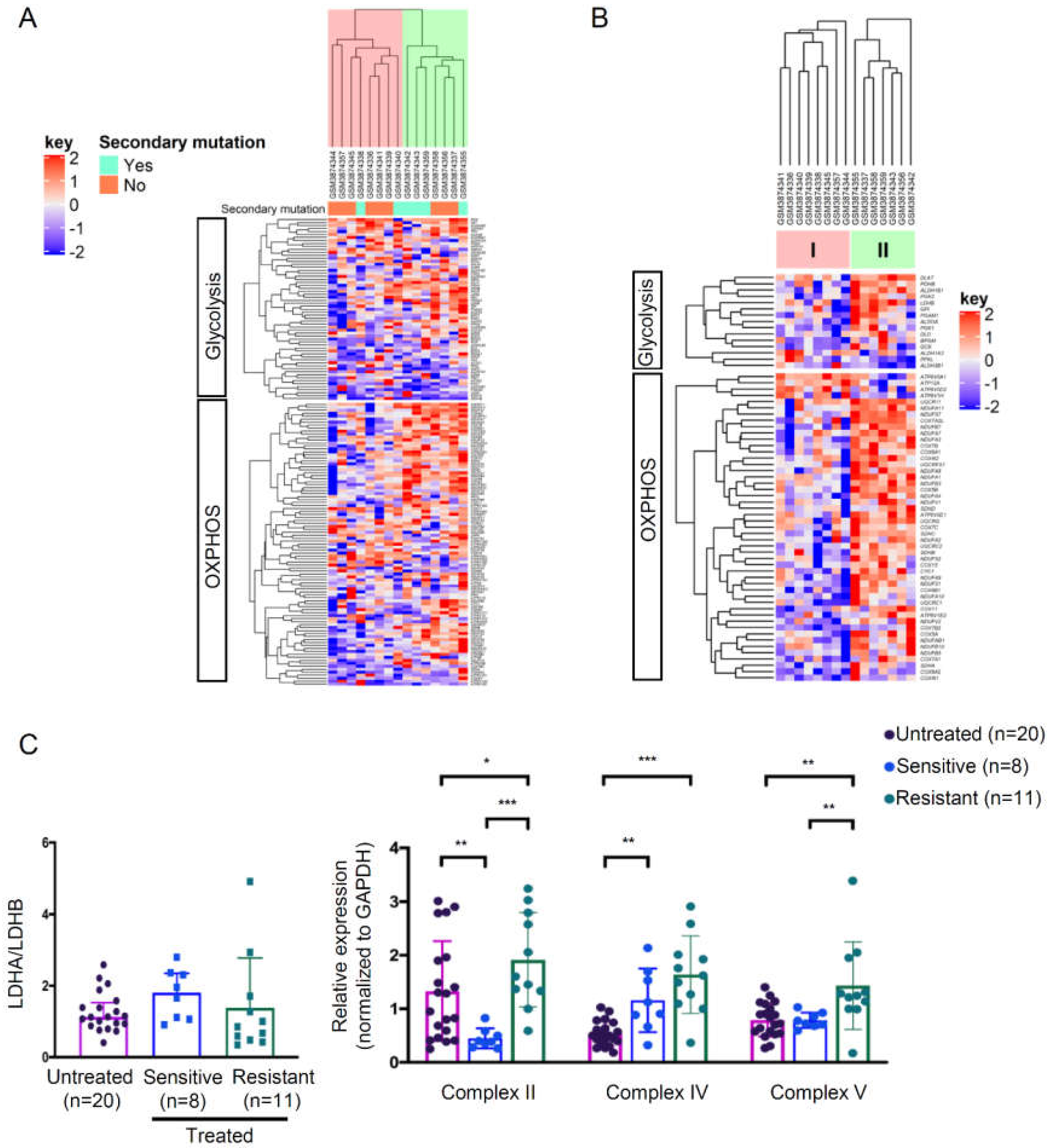
3.4. High Metabolically Active Phenotype in a Subset of Imatinib-Resistant GIST Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rubin, B.P.; Fletcher, J.A.; Fletcher, C.D. Molecular Insights into the Histogenesis and Pathogenesis of Gastrointestinal Stromal Tumors. Int. J. Surg. Pathol. 2000, 8, 5–10. [Google Scholar] [CrossRef]
- Blanke, C.D.; Demetri, G.D.; von Mehren, M.; Heinrich, M.C.; Eisenberg, B.; Fletcher, J.A.; Corless, C.L.; Fletcher, C.D.; Roberts, P.J.; Heinz, D.; et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J. Clin. Oncol. 2008, 26, 620–625. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Korbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [PubMed]
- Apicella, M.; Giannoni, E.; Fiore, S.; Ferrari, K.J.; Fernandez-Perez, D.; Isella, C.A.; Granchi, C.; Minutolo, F.; Sottile, A.; Comoglio, P.M.; et al. Increased Lactate Secretion by Cancer Cells Sustains Non-cell-autonomous Adaptive Resistance to MET and EGFR Targeted Therapies. Cell Metab. 2018, 28, 848–865. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.K.; Mustafi, S.B.; Xiong, X.; Dwivedi, S.K.D.; Nesin, V.; Saha, S.; Zhang, M.; Dhanasekaran, D.; Jayaraman, M.; Mannel, R.; et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat. Commun. 2017, 8, 14634. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Yang, C.; Tang, Z.; Yang, Y.; Tian, Y.; Yao, H.; Zhu, X.; Zhang, Z.; Ji, J.; Zheng, X. Adenylate kinase hCINAP determines self-renewal of colorectal cancer stem cells by facilitating LDHA phosphorylation. Nat. Commun. 2017, 8, 15308. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, G.A.; Medina, B.D.; Zeng, S.; Bowler, T.G.; Zhang, J.Q.; Loo, J.K.; Param, N.J.; Liu, M.; Moral, A.J.; Zhao, J.N.; et al. Mitochondrial Inhibition Augments the Efficacy of Imatinib by Resetting the Metabolic Phenotype of Gastrointestinal Stromal Tumor. Clin. Cancer Res. 2018, 24, 972–984. [Google Scholar] [CrossRef] [PubMed]
- Akcakaya, P.; Caramuta, S.; Ahlen, J.; Ghaderi, M.; Berglund, E.; Ostman, A.; Branstrom, R.; Larsson, C.; Lui, W.O. microRNA expression signatures of gastrointestinal stromal tumours: Associations with imatinib resistance and patient outcome. Br. J. Cancer 2014, 111, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Yu, L.K.; Demetri, G.D.; Fletcher, J.A. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006, 66, 9153–9161. [Google Scholar] [CrossRef]
- Huang, W.K.; Akcakaya, P.; Gangaev, A.; Lee, L.; Zeljic, K.; Hajeri, P.; Berglund, E.; Ghaderi, M.; Ahlen, J.; Branstrom, R.; et al. miR-125a-5p regulation increases phosphorylation of FAK that contributes to imatinib resistance in gastrointestinal stromal tumors. Exp. Cell Res. 2018, 371, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Urbanska, K.; Orzechowski, A. Unappreciated Role of LDHA and LDHB to Control Apoptosis and Autophagy in Tumor Cells. Int. J. Mol. Sci. 2019, 20, 85. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Ko, B.W.; Han, J.; Heo, J.Y.; Jang, Y.; Kim, S.J.; Kim, J.; Lee, M.J.; Ryu, M.J.; Song, I.C.; Jo, Y.S.; et al. Metabolic characterization of imatinib-resistant BCR-ABL T315I chronic myeloid leukemia cells indicates down-regulation of glycolytic pathway and low ROS production. Leuk. Lymphoma 2016, 57, 2180–2188. [Google Scholar] [CrossRef]
- Amirnasr, A.; Gits, C.M.M.; van Kuijk, P.F.; Smid, M.; Vriends, A.L.M.; Rutkowski, P.; Sciot, R.; Schoffski, P.; Debiec-Rychter, M.; Sleijfer, S.; et al. Molecular Comparison of Imatinib-Naive and Resistant Gastrointestinal Stromal Tumors: Differentially Expressed microRNAs and mRNAs. Cancers (Basel) 2019, 11, 882. [Google Scholar] [CrossRef]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef]
- Schurich, A.; Pallett, L.J.; Jajbhay, D.; Wijngaarden, J.; Otano, I.; Gill, U.S.; Hansi, N.; Kennedy, P.T.; Nastouli, E.; Gilson, R.; et al. Distinct Metabolic Requirements of Exhausted and Functional Virus-Specific CD8 T Cells in the Same Host. Cell Rep. 2016, 16, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.H.; Wang, R.; Wang, Y.; Kung, C.P.; Weber, J.D.; Patti, G.J. Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife 2019, 8, e41351. [Google Scholar] [CrossRef] [PubMed]
- Baltzer, C.; Tiefenbock, S.K.; Marti, M.; Frei, C. Nutrition controls mitochondrial biogenesis in the Drosophila adipose tissue through Delg and cyclin D/Cdk4. PLoS ONE 2009, 4, e6935. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef]
- Havas, K.M.; Milchevskaya, V.; Radic, K.; Alladin, A.; Kafkia, E.; Garcia, M.; Stolte, J.; Klaus, B.; Rotmensz, N.; Gibson, T.J.; et al. Metabolic shifts in residual breast cancer drive tumor recurrence. J. Clin. Investig. 2017, 127, 2091–2105. [Google Scholar] [CrossRef]
- Vellinga, T.T.; Borovski, T.; de Boer, V.C.; Fatrai, S.; van Schelven, S.; Trumpi, K.; Verheem, A.; Snoeren, N.; Emmink, B.L.; Koster, J.; et al. SIRT1/PGC1alpha-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin. Cancer Res. 2015, 21, 2870–2879. [Google Scholar] [CrossRef]
- Cruz-Bermudez, A.; Laza-Briviesca, R.; Vicente-Blanco, R.J.; Garcia-Grande, A.; Coronado, M.J.; Laine-Menendez, S.; Palacios-Zambrano, S.; Moreno-Villa, M.R.; Ruiz-Valdepenas, A.M.; Lendinez, C.; et al. Cisplatin resistance involves a metabolic reprogramming through ROS and PGC-1alpha in NSCLC which can be overcome by OXPHOS inhibition. Free Radic. Biol. Med. 2019, 135, 167–181. [Google Scholar] [CrossRef]
- Ruprecht, B.; Zaal, E.A.; Zecha, J.; Wu, W.; Berkers, C.R.; Kuster, B.; Lemeer, S. Lapatinib Resistance in Breast Cancer Cells Is Accompanied by Phosphorylation-Mediated Reprogramming of Glycolysis. Cancer Res. 2017, 77, 1842–1853. [Google Scholar] [CrossRef]
- Zhao, H.; Duan, Q.; Zhang, Z.; Li, H.; Wu, H.; Shen, Q.; Wang, C.; Yin, T. Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J. Cell Mol. Med. 2017, 21, 2055–2067. [Google Scholar] [CrossRef]
- Ye, M.; Wang, S.; Wan, T.; Jiang, R.; Qiu, Y.; Pei, L.; Pang, N.; Huang, Y.; Huang, Y.; Zhang, Z.; et al. Combined Inhibitions of Glycolysis and AKT/autophagy Can Overcome Resistance to EGFR-targeted Therapy of Lung Cancer. J. Cancer 2017, 8, 3774–3784. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, D.; Gaude, E.; Orso, G.; Giordano, C.; Guzzo, G.; Rasola, A.; Ragazzi, E.; Caparrotta, L.; Frezza, C.; Montopoli, M. Inhibition of glucose-6-phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget 2015, 6, 30102–30114. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Van den Abbeele, A.D. The lessons of GIST--PET and PET/CT: A new paradigm for imaging. Oncologist 2008, 13 (Suppl. 2), 8–13. [Google Scholar] [CrossRef]
- Dar, S.; Chhina, J.; Mert, I.; Chitale, D.; Buekers, T.; Kaur, H.; Giri, S.; Munkarah, A.; Rattan, R. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci. Rep. 2017, 7, 8760. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; McAllister, D.; Tsai, S.; Dwinell, M.B.; Kalyanaraman, B. Profiling and targeting of cellular bioenergetics: Inhibition of pancreatic cancer cell proliferation. Br. J. Cancer 2014, 111, 85–93. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef]
- Abou-Donia, M.B.; Dieckert, J.W. Metabolic fate of gossypol: The metabolism of [14-C]gossypol in swine. Toxicol. Appl. Pharmacol. 1975, 31, 32–46. [Google Scholar] [CrossRef]
- Myers, B.D.; Throneberry, G.O. Effect of gossypol on some oxidative respiratory enzymes. Plant Physiol. 1966, 41, 787–791. [Google Scholar] [CrossRef]
- Kitada, S.; Leone, M.; Sareth, S.; Zhai, D.; Reed, J.C.; Pellecchia, M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J. Med. Chem. 2003, 46, 4259–4264. [Google Scholar] [CrossRef]
- Oliver, C.L.; Bauer, J.A.; Wolter, K.G.; Ubell, M.L.; Narayan, A.; O’Connell, K.M.; Fisher, S.G.; Wang, S.; Wu, X.; Ji, M.; et al. In vitro effects of the BH3 mimetic, (-)-gossypol, on head and neck squamous cell carcinoma cells. Clin. Cancer Res. 2004, 10, 7757–7763. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Zhang, W.; Liu, J.; Hammoudi, N.; Dai, J.; Xu, R.H.; Pusztai, L.; Huang, P. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: Role of mTOR pathway and therapeutic potential. Breast Cancer Res. 2014, 16, 434. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, W.-K.; Gao, J.; Chen, Z.; Shi, H.; Yuan, J.; Cui, H.L.; Yeh, C.-N.; Bränström, R.; Larsson, C.; Li, S.; et al. Heterogeneity of Metabolic Vulnerability in Imatinib-Resistant Gastrointestinal Stromal Tumor. Cells 2020, 9, 1333. https://doi.org/10.3390/cells9061333
Huang W-K, Gao J, Chen Z, Shi H, Yuan J, Cui HL, Yeh C-N, Bränström R, Larsson C, Li S, et al. Heterogeneity of Metabolic Vulnerability in Imatinib-Resistant Gastrointestinal Stromal Tumor. Cells. 2020; 9(6):1333. https://doi.org/10.3390/cells9061333
Chicago/Turabian StyleHuang, Wen-Kuan, Jiwei Gao, Ziqing Chen, Hao Shi, Juan Yuan, Huanhuan L. Cui, Chun-Nan Yeh, Robert Bränström, Catharina Larsson, Shuijie Li, and et al. 2020. "Heterogeneity of Metabolic Vulnerability in Imatinib-Resistant Gastrointestinal Stromal Tumor" Cells 9, no. 6: 1333. https://doi.org/10.3390/cells9061333
APA StyleHuang, W.-K., Gao, J., Chen, Z., Shi, H., Yuan, J., Cui, H. L., Yeh, C.-N., Bränström, R., Larsson, C., Li, S., & Lui, W.-O. (2020). Heterogeneity of Metabolic Vulnerability in Imatinib-Resistant Gastrointestinal Stromal Tumor. Cells, 9(6), 1333. https://doi.org/10.3390/cells9061333