From Genetic Alterations to Tumor Microenvironment: The Ariadne’s String in Pancreatic Cancer

 , ,
, ,  , , , , , , and
, , , , , , and 
Abstract
1. Introduction
2. Driver Genes Alterations and Molecular Pathways to PC Development
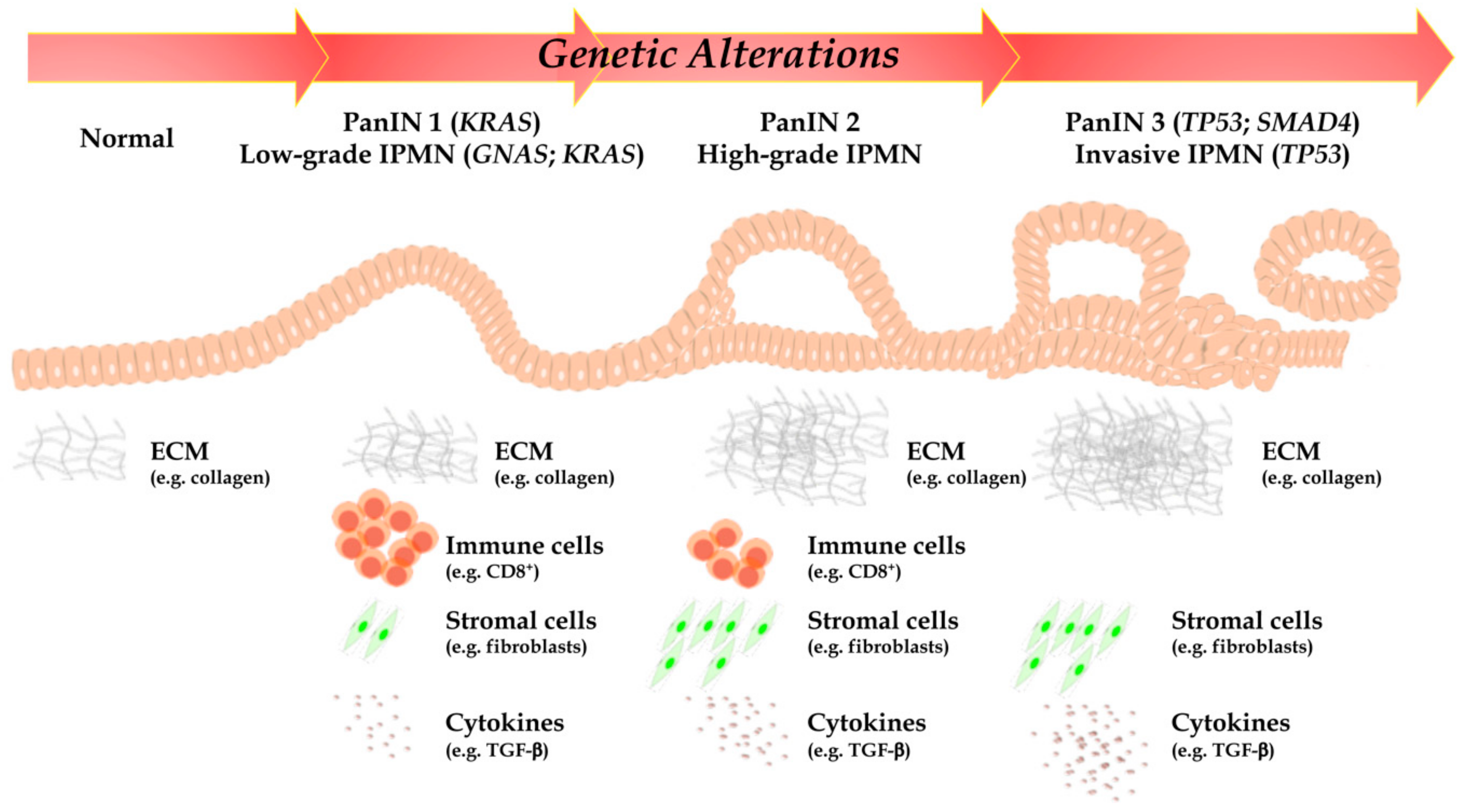
2.1. Precursor Lesions
2.2. Driver Genes Alterations
2.2.1. KRAS
2.2.2. TP53
2.2.3. CDKN2A
2.2.4. SMAD4
2.2.5. Other Mutations
2.3. Dysregulation of Core Signaling Pathways
2.4. Molecularly-Defined PC Subtypes
3. TME
3.1. Cellular Components
3.1.1. Stromal Cells
PSC and CAF
Endothelial Cells
3.1.2. Immune Cells
Macrophages
Neutrophils
Lymphocytes
MDSC
DC
3.2. Acellular Components
3.2.1. ECM
Collagens
Proteoglycans
Fibronectin
3.2.2. Cytokines
IL-1β
IL-6
IL-8
MIF
IL-10
TGF-β
TNF-α
4. Current Treatment Standard of Care and Novel Therapeutic Strategies
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- AIOM. I Numeri del Cancro in Italia 2019, 9th ed.; Intermedia EDITORE: Rodengo Saiano (BS), Italy, 2019; pp. 1–388. [Google Scholar]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Uzunparmak, B.; Sahin, I.H. Pancreatic cancer microenvironment: A current dilemma. Clin. Transl. Med. 2019, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.G.; Iacobuzio-Donahue, C.A. Pancreatic cancer: Pancreatic carcinogenesis - several small steps or one giant leap? Nat. Rev. Gastroenterol. Hepatol. 2016, 14, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, N.E.; Beniata, O.V.; Vitsos, P.; Tsitsilonis, O.; Samara, P. Harnessing the immune system to improve cancer therapy. Ann. Transl. Med. 2016, 4, 261. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019. [Google Scholar] [CrossRef]
- Ciuffreda, L.; Del Curatolo, A.; Falcone, I.; Conciatori, F.; Bazzichetto, C.; Cognetti, F.; Corbo, V.; Scarpa, A.; Milella, M. Lack of growth inhibitory synergism with combined MAPK/PI3K inhibition in preclinical models of pancreatic cancer. Ann. Oncol. 2017, 28, 2896–2898. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Matteoni, S.; Sacconi, A.; De Luca, T.; Bazzichetto, C.; Corbo, V.; Simbolo, M.; Sperduti, I.; et al. PTEN status is a crucial determinant of the functional outcome of combined MEK and mTOR inhibition in cancer. Sci. Rep. 2017, 7, 43013. [Google Scholar] [CrossRef]
- Andea, A.; Sarkar, F.; Adsay, V.N. Clinicopathological correlates of pancreatic intraepithelial neoplasia: A comparative analysis of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma. Mod. Pathol. 2003, 16, 996–1006. [Google Scholar] [CrossRef]
- WHO. Digestive System Tumours, 5th ed.; IARC: Lyon, France, 2019. [Google Scholar]
- Yokode, M.; Akita, M.; Fujikura, K.; Kim, M.J.; Morinaga, Y.; Yoshikawa, S.; Terada, T.; Matsukiyo, H.; Tajiri, T.; Abe-Suzuki, S.; et al. High-grade PanIN presenting with localised stricture of the main pancreatic duct: A clinicopathological and molecular study of 10 cases suggests a clue for the early detection of pancreatic cancer. Histopathology 2018, 73, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Klimstra, D.S.; Longnecker, D.S. K-ras mutations in pancreatic ductal proliferative lesions. Am. J. Pathol. 1994, 145, 1547–1550. [Google Scholar]
- Moskaluk, C.A.; Hruban, R.H.; Kern, S.E. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997, 57, 2140–2143. [Google Scholar] [PubMed]
- Hosoda, W.; Chianchiano, P.; Griffin, J.F.; Pittman, M.E.; Brosens, L.A.; Noe, M.; Yu, J.; Shindo, K.; Suenaga, M.; Rezaee, N.; et al. Genetic analyses of isolated high-grade pancreatic intraepithelial neoplasia (HG-PanIN) reveal paucity of alterations in TP53 and SMAD4. J. Pathol. 2017, 242, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M. Intraductal Papillary Mucinous Neoplasm of the Pancreas as the Main Focus for Early Detection of Pancreatic Adenocarcinoma. Pancreas 2018, 47, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Hatori, T.; Fujita, I.; Yamamoto, M.; Kobayashi, M.; Ohike, N.; Morohoshi, T.; Egawa, S.; Unno, M.; Takao, S.; et al. Prognostic relevance of morphological types of intraductal papillary mucinous neoplasms of the pancreas. Gut 2011, 60, 509–516. [Google Scholar] [CrossRef]
- Distler, M.; Kersting, S.; Niedergethmann, M.; Aust, D.E.; Franz, M.; Ruckert, F.; Ehehalt, F.; Pilarsky, C.; Post, S.; Saeger, H.D.; et al. Pathohistological subtype predicts survival in patients with intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann. Surg. 2013, 258, 324–330. [Google Scholar] [CrossRef]
- Wu, J.; Matthaei, H.; Maitra, A.; Dal Molin, M.; Wood, L.D.; Eshleman, J.R.; Goggins, M.; Canto, M.I.; Schulick, R.D.; Edil, B.H.; et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci. Transl. Med. 2011, 3, 92ra66. [Google Scholar] [CrossRef]
- Tan, M.C.; Basturk, O.; Brannon, A.R.; Bhanot, U.; Scott, S.N.; Bouvier, N.; LaFemina, J.; Jarnagin, W.R.; Berger, M.F.; Klimstra, D.; et al. GNAS and KRAS Mutations Define Separate Progression Pathways in Intraductal Papillary Mucinous Neoplasm-Associated Carcinoma. J. Am. Coll. Surg. 2015, 220, 845–854. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Pea, A.; Cheng, L.; Luchini, C. Dissecting the molecular landscape of pancreatic cancer: Towards a precision medicine approach. Exp. Rev. Prec. Med. Drug Develop. 2019, 4, 113–119. [Google Scholar] [CrossRef]
- Luchini, C.; Veronese, N.; Nottegar, A.; Cappelletti, V.; Daidone, M.G.; Smith, L.; Parris, C.; Brosens, L.A.A.; Caruso, M.G.; Cheng, L.; et al. Liquid Biopsy as Surrogate for Tissue for Molecular Profiling in Pancreatic Cancer: A Meta-Analysis Towards Precision Medicine. Cancers 2019, 11, 1152. [Google Scholar] [CrossRef] [PubMed]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53, 549–554. [Google Scholar] [CrossRef]
- Smit, V.T.; Boot, A.J.; Smits, A.M.; Fleuren, G.J.; Cornelisse, C.J.; Bos, J.L. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988, 16, 7773–7782. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef]
- Lou, K.; Steri, V.; Ge, A.Y.; Hwang, Y.C.; Yogodzinski, C.H.; Shkedi, A.R.; Choi, A.L.M.; Mitchell, D.C.; Swaney, D.L.; Hann, B.; et al. KRAS(G12C) inhibition produces a driver-limited state revealing collateral dependencies. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Riva, G.; Pea, A.; Pilati, C.; Fiadone, G.; Lawlor, R.T.; Scarpa, A.; Luchini, C. Histo-molecular oncogenesis of pancreatic cancer: From precancerous lesions to invasive ductal adenocarcinoma. World J. Gastrointest. Oncol. 2018, 10, 317–327. [Google Scholar] [CrossRef]
- Scarpa, A.; Capelli, P.; Mukai, K.; Zamboni, G.; Oda, T.; Iacono, C.; Hirohashi, S. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am. J. Pathol. 1993, 142, 1534–1543. [Google Scholar]
- Pellegata, N.S.; Sessa, F.; Renault, B.; Bonato, M.; Leone, B.E.; Solcia, E.; Ranzani, G.N. K-ras and p53 gene mutations in pancreatic cancer: Ductal and nonductal tumors progress through different genetic lesions. Cancer Res. 1994, 54, 1556–1560. [Google Scholar] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tsutsumi, S.; Kawaguchi, T.; Nagasaki, K.; Tatsuno, K.; Yamamoto, S.; Sang, F.; Sonoda, K.; Sugawara, M.; Saiura, A.; et al. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res. 2012, 22, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Caldas, C.; Hahn, S.A.; da Costa, L.T.; Redston, M.S.; Schutte, M.; Seymour, A.B.; Weinstein, C.L.; Hruban, R.H.; Yeo, C.J.; Kern, S.E. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat. Genet. 1994, 8, 27–32. [Google Scholar] [CrossRef]
- Ueki, T.; Toyota, M.; Sohn, T.; Yeo, C.J.; Issa, J.P.; Hruban, R.H.; Goggins, M. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000, 60, 1835–1839. [Google Scholar]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef]
- Hustinx, S.R.; Hruban, R.H.; Leoni, L.M.; Iacobuzio-Donahue, C.; Cameron, J.L.; Yeo, C.J.; Brown, P.N.; Argani, P.; Ashfaq, R.; Fukushima, N.; et al. Homozygous deletion of the MTAP gene in invasive adenocarcinoma of the pancreas and in periampullary cancer: A potential new target for therapy. Cancer Biol. Ther. 2005, 4, 83–86. [Google Scholar] [CrossRef]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef]
- Hahn, S.A.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Schutte, M.; Rozenblum, E.; Seymour, A.B.; Weinstein, C.L.; Yeo, C.J.; Hruban, R.H.; et al. Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res. 1996, 56, 490–494. [Google Scholar] [PubMed]
- Iacobuzio-Donahue, C.A.; Song, J.; Parmiagiani, G.; Yeo, C.J.; Hruban, R.H.; Kern, S.E. Missense mutations of MADH4: Characterization of the mutational hot spot and functional consequences in human tumors. Clin. Cancer Res. 2004, 10, 1597–1604. [Google Scholar] [CrossRef]
- Lawlor, R.T.; Veronese, N.; Nottegar, A.; Malleo, G.; Smith, L.; Demurtas, J.; Cheng, L.; Wood, L.D.; Silvestris, N.; Salvia, R.; et al. Prognostic Role of High-Grade Tumor Budding in Pancreatic Ductal Adenocarcinoma: A Systematic Review and Meta-Analysis with a Focus on Epithelial to Mesenchymal Transition. Cancers 2019, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Massague, J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253. [Google Scholar] [CrossRef]
- Goggins, M.; Offerhaus, G.J.; Hilgers, W.; Griffin, C.A.; Shekher, M.; Tang, D.; Sohn, T.A.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am. J. Pathol. 1998, 152, 1501–1507. [Google Scholar]
- Wilentz, R.E.; Goggins, M.; Redston, M.; Marcus, V.A.; Adsay, N.V.; Sohn, T.A.; Kadkol, S.S.; Yeo, C.J.; Choti, M.; Zahurak, M.; et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: A newly described and characterized entity. Am. J. Pathol. 2000, 156, 1641–1651. [Google Scholar] [CrossRef]
- Luchini, C.; Capelli, P.; Scarpa, A. Pancreatic Ductal Adenocarcinoma and Its Variants. Surg. Pathol. Clin. 2016, 9, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.Y.; Andre, F.; et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Amato, E.; Molin, M.D.; Mafficini, A.; Yu, J.; Malleo, G.; Rusev, B.; Fassan, M.; Antonello, D.; Sadakari, Y.; Castelli, P.; et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J. Pathol. 2014, 233, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, W.; Sasaki, E.; Murakami, Y.; Yamao, K.; Shimizu, Y.; Yatabe, Y. GNAS mutation is a frequent event in pancreatic intraductal papillary mucinous neoplasms and associated adenocarcinomas. Virchows Arch. 2015, 466, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Pea, A.; Lionheart, G.; Mafficini, A.; Nottegar, A.; Veronese, N.; Chianchiano, P.; Brosens, L.A.; Noe, M.; Offerhaus, G.J.A.; et al. Pancreatic undifferentiated carcinoma with osteoclast-like giant cells is genetically similar to, but clinically distinct from, conventional ductal adenocarcinoma. J. Pathol. 2017, 243, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Candido, J.B.; Morton, J.P.; Bailey, P.; Campbell, A.D.; Karim, S.A.; Jamieson, T.; Lapienyte, L.; Gopinathan, A.; Clark, W.; McGhee, E.J.; et al. CSF1R(+) Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Rep. 2018, 23, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 2018, 33, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [PubMed]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef]
- Yan, Z.; Ohuchida, K.; Fei, S.; Zheng, B.; Guan, W.; Feng, H.; Kibe, S.; Ando, Y.; Koikawa, K.; Abe, T.; et al. Inhibition of ERK1/2 in cancer-associated pancreatic stellate cells suppresses cancer-stromal interaction and metastasis. J. Exp. Clin. Cancer Res. 2019, 38, 221. [Google Scholar] [CrossRef]
- Amrutkar, M.; Aasrum, M.; Verbeke, C.S.; Gladhaug, I.P. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer 2019, 19, 596. [Google Scholar] [CrossRef]
- Xue, R.; Jia, K.; Wang, J.; Yang, L.; Wang, Y.; Gao, L.; Hao, J. A Rising Star in Pancreatic Diseases: Pancreatic Stellate Cells. Front. Physiol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Liu, W.; Yu, X.; Xu, J. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics 2018, 8, 5072–5087. [Google Scholar] [CrossRef] [PubMed]
- Katsuta, E.; Qi, Q.; Peng, X.; Hochwald, S.N.; Yan, L.; Takabe, K. Pancreatic adenocarcinomas with mature blood vessels have better overall survival. Sci. Rep. 2019, 9, 1310. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Invest. 2013, 93, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Fei, M.; Wu, Y.; Zheng, D.; Wan, D.; Wang, L.; Li, D. Distribution and clinical significance of Th17 cells in the tumor microenvironment and peripheral blood of pancreatic cancer patients. Int. J. Mol. Sci. 2011, 12, 7424–7437. [Google Scholar] [CrossRef]
- Drake, C.G.; Jaffee, E.; Pardoll, D.M. Mechanisms of immune evasion by tumors. Adv. Immunol. 2006, 90, 51–81. [Google Scholar] [CrossRef]
- Markowitz, J.; Brooks, T.R.; Duggan, M.C.; Paul, B.K.; Pan, X.; Wei, L.; Abrams, Z.; Luedke, E.; Lesinski, G.B.; Mundy-Bosse, B.; et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease. Cancer Immunol. Immunother. 2015, 64, 149–159. [Google Scholar] [CrossRef]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef]
- Liu, C.; Yu, S.; Kappes, J.; Wang, J.; Grizzle, W.E.; Zinn, K.R.; Zhang, H.G. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood 2007, 109, 4336–4342. [Google Scholar] [CrossRef]
- Lu, J.; Zhou, S.; Siech, M.; Habisch, H.; Seufferlein, T.; Bachem, M.G. Pancreatic stellate cells promote hapto-migration of cancer cells through collagen I-mediated signalling pathway. Br. J. Cancer 2014, 110, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.; Philippi, C.; Vogelmann, R.; Seidel, B.; Lutz, M.P.; Adler, G.; Wedlich, D. Down-regulation of E-cadherin gene expression by collagen type I and type III in pancreatic cancer cell lines. Cancer Res. 2001, 61, 3508–3517. [Google Scholar] [PubMed]
- Ohlund, D.; Franklin, O.; Lundberg, E.; Lundin, C.; Sund, M. Type IV collagen stimulates pancreatic cancer cell proliferation, migration, and inhibits apoptosis through an autocrine loop. BMC Cancer 2013, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, S.; Grunwald, B.; Kruger, A.; Reithmeier, A.; Hahl, T.; Cheng, T.; Feuchtinger, A.; Born, D.; Erkan, M.; Kleeff, J.; et al. Collagen type V promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Cancer Lett. 2015, 356, 721–732. [Google Scholar] [CrossRef]
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10, 316. [Google Scholar] [CrossRef]
- Yamamoto, T.; Matsuda, Y.; Kawahara, K.; Ishiwata, T.; Naito, Z. Secreted 70kDa lumican stimulates growth and inhibits invasion of human pancreatic cancer. Cancer Lett. 2012, 320, 31–39. [Google Scholar] [CrossRef]
- Javle, M.M.; Gibbs, J.F.; Iwata, K.K.; Pak, Y.; Rutledge, P.; Yu, J.; Black, J.D.; Tan, D.; Khoury, T. Epithelial-mesenchymal transition (EMT) and activated extracellular signal-regulated kinase (p-Erk) in surgically resected pancreatic cancer. Ann. Surg. Oncol. 2007, 14, 3527–3533. [Google Scholar] [CrossRef]
- Schmid, M.C.; Avraamides, C.J.; Foubert, P.; Shaked, Y.; Kang, S.W.; Kerbel, R.S.; Varner, J.A. Combined blockade of integrin-alpha4beta1 plus cytokines SDF-1alpha or IL-1beta potently inhibits tumor inflammation and growth. Cancer Res. 2011, 71, 6965–6975. [Google Scholar] [CrossRef]
- Mitsunaga, S.; Ikeda, M.; Shimizu, S.; Ohno, I.; Furuse, J.; Inagaki, M.; Higashi, S.; Kato, H.; Terao, K.; Ochiai, A. Serum levels of IL-6 and IL-1beta can predict the efficacy of gemcitabine in patients with advanced pancreatic cancer. Br. J. Cancer 2013, 108, 2063–2069. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A.; Yoshida, N.; Takikawa, T.; Shimosegawa, T. IL-6/STAT3 Plays a Regulatory Role in the Interaction Between Pancreatic Stellate Cells and Cancer Cells. Dig. Dis. Sci. 2016, 61, 1561–1571. [Google Scholar] [CrossRef]
- Feurino, L.W.; Zhang, Y.; Bharadwaj, U.; Zhang, R.; Li, F.; Fisher, W.E.; Brunicardi, F.C.; Chen, C.; Yao, Q.; Min, L. IL-6 stimulates Th2 type cytokine secretion and upregulates VEGF and NRP-1 expression in pancreatic cancer cells. Cancer Biol. Ther. 2007, 6, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Bellone, G.; Smirne, C.; Mauri, F.A.; Tonel, E.; Carbone, A.; Buffolino, A.; Dughera, L.; Robecchi, A.; Pirisi, M.; Emanuelli, G. Cytokine expression profile in human pancreatic carcinoma cells and in surgical specimens: Implications for survival. Cancer Immunol. Immunother. 2006, 55, 684–698. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Y.; Feurino, L.W.; Wang, H.; Fisher, W.E.; Brunicardi, F.C.; Chen, C.; Yao, Q. Interleukin-8 increases vascular endothelial growth factor and neuropilin expression and stimulates ERK activation in human pancreatic cancer. Cancer Sci. 2008, 99, 733–737. [Google Scholar] [CrossRef]
- Chen, L.; Fan, J.; Chen, H.; Meng, Z.; Chen, Z.; Wang, P.; Liu, L. The IL-8/CXCR1 axis is associated with cancer stem cell-like properties and correlates with clinical prognosis in human pancreatic cancer cases. Sci. Rep. 2014, 4, 5911. [Google Scholar] [CrossRef]
- Funamizu, N.; Hu, C.; Lacy, C.; Schetter, A.; Zhang, G.; He, P.; Gaedcke, J.; Ghadimi, M.B.; Ried, T.; Yfantis, H.G.; et al. Macrophage migration inhibitory factor induces epithelial to mesenchymal transition, enhances tumor aggressiveness and predicts clinical outcome in resected pancreatic ductal adenocarcinoma. Int. J. Cancer 2013, 132, 785–794. [Google Scholar] [CrossRef]
- Wang, D.; Wang, R.; Huang, A.; Fang, Z.; Wang, K.; He, M.; Xia, J.T.; Li, W. Upregulation of macrophage migration inhibitory factor promotes tumor metastasis and correlates with poor prognosis of pancreatic ductal adenocarcinoma. Oncol. Rep. 2018, 40, 2628–2636. [Google Scholar] [CrossRef]
- Chen, Y.; Ayaru, L.; Mathew, S.; Morris, E.; Pereira, S.P.; Behboudi, S. Expansion of anti-mesothelin specific CD4+ and CD8+ T cell responses in patients with pancreatic carcinoma. PLoS ONE 2014, 9, e88133. [Google Scholar] [CrossRef]
- Dima, S.O.; Tanase, C.; Albulescu, R.; Herlea, V.; Chivu-Economescu, M.; Purnichescu-Purtan, R.; Dumitrascu, T.; Duda, D.G.; Popescu, I. An exploratory study of inflammatory cytokines as prognostic biomarkers in patients with ductal pancreatic adenocarcinoma. Pancreas 2012, 41, 1001–1007. [Google Scholar] [CrossRef]
- Feng, L.; Qi, Q.; Wang, P.; Chen, H.; Chen, Z.; Meng, Z.; Liu, L. Serum levels of IL-6, IL-8, and IL-10 are indicators of prognosis in pancreatic cancer. J. Int. Med. Res. 2018, 46, 5228–5236. [Google Scholar] [CrossRef]
- Maier, H.J.; Wirth, T.; Beug, H. Epithelial-mesenchymal transition in pancreatic carcinoma. Cancers 2010, 2, 2058–2083. [Google Scholar] [CrossRef] [PubMed]
- Pu, N.; Zhao, G.; Gao, S.; Cui, Y.; Xu, Y.; Lv, Y.; Nuerxiati, A.; Wu, W. Neutralizing TGF-beta promotes anti-tumor immunity of dendritic cells against pancreatic cancer by regulating T lymphocytes. Cent. Eur. J. Immunol. 2018, 43, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Li, Y.; Tan, D.; Dong, X.; Chang, P.; Kar, S.; Li, D. Biomarkers of TGF-beta signaling pathway and prognosis of pancreatic cancer. PLoS ONE 2014, 9, e85942. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Bang, J.-H.; Nam, A.-R.; Park, J.E.; Kim, M.H.; Bang, Y.-J.; Oh, D.-Y. The role of soluble TGF-beta and its dynamics for predicting the prognosis in unresectable pancreatic cancer patients treated with chemotherapy. J. Clin. Oncol. 2018, 36, 288. [Google Scholar] [CrossRef]
- Egberts, J.H.; Cloosters, V.; Noack, A.; Schniewind, B.; Thon, L.; Klose, S.; Kettler, B.; von Forstner, C.; Kneitz, C.; Tepel, J.; et al. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008, 68, 1443–1450. [Google Scholar] [CrossRef]
- Helm, O.; Held-Feindt, J.; Grage-Griebenow, E.; Reiling, N.; Ungefroren, H.; Vogel, I.; Kruger, U.; Becker, T.; Ebsen, M.; Rocken, C.; et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int. J. Cancer 2014, 135, 843–861. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Qu, C.; Wang, Q.; Meng, Z.; Wang, P. Cancer-Associated Fibroblasts in Pancreatic Cancer: Should They Be Deleted or Reeducated? Integr. Cancer Ther. 2018, 17, 1016–1019. [Google Scholar] [CrossRef]
- Parente, P.; Parcesepe, P.; Covelli, C.; Olivieri, N.; Remo, A.; Pancione, M.; Latiano, T.P.; Graziano, P.; Maiello, E.; Giordano, G. Crosstalk between the Tumor Microenvironment and Immune System in Pancreatic Ductal Adenocarcinoma: Potential Targets for New Therapeutic Approaches. Gastroenterol. Res. Pract. 2018, 2018, 7530619. [Google Scholar] [CrossRef]
- Bynigeri, R.R.; Jakkampudi, A.; Jangala, R.; Subramanyam, C.; Sasikala, M.; Rao, G.V.; Reddy, D.N.; Talukdar, R. Pancreatic stellate cell: Pandora’s box for pancreatic disease biology. World J. Gastroenterol. 2017, 23, 382–405. [Google Scholar] [CrossRef]
- Wei, L.; Ye, H.; Li, G.; Lu, Y.; Zhou, Q.; Zheng, S.; Lin, Q.; Liu, Y.; Li, Z.; Chen, R. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 2018, 9, 1065. [Google Scholar] [CrossRef]
- McCarroll, J.A.; Naim, S.; Sharbeen, G.; Russia, N.; Lee, J.; Kavallaris, M.; Goldstein, D.; Phillips, P.A. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 2014, 5, 141. [Google Scholar] [CrossRef]
- Liu, S.L.; Cao, S.G.; Li, Y.; Sun, B.; Chen, D.; Wang, D.S.; Zhou, Y.B. Pancreatic stellate cells facilitate pancreatic cancer cell viability and invasion. Oncol. Lett. 2019, 17, 2057–2062. [Google Scholar] [CrossRef]
- Bolm, L.; Cigolla, S.; Wittel, U.A.; Hopt, U.T.; Keck, T.; Rades, D.; Bronsert, P.; Wellner, U.F. The Role of Fibroblasts in Pancreatic Cancer: Extracellular Matrix Versus Paracrine Factors. Transl. Oncol. 2017, 10, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Hirota, M.; Shimosegawa, T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am. J. Physiol. Gastrointest Liver Physiol. 2008, 295, G709–G717. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Deucker, S.; Sauliunaite, D.; Streit, S.; Esposito, I.; Friess, H.; Kleeff, J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 2009, 11, 497–508. [Google Scholar] [CrossRef]
- Craven, K.E.; Gore, J.; Wilson, J.L.; Korc, M. Angiogenic gene signature in human pancreatic cancer correlates with TGF-beta and inflammatory transcriptomes. Oncotarget 2016, 7, 323–341. [Google Scholar] [CrossRef]
- Dineen, S.P.; Lynn, K.D.; Holloway, S.E.; Miller, A.F.; Sullivan, J.P.; Shames, D.S.; Beck, A.W.; Barnett, C.C.; Fleming, J.B.; Brekken, R.A. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008, 68, 4340–4346. [Google Scholar] [CrossRef]
- Barros, M.H.; Hauck, F.; Dreyer, J.H.; Kempkes, B.; Niedobitek, G. Macrophage polarisation: An immunohistochemical approach for identifying M1 and M2 macrophages. PLoS ONE 2013, 8, e80908. [Google Scholar] [CrossRef]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat. Commun. 2015, 6, 7158. [Google Scholar] [CrossRef] [PubMed]
- Looi, C.K.; Chung, F.F.; Leong, C.O.; Wong, S.F.; Rosli, R.; Mai, C.W. Therapeutic challenges and current immunomodulatory strategies in targeting the immunosuppressive pancreatic tumor microenvironment. J. Exp. Clin. Cancer Res. 2019, 38, 162. [Google Scholar] [CrossRef]
- Glazer, E.S.; Reed, L.F.; Freeburg, C.J.; Husain, S.M.; Deneve, J.L.; Dickson, P.V.; Shibata, D. Effect of macrophages on pancreatic cancer. J. Clin. Oncol. 2018, 36, 324. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wei, Q.; Fan, J.; Cheng, S.; Ding, W.; Hua, Z. Prognostic role of the neutrophil-to-lymphocyte ratio in pancreatic cancer: A meta-analysis containing 8252 patients. Clin. Chim. Acta 2018, 479, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A., 3rd; Yarchoan, M.; Lee, V.; Laheru, D.A.; Jaffee, E.M. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin. Cancer Res. 2017, 23, 1656–1669. [Google Scholar] [CrossRef]
- Chang, J.H.; Jiang, Y.; Pillarisetty, V.G. Role of immune cells in pancreatic cancer from bench to clinical application: An updated review. Medicine 2016, 95, e5541. [Google Scholar] [CrossRef]
- Bang, S.; Kim, H.S.; Choo, Y.S.; Park, S.W.; Chung, J.B.; Song, S.Y. Differences in immune cells engaged in cell-mediated immunity after chemotherapy for far advanced pancreatic cancer. Pancreas 2006, 32, 29–36. [Google Scholar] [CrossRef]
- Fukunaga, A.; Miyamoto, M.; Cho, Y.; Murakami, S.; Kawarada, Y.; Oshikiri, T.; Kato, K.; Kurokawa, T.; Suzuoki, M.; Nakakubo, Y.; et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas 2004, 28, E26–E31. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- De Monte, L.; Reni, M.; Tassi, E.; Clavenna, D.; Papa, I.; Recalde, H.; Braga, M.; Di Carlo, V.; Doglioni, C.; Protti, M.P. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J. Exp. Med. 2011, 208, 469–478. [Google Scholar] [CrossRef]
- Tassi, E.; Braga, M.; Longhi, R.; Gavazzi, F.; Parmiani, G.; Di Carlo, V.; Protti, M.P. Non-redundant role for IL-12 and IL-27 in modulating Th2 polarization of carcinoembryonic antigen specific CD4 T cells from pancreatic cancer patients. PLoS ONE 2009, 4, e7234. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Mignot, G.; Bruchard, M.; Chevriaux, A.; Vegran, F.; Hichami, A.; Ladoire, S.; Derangere, V.; Vincent, J.; Masson, D.; et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 2012, 36, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Xu, X.; Guo, S.; Zhang, C.; Tang, Y.; Tian, Y.; Ni, B.; Lu, B.; Wang, H. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS ONE 2014, 9, e91551. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yanagimoto, H.; Satoi, S.; Toyokawa, H.; Hirooka, S.; Yamaki, S.; Yui, R.; Yamao, J.; Kim, S.; Kwon, A.H. Circulating CD4+CD25+ regulatory T cells in patients with pancreatic cancer. Pancreas 2012, 41, 409–415. [Google Scholar] [CrossRef]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.; et al. Regulatory T cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Wormann, S.M.; Diakopoulos, K.N.; Lesina, M.; Algul, H. The immune network in pancreatic cancer development and progression. Oncogene 2014, 33, 2956–2967. [Google Scholar] [CrossRef]
- Hou, Y.C.; Chao, Y.J.; Hsieh, M.H.; Tung, H.L.; Wang, H.C.; Shan, Y.S. Low CD8(+) T Cell Infiltration and High PD-L1 Expression Are Associated with Level of CD44(+)/CD133(+) Cancer Stem Cells and Predict an Unfavorable Prognosis in Pancreatic Cancer. Cancers 2019, 11, 541. [Google Scholar] [CrossRef]
- Ademmer, K.; Ebert, M.; Muller-Ostermeyer, F.; Friess, H.; Buchler, M.W.; Schubert, W.; Malfertheiner, P. Effector T lymphocyte subsets in human pancreatic cancer: Detection of CD8+CD18+ cells and CD8+CD103+ cells by multi-epitope imaging. Clin. Exp. Immunol. 1998, 112, 21–26. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Pergamo, M.; Miller, G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2017, 24, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G.W. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. 2011, 60, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Song, X.; Yu, H.; Zhang, M.; Li, F.; Cao, C.; Jiang, Q. Dendritic cell-based cancer immunotherapy for pancreatic cancer. Arab. J. Gastroenterol. 2018, 19, 1–6. [Google Scholar] [CrossRef]
- Dallal, R.M.; Christakos, P.; Lee, K.; Egawa, S.; Son, Y.I.; Lotze, M.T. Paucity of dendritic cells in pancreatic cancer. Surgery 2002, 131, 135–138. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yanagimoto, H.; Satoi, S.; Toyokawa, H.; Yamao, J.; Kim, S.; Terakawa, N.; Takahashi, K.; Kwon, A.H. Circulating myeloid dendritic cells as prognostic factors in patients with pancreatic cancer who have undergone surgical resection. J. Surg. Res. 2012, 173, 299–308. [Google Scholar] [CrossRef]
- Hirooka, S.; Yanagimoto, H.; Satoi, S.; Yamamoto, T.; Toyokawa, H.; Yamaki, S.; Yui, R.; Inoue, K.; Michiura, T.; Kwon, A.H. The role of circulating dendritic cells in patients with unresectable pancreatic cancer. Anticancer Res. 2011, 31, 3827–3834. [Google Scholar]
- Bauer, C.; Bauernfeind, F.; Sterzik, A.; Orban, M.; Schnurr, M.; Lehr, H.A.; Endres, S.; Eigler, A.; Dauer, M. Dendritic cell-based vaccination combined with gemcitabine increases survival in a murine pancreatic carcinoma model. Gut 2007, 56, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Mayanagi, S.; Kitago, M.; Sakurai, T.; Matsuda, T.; Fujita, T.; Higuchi, H.; Taguchi, J.; Takeuchi, H.; Itano, O.; Aiura, K.; et al. Phase I pilot study of Wilms tumor gene 1 peptide-pulsed dendritic cell vaccination combined with gemcitabine in pancreatic cancer. Cancer Sci. 2015, 106, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, R.; Koizumi, T.; Koya, T.; Sano, K.; Koido, S.; Nagai, K.; Kobayashi, M.; Okamoto, M.; Sugiyama, H.; Shimodaira, S. WT1-pulsed Dendritic Cell Vaccine Combined with Chemotherapy for Resected Pancreatic Cancer in a Phase I Study. Anticancer Res. 2018, 38, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, T.E.; Fosang, A.J. Proteoglycans: Many forms and many functions. FASEB J. 1992, 6, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.A.; McCarroll, J.A.; Park, S.; Wu, M.J.; Pirola, R.; Korsten, M.; Wilson, J.S.; Apte, M.V. Rat pancreatic stellate cells secrete matrix metalloproteinases: Implications for extracellular matrix turnover. Gut 2003, 52, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: A starring role in normal and diseased pancreas. Front. Physiol. 2012, 3, 344. [Google Scholar] [CrossRef]
- Mekapogu, A.R.; Pothula, S.P.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Multifunctional role of pancreatic stellate cells in pancreatic cancer. Ann. Pancreat. Cancer 2019, 2, 10. [Google Scholar] [CrossRef]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef]
- Linder, S.; Castanos-Velez, E.; von Rosen, A.; Biberfeld, P. Immunohistochemical expression of extracellular matrix proteins and adhesion molecules in pancreatic carcinoma. Hepatogastroenterology 2001, 48, 1321–1327. [Google Scholar]
- Yang, J.C.; Zhang, Y.; He, S.J.; Li, M.M.; Cai, X.L.; Wang, H.; Xu, L.M.; Cao, J. TM4SF1 Promotes Metastasis of Pancreatic Cancer via Regulating the Expression of DDR1. Sci. Rep. 2017, 7, 45895. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yang, J.C.; Ramachandran, V.; Arumugam, T.; Deng, D.F.; Li, Z.S.; Xu, L.M.; Logsdon, C.D. TM4SF1 Regulates Pancreatic Cancer Migration and Invasion In Vitro and In Vivo. Cell Physiol. Biochem. 2016, 39, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Clementz, A.G.; Mutolo, M.J.; Leir, S.H.; Morris, K.J.; Kucybala, K.; Harris, H.; Harris, A. Collagen XV inhibits epithelial to mesenchymal transition in pancreatic adenocarcinoma cells. PLoS ONE 2013, 8, e72250. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Pan, S.; Brentnall, T.A.; Chen, R. Glycoproteins and glycoproteomics in pancreatic cancer. World J. Gastroenterol. 2016, 22, 9288–9299. [Google Scholar] [CrossRef]
- Yang, V.W.; LaBrenz, S.R.; Rosenberg, L.C.; McQuillan, D.; Hook, M. Decorin is a Zn2+ metalloprotein. J. Biol. Chem. 1999, 274, 12454–12460. [Google Scholar] [CrossRef]
- Goldoni, S.; Owens, R.T.; McQuillan, D.J.; Shriver, Z.; Sasisekharan, R.; Birk, D.E.; Campbell, S.; Iozzo, R.V. Biologically active decorin is a monomer in solution. J. Biol. Chem. 2004, 279, 6606–6612. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Mann, D.M.; Ruoslahti, E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature 1990, 346, 281–284. [Google Scholar] [CrossRef]
- Santra, M.; Eichstetter, I.; Iozzo, R.V. An anti-oncogenic role for decorin. Down-regulation of ErbB2 leads to growth suppression and cytodifferentiation of mammary carcinoma cells. J. Biol. Chem. 2000, 275, 35153–35161. [Google Scholar] [CrossRef]
- Csordas, G.; Santra, M.; Reed, C.C.; Eichstetter, I.; McQuillan, D.J.; Gross, D.; Nugent, M.A.; Hajnoczky, G.; Iozzo, R.V. Sustained down-regulation of the epidermal growth factor receptor by decorin. A mechanism for controlling tumor growth in vivo. J. Biol. Chem. 2000, 275, 32879–32887. [Google Scholar] [CrossRef]
- Mauviel, A.; Santra, M.; Chen, Y.Q.; Uitto, J.; Iozzo, R.V. Transcriptional regulation of decorin gene expression. Induction by quiescence and repression by tumor necrosis factor-alpha. J. Biol. Chem. 1995, 270, 11692–11700. [Google Scholar] [CrossRef] [PubMed]
- Koninger, J.; Giese, N.A.; di Mola, F.F.; Berberat, P.; Giese, T.; Esposito, I.; Bachem, M.G.; Buchler, M.W.; Friess, H. Overexpressed decorin in pancreatic cancer: Potential tumor growth inhibition and attenuation of chemotherapeutic action. Clin. Cancer Res. 2004, 10, 4776–4783. [Google Scholar] [CrossRef] [PubMed]
- Kolb, M.; Margetts, P.J.; Sime, P.J.; Gauldie, J. Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L1327–L1334. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Truty, M.A.; Kang, Y.; Chopin-Laly, X.; Zhang, R.; Roife, D.; Chatterjee, D.; Lin, E.; Thomas, R.M.; Wang, H.; et al. Extracellular lumican inhibits pancreatic cancer cell growth and is associated with prolonged survival after surgery. Clin. Cancer Res. 2014, 20, 6529–6540. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Ansari, D.; Zhou, Q.; Sasor, A.; Said Hilmersson, K.; Andersson, R. Stromal fibronectin expression in patients with resected pancreatic ductal adenocarcinoma. World J. Surg. Oncol. 2019, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Topalovski, M.; Brekken, R.A. Matrix control of pancreatic cancer: New insights into fibronectin signaling. Cancer Lett. 2016, 381, 252–258. [Google Scholar] [CrossRef]
- Durymanov, M.; Kroll, C.; Permyakova, A.; O’Neill, E.; Sulaiman, R.; Person, M.; Reineke, J. Subcutaneous Inoculation of 3D Pancreatic Cancer Spheroids Results in Development of Reproducible Stroma-Rich Tumors. Transl. Oncol. 2019, 12, 180–189. [Google Scholar] [CrossRef]
- Cortes, E.; Lachowski, D.; Robinson, B.; Sarper, M.; Teppo, J.S.; Thorpe, S.D.; Lieberthal, T.J.; Iwamoto, K.; Lee, D.A.; Okada-Hatakeyama, M.; et al. Tamoxifen mechanically reprograms the tumor microenvironment via HIF-1A and reduces cancer cell survival. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Jagadeeshan, S.; Krishnamoorthy, Y.R.; Singhal, M.; Subramanian, A.; Mavuluri, J.; Lakshmi, A.; Roshini, A.; Baskar, G.; Ravi, M.; Joseph, L.D.; et al. Transcriptional regulation of fibronectin by p21-activated kinase-1 modulates pancreatic tumorigenesis. Oncogene 2015, 34, 455–464. [Google Scholar] [CrossRef]
- Ma, Y.P.; Peng, L.; Luo, X.; Xu, J.; Peng, Y.; Tang, X. Identification of potential hub genes associated with the pathogenesis and prognosis of pancreatic duct adenocarcinoma using bioinformatics meta-analysis of multi-platform datasets. Oncol. Lett. 2019, 18, 6741–6751. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Bazzichetto, C.; Conciatori, F.; Falcone, I.; Cognetti, F.; Milella, M.; Ciuffreda, L. Advances in Tumor-Stroma Interactions: Emerging Role of Cytokine Network in Colorectal and Pancreatic Cancer. J. Oncol. 2019, 2019, 5373580. [Google Scholar] [CrossRef] [PubMed]
- Roshani, R.; McCarthy, F.; Hagemann, T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 2014, 345, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Farajzadeh Valilou, S.; Keshavarz-Fathi, M.; Silvestris, N.; Argentiero, A.; Rezaei, N. The role of inflammatory cytokines and tumor associated macrophages (TAMs) in microenvironment of pancreatic cancer. Cytokine Growth Factor Rev. 2018, 39, 46–61. [Google Scholar] [CrossRef]
- Yako, Y.Y.; Kruger, D.; Smith, M.; Brand, M. Cytokines as Biomarkers of Pancreatic Ductal Adenocarcinoma: A Systematic Review. PLoS ONE 2016, 11, e0154016. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.J.; Houston, A.; Brint, E. IL-1 Family Members in Cancer; Two Sides to Every Story. Front. Immunol. 2019, 10, 1197. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Vorndamm, J.; Muerkoster, S.; Yu, H.; Schmidt, W.E.; Folsch, U.R.; Schafer, H. Autocrine production of interleukin 1beta confers constitutive nuclear factor kappaB activity and chemoresistance in pancreatic carcinoma cell lines. Cancer Res. 2002, 62, 910–916. [Google Scholar]
- Verma, G.; Bhatia, H.; Datta, M. Gene expression profiling and pathway analysis identify the integrin signaling pathway to be altered by IL-1beta in human pancreatic cancer cells: Role of JNK. Cancer Lett. 2012, 320, 86–95. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Muller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Kloppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 Axis in Cancer and Inflammatory Diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Ochi, N.; Sawai, H.; Yasuda, A.; Takahashi, H.; Funahashi, H.; Takeyama, H.; Tong, Z.; Guha, S. CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer 2009, 124, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Kuwada, Y.; Sasaki, T.; Morinaka, K.; Kitadai, Y.; Mukaida, N.; Chayama, K. Potential involvement of IL-8 and its receptors in the invasiveness of pancreatic cancer cells. Int. J. Oncol. 2003, 4, 765–771. [Google Scholar] [CrossRef]
- Chen, Y.; Shi, M.; Yu, G.Z.; Qin, X.R.; Jin, G.; Chen, P.; Zhu, M.H. Interleukin-8, a promising predictor for prognosis of pancreatic cancer. World J. Gastroenterol. 2012, 18, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Calandra, T.; Bernhagen, J.; Mitchell, R.A.; Bucala, R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J. Exp. Med. 1994, 179, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Jankauskas, S.S.; Wong, D.W.L.; Bucala, R.; Djudjaj, S.; Boor, P. Evolving complexity of MIF signaling. Cell Signal. 2019, 57, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Denz, A.; Pilarsky, C.; Muth, D.; Ruckert, F.; Saeger, H.D.; Grutzmann, R. Inhibition of MIF leads to cell cycle arrest and apoptosis in pancreatic cancer cells. J. Surg. Res. 2010, 160, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.R. The molecular basis of IL-10 function: From receptor structure to the onset of signaling. Curr. Top. Microbiol. Immunol. 2014, 380, 191–212. [Google Scholar] [CrossRef]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef]
- Batchu, R.B.; Gruzdyn, O.V.; Mahmud, E.M.; Chukr, F.; Dachepalli, R.; Manmari, S.K.; Mostafa, G.; Weaver, D.W.; Gruber, S.A. Inhibition of Interleukin-10 in the tumor microenvironment can restore mesothelin chimeric antigen receptor T cell activity in pancreatic cancer in vitro. Surgery 2018, 163, 627–632. [Google Scholar] [CrossRef]
- Shen, W.; Tao, G.Q.; Zhang, Y.; Cai, B.; Sun, J.; Tian, Z.Q. TGF-beta in pancreatic cancer initiation and progression: Two sides of the same coin. Cell Biosci. 2017, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFbeta in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Bradshaw, A.D.; Gera, S.; Dewan, M.Z.; Xu, R. The TGF-beta/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med. 2017, 6, 5. [Google Scholar] [CrossRef]
- Ellenrieder, V.; Hendler, S.F.; Boeck, W.; Seufferlein, T.; Menke, A.; Ruhland, C.; Adler, G.; Gress, T.M. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. 2001, 61, 4222–4228. [Google Scholar]
- Zhao, X.; Fan, W.; Xu, Z.; Chen, H.; He, Y.; Yang, G.; Yang, G.; Hu, H.; Tang, S.; Wang, P.; et al. Inhibiting tumor necrosis factor-alpha diminishes desmoplasia and inflammation to overcome chemoresistance in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 81110–81122. [Google Scholar] [CrossRef]
- Chopra, M.; Lang, I.; Salzmann, S.; Pachel, C.; Kraus, S.; Bauerlein, C.A.; Brede, C.; Garrote, A.L.; Mattenheimer, K.; Ritz, M.; et al. Tumor necrosis factor induces tumor promoting and anti-tumoral effects on pancreatic cancer via TNFR1. PLoS ONE 2013, 8, e75737. [Google Scholar] [CrossRef]
- Shadhu, K.; Xi, C. Inflammation and pancreatic cancer: An updated review. Saudi. J. Gastroenterol. 2019, 25, 3–13. [Google Scholar] [CrossRef]
- Ham, B.; Fernandez, M.C.; D’Costa, Z.; Brodt, P. The diverse roles of the TNF axis in cancer progression and metastasis. Trends Cancer Res. 2016, 11, 1–27. [Google Scholar]
- Talar-Wojnarowska, R.; Gasiorowska, A.; Smolarz, B.; Romanowicz-Makowska, H.; Kulig, A.; Malecka-Panas, E. Tumor necrosis factor alpha and interferon gamma genes polymorphisms and serum levels in pancreatic adenocarcinoma. Neoplasma 2009, 56, 56–62. [Google Scholar] [CrossRef]
- Murugesan, S.R.; King, C.R.; Osborn, R.; Fairweather, W.R.; O’Reilly, E.M.; Thornton, M.O.; Wei, L.L. Combination of human tumor necrosis factor-alpha (hTNF-alpha) gene delivery with gemcitabine is effective in models of pancreatic cancer. Cancer Gene Ther. 2009, 16, 841–847. [Google Scholar] [CrossRef]
- Furukawa, K.; Ohashi, T.; Haruki, K.; Fujiwara, Y.; Iida, T.; Shiba, H.; Uwagawa, T.; Kobayashi, H.; Yanaga, K. Combination treatment using adenovirus vector-mediated tumor necrosis factor-alpha gene transfer and a NF-kappaB inhibitor for pancreatic cancer in mice. Cancer Lett. 2011, 306, 92–98. [Google Scholar] [CrossRef]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef]
- O’Neil, B.H.; Scott, A.J.; Ma, W.W.; Cohen, S.J.; Leichman, L.; Aisner, D.L.; Menter, A.R.; Tejani, M.A.; Cho, J.K.; Granfortuna, J.; et al. A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. Ann. Oncol. 2015, 26, 2505. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Kindler, H.L.; Niedzwiecki, D.; Hollis, D.; Sutherland, S.; Schrag, D.; Hurwitz, H.; Innocenti, F.; Mulcahy, M.F.; O’Reilly, E.; Wozniak, T.F.; et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: Phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J. Clin. Oncol. 2010, 28, 3617–3622. [Google Scholar] [CrossRef] [PubMed]
- Reni, M.; Cereda, S.; Milella, M.; Novarino, A.; Passardi, A.; Mambrini, A.; Di Lucca, G.; Aprile, G.; Belli, C.; Danova, M.; et al. Maintenance sunitinib or observation in metastatic pancreatic adenocarcinoma: A phase II randomised trial. Eur. J. Cancer 2013, 49, 3609–3615. [Google Scholar] [CrossRef]
- Sehdev, A.; Gbolahan, O.; Hancock, B.A.; Stanley, M.; Shahda, S.; Wan, J.; Wu, H.H.; Radovich, M.; O’Neil, B.H. Germline and Somatic DNA Damage Repair Gene Mutations and Overall Survival in Metastatic Pancreatic Adenocarcinoma Patients Treated with FOLFIRINOX. Clin. Cancer Res. 2018, 24, 6204–6211. [Google Scholar] [CrossRef]
- Fogelman, D.R.; Wolff, R.A.; Kopetz, S.; Javle, M.; Bradley, C.; Mok, I.; Cabanillas, F.; Abbruzzese, J.L. Evidence for the efficacy of Iniparib, a PARP-1 inhibitor, in BRCA2-associated pancreatic cancer. Anticancer Res. 2011, 31, 1417–1420. [Google Scholar]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Lu, M.; Lin, S.C.; Huang, Y.; Kang, Y.J.; Rich, R.; Lo, Y.C.; Myszka, D.; Han, J.; Wu, H. XIAP induces NF-kappaB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol. Cell 2007, 26, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Carbone, C.; Piro, G.; Chiao, P.J.; Melisi, D. TAK-ing aim at chemoresistance: The emerging role of MAP3K7 as a target for cancer therapy. Drug Resist. Updat. 2017, 33–35, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Melisi, D. NF-kappaB as a target for pancreatic cancer therapy. Expert Opin. Ther. Targets 2012, 16 (Suppl. 2), S1–S10. [Google Scholar] [CrossRef]
- Melisi, D.; Chiao, P.J. NF-kappa B as a target for cancer therapy. Expert Opin. Ther. Targets 2007, 11, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Xia, Q.; Paradiso, G.; Ling, J.; Moccia, T.; Carbone, C.; Budillon, A.; Abbruzzese, J.L.; Chiao, P.J. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J. Natl. Cancer Inst. 2011, 103, 1190–1204. [Google Scholar] [CrossRef]
- Santoro, R.; Zanotto, M.; Simionato, F.; Zecchetto, C.; Merz, V.; Cavallini, C.; Piro, G.; Sabbadini, F.; Boschi, F.; Scarpa, A.; et al. Modulating TAK1 expression inhibits YAP and TAZ oncogenic functions in pancreatic cancer. Mol. Cancer Ther. 2019. [Google Scholar] [CrossRef]
- Schlingensiepen, K.H.; Jaschinski, F.; Lang, S.A.; Moser, C.; Geissler, E.K.; Schlitt, H.J.; Kielmanowicz, M.; Schneider, A. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011, 102, 1193–1200. [Google Scholar] [CrossRef]
- Rowland-Goldsmith, M.A.; Maruyama, H.; Kusama, T.; Ralli, S.; Korc, M. Soluble type II transforming growth factor-beta (TGF-beta) receptor inhibits TGF-beta signaling in COLO-357 pancreatic cancer cells in vitro and attenuates tumor formation. Clin. Cancer Res. 2001, 7, 2931–2940. [Google Scholar]
- Rowland-Goldsmith, M.A.; Maruyama, H.; Matsuda, K.; Idezawa, T.; Ralli, M.; Ralli, S.; Korc, M. Soluble type II transforming growth factor-beta receptor attenuates expression of metastasis-associated genes and suppresses pancreatic cancer cell metastasis. Mol. Cancer Ther. 2002, 1, 161–167. [Google Scholar] [PubMed]
- Subramanian, G.; Schwarz, R.E.; Higgins, L.; McEnroe, G.; Chakravarty, S.; Dugar, S.; Reiss, M. Targeting endogenous transforming growth factor beta receptor signaling in SMAD4-deficient human pancreatic carcinoma cells inhibits their invasive phenotype1. Cancer Res. 2004, 64, 5200–5211. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, N.J.; Li, L.; Kapoun, A.M.; Medicherla, S.; Reddy, M.; Li, G.; O’Young, G.; Quon, D.; Henson, M.; Damm, D.L.; et al. Inhibition of transforming growth factor beta signaling reduces pancreatic adenocarcinoma growth and invasiveness. Mol. Pharmacol. 2007, 72, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-G.; Sergeeva, O.; Luo, G.; Choi, S.H.; Lee, Z.; Kim, S.-J.; Letterio, J.; Malek, E. Abstract 2647: TGF-β type I receptor inhibitor (TEW-7197) diminishes myeloma progression by multiple immunomodulatory mechanisms in combination with ixazomib. Cancer Res. 2017, 77, 2647. [Google Scholar] [CrossRef]
- Kovacs, R.J.; Maldonado, G.; Azaro, A.; Fernandez, M.S.; Romero, F.L.; Sepulveda-Sanchez, J.M.; Corretti, M.; Carducci, M.; Dolan, M.; Gueorguieva, I.; et al. Cardiac Safety of TGF-beta Receptor I Kinase Inhibitor LY2157299 Monohydrate in Cancer Patients in a First-in-Human Dose Study. Cardiovasc. Toxicol. 2015, 15, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Kozloff, M.; Simionato, F.; Cleverly, A.; et al. TGFbeta receptor inhibitor galunisertib is linked to inflammation- and remodeling-related proteins in patients with pancreatic cancer. Cancer Chemother. Pharmacol. 2019, 83, 975–991. [Google Scholar] [CrossRef]
- Piro, G.; Simionato, F.; Carbone, C.; Frizziero, M.; Malleo, G.; Zanini, S.; Casolino, R.; Santoro, R.; Mina, M.M.; Zecchetto, C.; et al. A circulating TH2 cytokines profile predicts survival in patients with resectable pancreatic adenocarcinoma. Oncoimmunology 2017, 6, e1322242. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.J.; Fisher, G.A.; Hezel, A.F.; Chang, S.C.; Vlahovic, G.; et al. A randomized phase 2 study of durvalumab monotherapy and in combination with tremelimumab in patients with metastatic pancreatic ductal adenocarcinoma (mPDAC): ALPS study. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef]
- Principe, D.R.; DeCant, B.; Mascarinas, E.; Wayne, E.A.; Diaz, A.M.; Akagi, N.; Hwang, R.; Pasche, B.; Dawson, D.W.; Fang, D.; et al. TGFbeta Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res. 2016, 76, 2525–2539. [Google Scholar] [CrossRef]
- Melisi, D.; Hollebecque, A.; Oh, D.-Y.; Calvo, E.; Varghese, A.M.; Borazanci, E.H.; Mercade, T.M.; Simionato, F.; Park, J.O.; Bendell, J.C.; et al. A phase Ib dose-escalation and cohort-expansion study of safety and activity of the transforming growth factor (TGF) β receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Clin. Oncol. 2019, 37, 4124. [Google Scholar] [CrossRef]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef]
- Song, M.; Chen, D.; Lu, B.; Wang, C.; Zhang, J.; Huang, L.; Wang, X.; Timmons, C.L.; Hu, J.; Liu, B.; et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PLoS ONE 2013, 8, e65821. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, C.; Liu, F.; Zhao, Y.; Liu, J.; Hua, Y.; Liu, J.; Huang, J.; Ge, C. A blockade of PD-L1 produced antitumor and antimetastatic effects in an orthotopic mouse pancreatic cancer model via the PI3K/Akt/mTOR signaling pathway. Onco Targets Ther. 2017, 10, 2115–2126. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Prakash, J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer 2019, 5, 128–142. [Google Scholar] [CrossRef]
- Tang, D.; Wang, D.; Yuan, Z.; Xue, X.; Zhang, Y.; An, Y.; Chen, J.; Tu, M.; Lu, Z.; Wei, J.; et al. Persistent activation of pancreatic stellate cells creates a microenvironment favorable for the malignant behavior of pancreatic ductal adenocarcinoma. Int. J. Cancer 2013, 132, 993–1003. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Name of the Pathway | Most Important Mutated Gene (s) | |
|---|---|---|
| Oncogenes | Tumor Suppressor | |
| MAPK signaling | KRAS | |
| DNA damage control | TP53, BRCA1, BRCA2 | |
| Control of G1/S Phase transition | CDKN2A | |
| TGF-β signaling | TGF-βRII | SMAD4 |
| Apoptosis | CASP10, VCP | |
| Hedgehog signaling | SOX3, GLI1, GLI3 | |
| Homophilic cell adhesion | CDH1, FAT | |
| Integrin signaling | ITGA4, ITGA9, LAMA1, LAMA4, LAMA5 | |
| JNK signaling | MAP4K3, TNF | |
| SWI/SNF | ARID1A | |
| Small GTPase signaling | RP1 | |
| WNT/Notch signaling | MYC | TSC2 |
| Axon guidance | SLIT2, ROBO2 | |
| Study Drug | Sponsor | Treatment Setting | Combination Partner | Study Phase | Clinical Trials.gov ID |
|---|---|---|---|---|---|
| ERK1/2 Inhibitors | |||||
| Ulixertinib | University of Washington School of Medicine | First Line | Nab-paclitaxel plus gemcitabine | I | NCT02608229 |
| UNC Lineberger Comprehensive Cancer Center | Second Line | Palbociclib | I | NCT03454035 | |
| KO-947 | Kura Oncology | Second Line | - | I | NCT03051035 |
| CDK 4/6 Inhibitors | |||||
| PD-0332991 | Dana Farber Institute | Any | Gedatolisib | I | NCT03065062 |
| Palbociclib | Pfizer | Any | Nab-paclitaxel | I | NCT02501902 |
| KRASG12C Inhibitors | |||||
| AMG 510 | Amgen | Any | - | I | NCT03600883 |
| ALK Inhibitors | |||||
| Ceritinib | Roswell Park Cancer Institute | Any | Gemcitabine ± nab-paclitaxel or cisplatin | I | NCT02227940 |
| RTK Inhibitors | |||||
| Entrectinib | Hoffmann-La Roche | Any | - | II | NCT02568267 |
| Larotrectinib | Loxo Oncology | Second Line | - | II | NCT02576431 |
| TME Components | Biological Mechanisms | Implication in PC | Ref. | ||
|---|---|---|---|---|---|
| Cellular | Stromal cells | aPSC | ↑tumor proliferation, ECM production, EMT, proangiogenic soluble factors secretion; ↓CD8+ T cells migration | ↑chemoresistance; ↑antitumor immune response | [67,68,69,70] |
| CAF | ↑tumor proliferation, production of proangiogenic soluble factors, M2 polarization | ↑chemoresistance; ↓OS | [71,72,73] | ||
| Endothelial | ↑antitumor immune cells | ↑OS | [74] | ||
| Immune cells | M2 | ↑Snail, vimentin and EMT; ↓E-cadherin | ↓patient prognosis | [75,76] | |
| Neutrophils | ↓immunoresponse | ↑chemoresistance | [77] | ||
| CD4+/CD25+/Foxp3+ Treg | ↑immunosuppressive TME; ↓antitumor immune cells | ↑TNM stage | [78,79] | ||
| MDSC | ↑immunosuppressive TME | ↑tumor growth invasion and angiogenesis | [80,81,82] | ||
| Acellular | ECM | Collagen I, IV, V | ↑tumor proliferation and EMT | ↓patient prognosis | [83,84,85,86,87] |
| Lumican | ↑tumor proliferation | ↓patient prognosis | [88] | ||
| Fibronectin | ↑EMT | ↓patient prognosis and OS; ↑chemoresistance | [69,89] | ||
| Cytokines | IL-1β | ↑inflammation, migration | ↑chemoresistance; ↓PFS | [90,91] | |
| IL-6 | ↑tumor proliferation, angiogenesis, EMT and immunosuppressive TME | ↓patient prognosis | [92,93,94] | ||
| IL-8 | ↑tumor proliferation, angiogenesis, CSC properties, ECM disruption and migration | ↓patient prognosis | [95,96,97] | ||
| MIF | ↑cell proliferation and EMT | ↑chemoresistance; ↓OS | [98,99] | ||
| IL-10 | ↓CD4+ T-cell response | ↓OS | [100,101,102] | ||
| TGF-β | ↑EMT; ↓CD8+ T-cell apoptosis | ↓OS | [103,104,105,106] | ||
| TNF-α | ↑tumor proliferation, EMT and migration | ↓patient prognosis | [107,108] | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazzichetto, C.; Conciatori, F.; Luchini, C.; Simionato, F.; Santoro, R.; Vaccaro, V.; Corbo, V.; Falcone, I.; Ferretti, G.; Cognetti, F.; et al. From Genetic Alterations to Tumor Microenvironment: The Ariadne’s String in Pancreatic Cancer. Cells 2020, 9, 309. https://doi.org/10.3390/cells9020309
Bazzichetto C, Conciatori F, Luchini C, Simionato F, Santoro R, Vaccaro V, Corbo V, Falcone I, Ferretti G, Cognetti F, et al. From Genetic Alterations to Tumor Microenvironment: The Ariadne’s String in Pancreatic Cancer. Cells. 2020; 9(2):309. https://doi.org/10.3390/cells9020309
Chicago/Turabian StyleBazzichetto, Chiara, Fabiana Conciatori, Claudio Luchini, Francesca Simionato, Raffaela Santoro, Vanja Vaccaro, Vincenzo Corbo, Italia Falcone, Gianluigi Ferretti, Francesco Cognetti, and et al. 2020. "From Genetic Alterations to Tumor Microenvironment: The Ariadne’s String in Pancreatic Cancer" Cells 9, no. 2: 309. https://doi.org/10.3390/cells9020309
APA StyleBazzichetto, C., Conciatori, F., Luchini, C., Simionato, F., Santoro, R., Vaccaro, V., Corbo, V., Falcone, I., Ferretti, G., Cognetti, F., Melisi, D., Scarpa, A., Ciuffreda, L., & Milella, M. (2020). From Genetic Alterations to Tumor Microenvironment: The Ariadne’s String in Pancreatic Cancer. Cells, 9(2), 309. https://doi.org/10.3390/cells9020309