Hormone Receptor Loss in Breast Cancer: Molecular Mechanisms, Clinical Settings, and Therapeutic Implications


 , , ,
, , ,
Abstract
1. Introduction
2. Molecular Mechanisms Underlying Loss of HR Expression in BC Cells
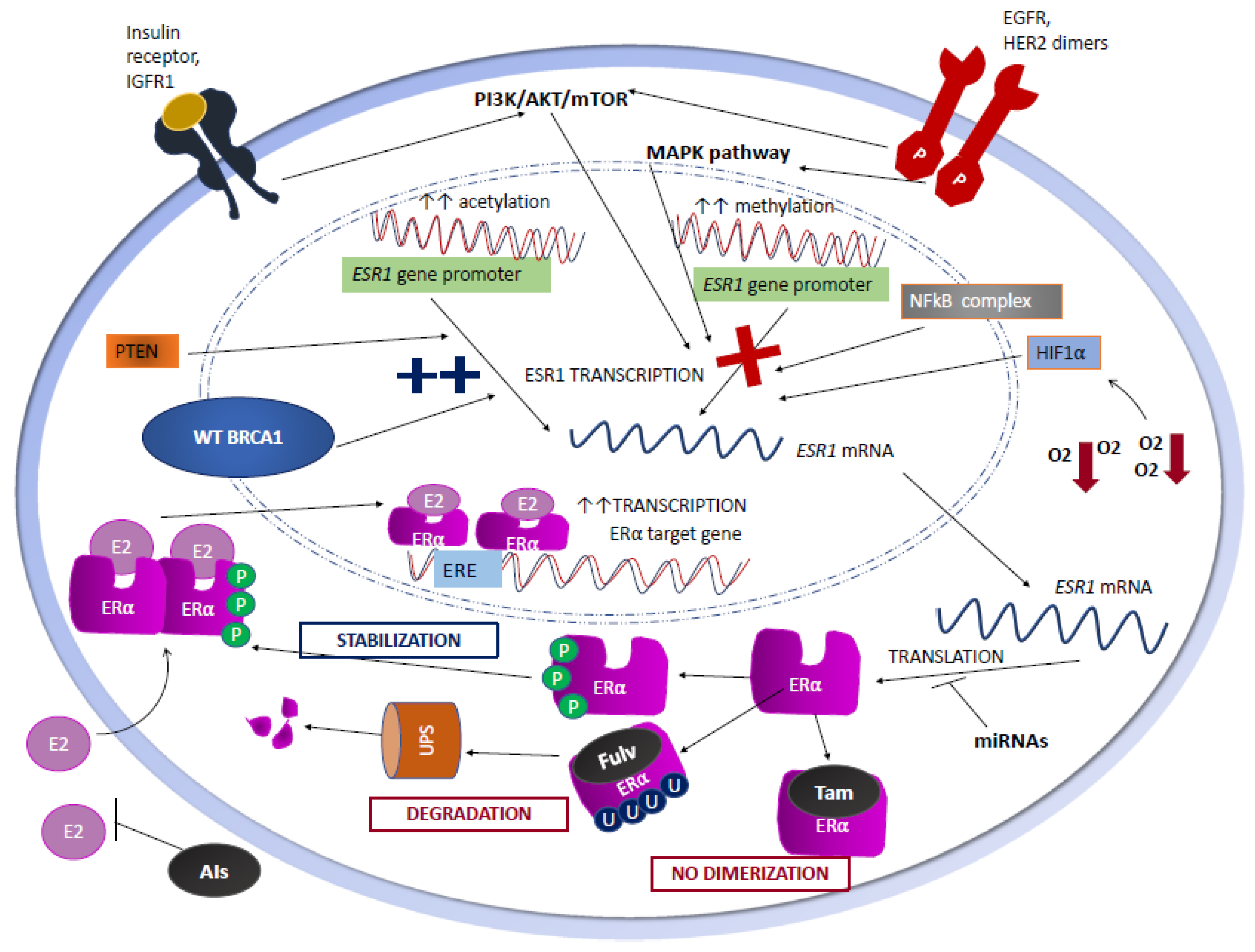
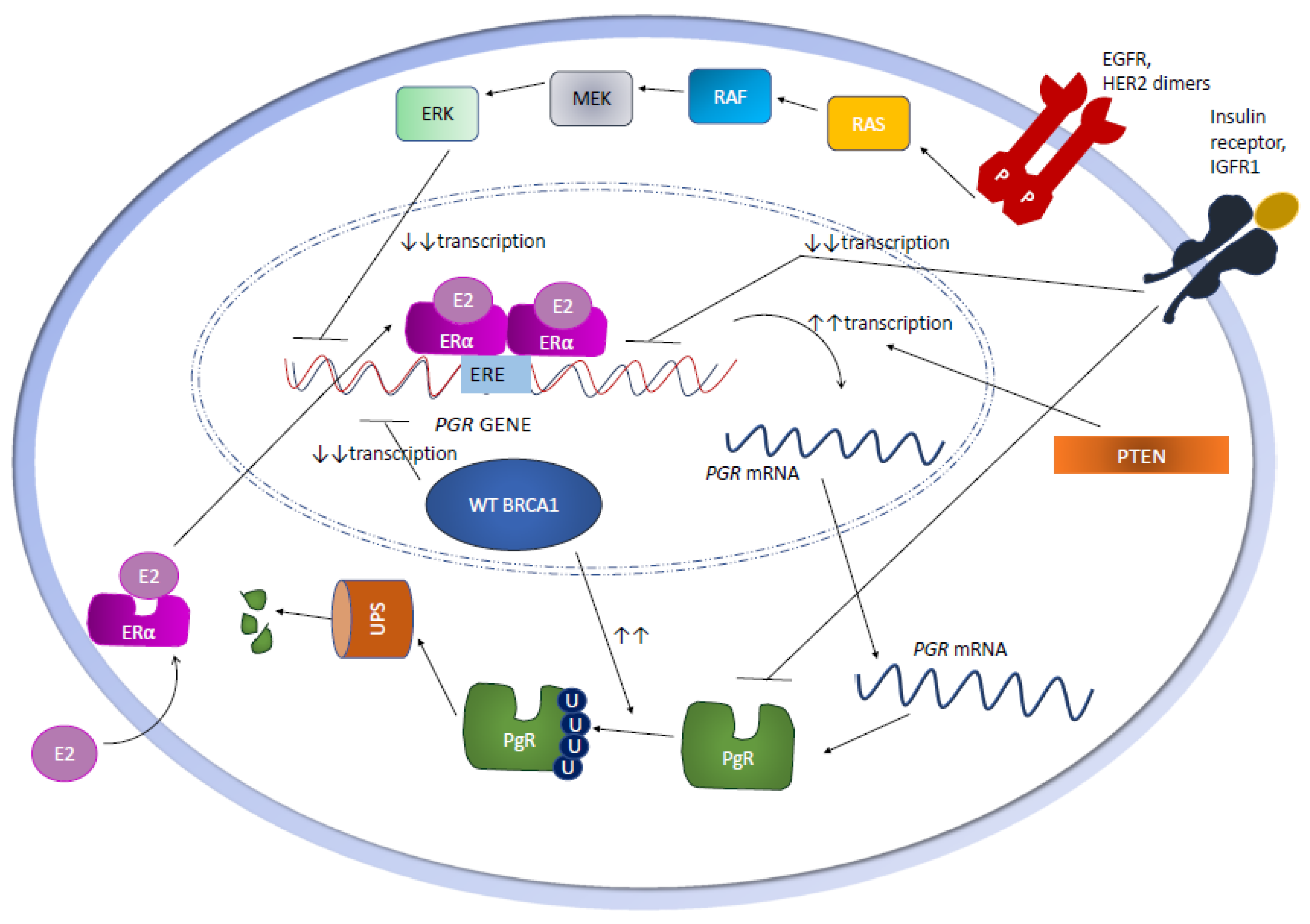
2.1. Genetic Mechanisms
2.2. Epigenetic Mechanisms
2.3. Growth Factor Signaling
2.4. Post-Transcriptional Regulation of ERαExpression
2.5. Post-Translational Regulation of ERαExpression
2.6. The Role of Hypoxia
2.7. The Role of BRCA1
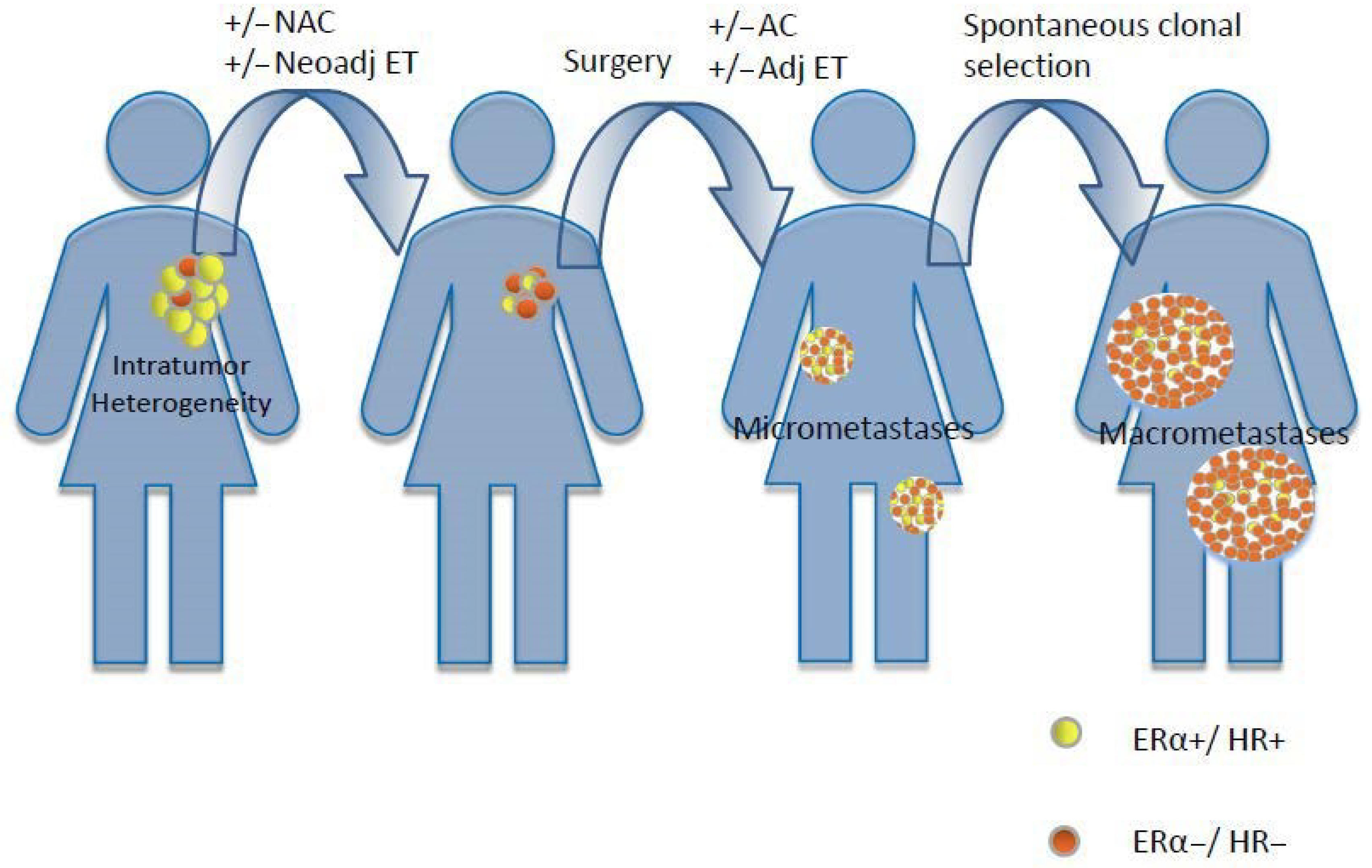
2.8. Intratumor Heterogeneity
3. HR Loss in Specific Clinical Settings
3.1. HR Loss after Neoadjuvant Treatments
3.2. HR Loss in Recurrent/Metastatic BC

{kind=link}
{kind=link}
{kind=link}
| Author | Methods | Site | Previous Treatments | ERα (%) | PgR (%) | HR Status (%) |
|---|---|---|---|---|---|---|
| Kuukasjärvi T. et al. (1996) [81] | Retrospective N = 50 pts | Local recurrence/Distant metastases | None | 24 | 24 | 36 |
| Lower E.E. et al. (2005) [118] | Retrospective N = 200 pts | Distant metastases | Tamoxifen only/chemotherapy only/chemotherapy+ tamoxifen | 30 | 39.3 | - |
| Simmons C. et al. (2009) [115] | Prospective N = 25 pts | Distant metastases | Various | - | - | 40 |
| Liedtke C. et al. (2009) [121] | Retrospective N = 789 pts | Local recurrence/ Distant metastases | Taxanes vs. endocrine therapy | 18.4 | 40.3 | 14–40 |
| Thompson A.M. et al. (2010) [122] | Prosepective N = 137 pts | Local recurrence/ Distant metastases | Various | 10 | 25 | 15 |
| Amir E. et al. (2012) [116] | Prospective N = 117 pts | Distant metastases | Various | 16 | 40 | 37.6 |
| Ongaro E. et al. (2018) [120] | Retrospective N = 232 pts | Distant metastases | Anthracyclines (50.4%); taxanes (33.6%); antiestrogens (49.1%); AI (39.2%) | 12.7 | 49.7 | - |
| Schrijver W.A.M.E. et al. (2018) [16] | Metanalysis N = 39 studies | Distant metastases | Various | 19.3 | 30.9 | - |
| Stueber T. et al. (2019) [119] | Retrospective N = 196 pts | Local recurrence/ Distant metastases | Various | 36.8 | 75.4 | - |
4. Therapeutic Implications
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perou, C.M.; Sørile, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Ress, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.L.; Hartig, S.M.; Edwards, D.P. Progesterone Receptor Signaling Mechanisms. J. Mol. Biol. 2016, 428, 3831–3849. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. Integration of the extranuclear and nuclear actions of estrogen. Mol. Endocrinol. 2005, 19, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, M.K.; Rare, S.M.; Skrtic, S.; Gao, H.; Dahlman-Wright, K.; Gustafsson, J.-Å.; Ohlsson, C. Estrogen Receptor (ER)-Reduces ER-Regulated Gene Transcription, Supporting a “Ying Yang” Relationship between ER and ER in Mice. Mol. Endocrinol. 2003, 17, 203–208. [Google Scholar] [CrossRef]
- Paruthiyil, S.; Parmar, H.; Kerekatte, V.; Cunha, G.R.; Firestone, G.L.; Leitmant, D.C. Estrogen Receptor β Inhibits Human Breast Cancer Cell Proliferation and Tumor Formation by Causing a G2 Cell Cycle Arrest. Cancer Res. 2004, 64, 423–428. [Google Scholar] [CrossRef]
- Chang, E.C.; Frasor, J.; Komm, B.; Katzenellenbogen, B.S. Impact of Estrogen Receptor on Gene Networks Regulated by Estrogen Receptor in Breast Cancer Cells. Endocrinology 2006, 147, 4831–4832. [Google Scholar] [CrossRef]
- Gruvberger-Saal, S.K.; Bendahl, P.O.; Saal, L.H.; Laakso, M.; Hegardt, C.; Edén, P.; Peterson, C.; Malmström, P.; Isola, J.; Borg, Å.; et al. Estrogen receptor β expression is associated with tamoxifen response in ERα-negative breast carcinoma. Clin. Cancer Res. 2007, 13, 1987–1994. [Google Scholar] [CrossRef]
- Onitilo, A.A.; Engel, J.M.; Greenlee, R.T.; Mukesh, B.N. Breast Cancer Subtypes Based on ER/PR and Her2 Expression: Comparison of Clinicopathologic Features and Survival. Clin. Med. Res. 2009, 7, 4–13. [Google Scholar] [CrossRef]
- Cortazar, P.; Zhang, L.; Untch, M.; Mehta, K.; Costantino, J.P.; Wolmark, N.; Bonnefoi, H.; Cameron, D.; Gianni, L.; Valagussa, P.; et al. Pathological complete response and long-term clinical benefit in breast cancer: The CTNeoBC pooled analysis. Lancet 2014, 384, 164–172. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Lurie, R.H.; Anderson, B.O.; Abraham, J.; Aft, R.; Agnese, D.; Allison, K.H.; Blair, S.L.; Burstein, H.J.; Dang, C.; Elias, A.D.; et al. NCCN Guidelines Version 6.2020 Breast Cancer; National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2020. [Google Scholar]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Swift, C.; Kilburn, L.; Fribbens, C.; Beaney, M.; Garcia-Murillas, I.; Budzar, A.U.; Robertson, J.F.R.; Gradishar, W.; Piccart, M.; et al. ESR1 Mutations and Overall Survival on Fulvestrant versus Exemestane in Advanced Hormone Receptor–Positive Breast Cancer: A Combined Analysis of the Phase III SoFEA and EFECT Trials. Clin. Cancer Res. 2020, 26, 5172–5177. [Google Scholar] [CrossRef] [PubMed]
- Allred, D.C.; Mohsin, S.K.; Fuqua, S.A.W. Histological and biological evolution of human premalignant breast disease. Endocr. Relat Cancer 2001, 8, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Aurilio, G.; Disalvatore, D.; Pruneri, G.; Bagnardi, V.; Viale, G.; Curigliano, G.; Adamoli, L.; Munzone, E.; Sciandivasci, A.; De Vita, F.; et al. A meta-analysis of oestrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 discordance between primary breast cancer and metastases. Eur. J. Cancer 2014, 50, 277–289. [Google Scholar] [CrossRef]
- Schrijver, W.A.M.E.M.E.; Suijkerbuijk, K.P.M.M.; van Gils, C.H.; van der Wall, E.; Moelans, C.B.; van Diest, P.J. Receptor Conversion in Distant Breast Cancer Metastases: A Systematic Review and Meta-analysis. J. Natl. Cancer Inst. 2018, 110, 568–580. [Google Scholar] [CrossRef]
- Rose, D.P. Effects of Adjuvant Chemohormonal Therapy on the Ovarian and Adrenal Function of Breast Cancer Patients. Cancer Res. 1980, 40, 4043–4047. [Google Scholar]
- Galli, G.; Bregni, G.; Cavalieri, S.; Porcu, L.; Baili, P.; Hade, A.; Di Salvo, F.; Sant, M.; Agresti, R.; Gennaro, M.; et al. Neoadjuvant Chemotherapy Exerts Selection Pressure Towards Luminal Phenotype Breast Cancer. Breast Care 2017, 12, 391–394. [Google Scholar] [CrossRef]
- Huang, W.; Peng, Y.; Kiselar, J.; Zhao, X.; Albaqami, A.; Mendez, D.; Chen, Y.; Chakravarthy, S.; Gupta, S.; Ralston, C.; et al. Multidomain architecture of estrogen receptor reveals interfacial cross-talk between its DNA-binding and ligand-binding domains. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Press, M.F.; Nousek-Goebl, N.A.; Bur, M.; Greene, G.L. Estrogen receptor localization in the female genital tract. Am. J. Pathol. 1986, 123, 280–292. [Google Scholar] [CrossRef][Green Version]
- Press, M.F.; Xu, S.-H.; Wang, J.-D.; Greene, G.L. Subcellular distribution of estrogen receptor and progesterone receptor with and without specific ligand. Am. J. Pathol. 1989, 135, 857–864. [Google Scholar]
- Tecalco-Cruz, A.C.; Pérez-Alvarado, I.A.; Ramírez-Jarquín, J.O.; Rocha-Zavaleta, L. Nucleo-cytoplasmic transport of estrogen receptor alpha in breast cancer cells. Cell. Signal. 2017, 34, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Wärnmark, A.; Treuter, E.; Wright, A.P.H.H.; Gustafsson, J.-A.Å.; Wärnmark, A.; Treuter, E.; Wright, A.P.H.H.; Gustafsson, J.-A.Å. Activation Functions 1 and 2 of Nuclear Receptors: Molecular Strategies for Transcriptional Activation. Mol. Endocrinol. 2003, 17, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Riedl, T.; Washbrook, E.; Pace, P.E.; Coombes, R.C.; Egly, J.M.; Ali, S. Activation of estrogen receptor α by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol. Cell 2000, 6, 127–137. [Google Scholar] [CrossRef]
- Pietras, R.J.; Márquez-Garbán, D.C. Membrane-associated estrogen receptor signaling pathways in human cancers. Clin. Cancer Res. 2007, 13, 4672–4676. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Wang, J.; Santen, R.J.; Yue, W. Long-term treatment with tamoxifen facilitates translocation of estrogen receptor α out of the nucleus and enhances its interaction with EGFR in MCF-7 breast cancer cells. Cancer Res. 2007, 67, 1352–1360. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Buchwalter, G.; De Angelis, C.; Brown, M.; Schiff, R. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef]
- Zheng, W.Q.; Zheng, J.M.; Lu, J.; Hu, F.X. Loss of heterozygosity of ER gene in breast cancer and its clinical significance. Chin. J. Cancer Res. 2002, 14, 122–125. [Google Scholar] [CrossRef]
- Herynk, M.H.; Fuqua, S.A.W. Estrogen receptors in resistance to hormone therapy. Adv. Exp. Med. Biol. 2007, 608, 130–143. [Google Scholar]
- Lapidus, R.G.; Nass, S.J.; Davidson, N.E. The Loss of Estrogen and Progesterone Receptor Gene Expression in Human Breast Cancer. J. Mammary Gland Biol. Neoplasia 1998, 3, 85–94. [Google Scholar] [CrossRef]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjöld, M.; Gustafsson, J.Å. Human estrogen receptor β-gene structure, chromosomal localization, and expression pattern. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates ERα action in breast cancer. Nature 2015, 523, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Schiff, R.; Arpino, G.; Osborne, C.K.; Lee, A.V. Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J. Clin. Oncol. 2005, 23, 7721–7735. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.M.; Nicolai, H.; Solomon, E.; Boomer, W.F. The frequency and mechanism of loss of heterozygosity on chromosome 11q in breast cancer. J. Pathol. 1996, 180, 38–43. [Google Scholar] [CrossRef]
- Kangaspeska, S.; Stride, B.; Métivier, R.; Polycarpou-Schwarz, M.; Ibberson, D.; Carmouche, R.P.; Benes, V.; Gannon, F.; Reid, G. Transient cyclical methylation of promoter DNA. Nature 2008, 452, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, Y.L.; Issa, J.-P.; Parl, F.F.; Smith, H.S.; Baylin, S.B.; Davidson, N.E. Methylation of the Estrogen Receptor Gene CpG Island Marks Loss of Estrogen Receptor Expression in Human Breast Cancer Cells. Cancer Res. 1994, 54, 2552–2555. [Google Scholar] [PubMed]
- Yang, X.; Phillips, D.L.; Ferguson, A.T.; Nelson, W.G.; Herman, J.G.; Davidson, N.E. Synergistic activation of functional estrogen receptor (ER)-α by DNA methyltransferase and histone deacetylase inhibition in human ER-α-negative breast cancer cells. Cancer Res. 2001, 61, 7025–7029. [Google Scholar]
- Tsuboi, K.; Nagatomo, T.; Gohno, T.; Higuchi, T.; Sasaki, S.; Fujiki, N.; Kurosumi, M.; Takei, H.; Yamaguchi, Y.; Niwa, T.; et al. Single CpG site methylation controls estrogen receptor gene transcription and correlates with hormone therapy resistance. J. Steroid Biochem. Mol. Biol. 2017, 171, 209–217. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, C.; Jiang, H.; Liang, L.; Shi, W.; Zhang, Q.; Sun, P.; Xiang, R.; Wang, Y.; Yang, S. ZEB1 induces ER-α promoter hypermethylation and confers antiestrogen resistance in breast cancer. Cell Death Dis. 2017, 8, e2732. [Google Scholar] [CrossRef]
- Lapidus, R.G.; Ferguson, A.T.; Ottaviano, Y.L.; Parl, F.F.; Smith, H.S.; Weitzman, S.A.; Baylin, S.B.; Issa, J.P.J.; Davidson, N.E. Methylation of estrogen and progesterone receptor gene 5′ CpG islands correlates with lack of estrogen and progesterone receptor gene expression in breast tumors. Clin. Cancer Res. 1996, 2, 805–810. [Google Scholar]
- Leu, Y.W.; Yan, P.S.; Fan, M.; Jin, V.X.; Liu, J.C.; Curran, E.M.; Welshons, W.V.; Wei, S.H.; Davuluri, R.V.; Plass, C.; et al. Loss of estrogen receptor signaling triggers epigenetic silencing of downstream targets in breast cancer. Cancer Res. 2004, 64, 8184–8192. [Google Scholar] [CrossRef]
- Perren, A.; Weng, L.P.; Boag, A.H.; Ziebold, U.; Thakore, K.; Dahia, P.L.M.; Komminoth, P.; Lees, J.A.; Mulligan, L.M.; Mutter, G.L.; et al. Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am. J. Pathol. 1999, 155, 1253–1260. [Google Scholar] [CrossRef]
- Garcia, J.M.; Silva, J.M.; Dominguez, G.; Gonzalez, R.; Navarro, A.; Carretero, L.; Provencio, M.; España, P.; Bonilla, F. Allelic loss of the PTEN region (10q23) in breast carcinomas of poor pathophenotype. Breast Cancer Res. Treat. 1999, 57, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Zhang, P.; Deng, W.; Oesterreich, S.; Lu, Y.; Mills, G.B.; Lee, A.V. Insulin-like growth factor-I inhibits progesterone receptor expression in breast cancer cells via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway: Progesterone receptor as a potential indicator of growth factor activity in breast cancer. Mol. Endocrinol. 2003, 17, 575–588. [Google Scholar] [PubMed]
- Oh, A.S.; Lorant, L.A.; Holloway, J.N.; Miller, D.L.; Kern, F.G.; El-Ashry, D. Hyperactivation of MAPK Induces Loss of ERα Expression in Breast Cancer Cells. Mol. Endocrinol. 2001, 15, 1344–1359. [Google Scholar] [CrossRef]
- Sheikh, M.S.; Shao, Z.-M.; Chen, J.-C.; Li, X.-S.; Hussain, A.; Fontana, J.A. Expression of estrogen receptors in estrogen receptor–negative human breast carcinoma cells: Modulation of epidermal growth factor-receptor (EGF-R) and transforming growth factor α (TGFα) gene expression. J. Cell. Biochem. 1994, 54, 289–298. [Google Scholar] [CrossRef]
- Montemurro, F.; Di Cosimo, S.; Arpino, G. Human epidermal growth factor receptor 2 (her2)-positive and hormone receptor-positive breast cancer: New insights into molecular interactions and clinical implications. Ann. Oncol. 2013, 24, 2715–2724. [Google Scholar] [CrossRef]
- Guo, S.; Sonenshein, G.E. Forkhead Box Transcription Factor FOXO3a Regulates Estrogen Receptor Alpha Expression and Is Repressed by the Her-2/neu/Phosphatidylinositol 3-Kinase/Akt Signaling Pathway. Mol. Cell. Biol. 2004, 24, 8681–8690. [Google Scholar] [CrossRef]
- Lal, P.; Tan, L.K.; Chen, B. Correlation of HER-2 Status With Estrogen and Progesterone Receptors and Histologic Features in 3,655 Invasive Breast Carcinomas. Am. J. Clin. Pathol. 2005, 123, 541–546. [Google Scholar] [CrossRef]
- Creighton, C.J.; Hilger, A.M.; Murthy, S.; Rae, J.M.; Chinnaiyan, A.M.; El-Ashry, D. Activation of mitogen-activated protein kinase in estrogen receptor α-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor α-negative human breast tumors. Cancer Res. 2006, 66, 3903–3911. [Google Scholar] [CrossRef]
- Zhou, Y.; Eppenberger-Castori, S.; Eppenberger, U.; Benz, C.C. The NFκB pathway and endocrine-resistant breast cancer. Endocr. Relat. Cancer 2005, 12, S37–S46. [Google Scholar] [CrossRef]
- Zhou, Y.; Eppenberger-Castori, S.; Marx, C.; Yau, C.; Scott, G.K.; Eppenberger, U.; Benz, C.C. Activation of nuclear factor-κB (NFκB) identifies a high-risk subset of hormone-dependent breast cancers. Int. J. Biochem. Cell Biol. 2005, 37, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Holloway, J.N.; Murthy, S.; El-Ashry, D. A cytoplasmic substrate of mitogen-activated protein kinase is responsible for estrogen receptor-α down-regulation in breast cancer cells: The role of nuclear factor-κB. Mol. Endocrinol. 2004, 18, 1396–1410. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pianetti, S.; Arsura, M.; Romieu-Mourez, R.; Coffey, R.J.; Sonenshein, G.E. Her-2/neu overexpression induces NF-κB via a PI3-kinase/Akt pathway involving calpain-mediated degradation of IκB-α that can be inhibited by the tumor suppressor PTEN. Oncogene 2001, 20, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into molecular classifications of triple-negative breast cancer: Improving patient selection for treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [PubMed]
- Kenealy, M.R.; Flouriot, G.; Sonntag-Buck, V.; Dandekar, T.; Brand, H.; Gannon, F. The 3′-untranslated region of the human estrogen receptor α gene mediates rapid messenger ribonucleic acid turnover. Endocrinology 2000, 141, 2805–2813. [Google Scholar] [CrossRef]
- Carmeci, C.; DeConinck, E.C.; Lawton, T.; Bloch, D.A.; Weigel, R.J. Analysis of estrogen receptor messenger RNA in breast carcinomas from archival specimens is predictive of tumor biology. Am. J. Pathol. 1997, 150, 1563–1570. [Google Scholar]
- Liang, Y.K.; Lin, H.Y.; Dou, X.W.; Chen, M.; Wei, X.L.; Zhang, Y.Q.; Wu, Y.; Chen, C.F.; Bai, J.W.; Xiao, Y.S.; et al. MiR-221/222 promote epithelial-mesenchymal transition by targeting Notch3 in breast cancer cell lines. NPJ Breast Cancer 2018, 4, 1–9. [Google Scholar] [CrossRef]
- Al-Nakhle, H.; Burns, P.A.; Cummings, M.; Hanby, A.M.; Hughes, T.A.; Satheesha, S.; Shaaban, A.M.; Smith, L.; Speirs, V. Estrogen receptor β1 expression is regulated by miR-92 in breast cancer. Cancer Res. 2010, 70, 4778–4784. [Google Scholar] [CrossRef]
- Li, X.; Mertens-Talcott, S.U.; Zhang, S.; Kim, K.H.; Ball, J.; Safe, S. MicroRNA-27a indirectly regulates estrogen receptor α expression and hormone responsiveness in MCF-7 breast cancer cells. Endocrinology 2010, 151, 2462–2473. [Google Scholar] [CrossRef]
- Tecalco-Cruz, A.C.; Ramírez-Jarquín, J.O. Mechanisms that Increase Stability of Estrogen Receptor Alpha in Breast Cancer. Clin. Breast Cancer 2017, 17, 1–10. [Google Scholar] [CrossRef]
- Osborne, C.K.; Wakeling, A.; Nicholson, R.I. Fulvestrant: An oestrogen receptor antagonist with a novel mechanism of action. Br. J. Cancer 2004, 90, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Wardell, S.E.; Marks, J.R.; McDonnell, D.P. The turnover of estrogen receptor α by the selective estrogen receptor degrader (SERD) fulvestrant is a saturable process that is not required for antagonist efficacy. Biochem. Pharmacol. 2011, 82, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Zhou, W.; Hafner, M.; Blake, R.A.; Chalouni, C.; Chen, I.P.; De Bruyn, T.; Giltnane, J.M.; Hartman, S.J.; Heidersbach, A.; et al. Therapeutic Ligands Antagonize Estrogen Receptor Function by Impairing Its Mobility. Cell 2019, 178, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Castoria, G.; Giovannelli, P.; Lombardi, M.; De Rosa, C.; Giraldi, T.; De Falco, A.; Barone, M.V.; Abbondanza, C.; Migliaccio, A.; Auricchio, F. Tyrosine phosphorylation of estradiol receptor by Src regulates its hormone-dependent nuclear export and cell cycle progression in breast cancer cells. Oncogene 2012, 31, 4868–4877. [Google Scholar] [CrossRef]
- Kufe, D.W. MUC1-C oncoprotein as a target in breast cancer: Activation of signaling pathways and therapeutic approaches. Oncogene 2013, 32, 1073–1081. [Google Scholar] [CrossRef]
- Rajbhandari, P.; Schalper, K.A.; Solodin, N.M.; Ellison-Zelski, S.J.; Ping Lu, K.; Rimm, D.L.; Alarid, E.T. Pin1 modulates ERα levels in breast cancer through inhibition of phosphorylation-dependent ubiquitination and degradation. Oncogene 2014, 33, 1438–1447. [Google Scholar] [CrossRef]
- Caligiuri, I.; Rizzolio, F.; Boffo, S.; Giordano, A.; Toffoli, G. Critical choices for modeling breast cancer in transgenic mouse models. J. Cell Physiol. 2012, 227, 2988–2991. [Google Scholar] [CrossRef]
- Zhu, J.; Zhao, C.; Kharman-Biz, A.; Zhuang, T.; Jonsson, P.; Liang, N.; Williams, C.; Lin, C.Y.; Qiao, Y.; Zendehdel, K.; et al. The atypical ubiquitin ligase RNF31 stabilizes estrogen receptor α and modulates estrogen-stimulated breast cancer cell proliferation. Oncogene 2014, 33, 4340–4351. [Google Scholar] [CrossRef]
- la Rosa, P.; Pesiri, V.; Leclercq, G.; Marino, M.; Acconcia, F. Palmitoylation regulates 17β-estradiol-induced estrogen receptor-α degradation and transcriptional activity. Mol. Endocrinol. 2012, 26, 762–774. [Google Scholar] [CrossRef]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef]
- Stoner, M.; Saville, B.; Wormke, M.; Dean, D.; Burghardt, R.; Safe, S. Hypoxia induces proteasome-dependent degradation of estrogen receptor α in ZR-75 breast cancer cells. Mol. Endocrinol. 2002, 16, 2231–2242. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.; Park, C.; Lee, Y.J. Hypoxia-inducible factor 1 alpha represses the transcription of the estrogen receptor alpha gene in human breast cancer cells. Biochem. Biophys. Res. Commun. 2011, 407, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.; Kosyna, F.K.; Dunst, J.; Jelkmann, W.; Depping, R. Impact of hypoxia inducible factors on estrogen receptor expression in breast cancer cells. Arch. Biochem. Biophys. 2017, 613, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Militello, A.M.; Zielli, T.; Boggiani, D.; Michiara, M.; Naldi, N.; Bortesi, B.; Zanelli, P.; Uliana, V.; Giuliotti, S.; Musolino, A. Mechanism of action and clinical efficacy of CDK4/6 inhibitors in BRCA-mutated, estrogen receptor-positive breast cancers: Case report and literature review. Front. Oncol. 2019, 9, 759. [Google Scholar] [CrossRef]
- Goodwin, P.J.; Phillips, K.A.; West, D.W.; Ennis, M.; Hopper, J.L.; John, E.M.; O’Malley, F.P.; Milne, R.L.; Andrulis, I.L.; Friedlander, M.L.; et al. Breast cancer prognosis in BRCA1 and BRCA2 mutation carriers: An international prospective breast cancer family registry population-based cohort study. J. Clin. Oncol. 2012, 30, 19–26. [Google Scholar] [CrossRef]
- Anderson, S.F.; Schlegel, B.P.; Nakajima, T.; Wolpin, E.S.; Parvin, J.D. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat. Genet. 1998, 19, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Hosey, A.M.; Gorski, J.J.; Murray, M.M.; Quinn, J.E.; Chung, W.Y.; Stewart, G.E.; James, C.R.; Farragher, S.M.; Mulligan, J.M.; Scott, A.N.; et al. Molecular basis for estrogen receptor α deficiency in BRCA1-linked breast cancer. J. Natl. Cancer Inst. 2007, 99, 1683–1694. [Google Scholar] [CrossRef]
- Roldán, G.; Delgado, L.; Musé, I.M. Tumoral expression of BRCA1, estrogen receptor alpha and ID4 protein in patients with sporadic breast cancer. Cancer Biol. Ther. 2006, 5, 505–510. [Google Scholar] [CrossRef]
- Calvo, V.; Beato, M. BRCA1 counteracts progesterone action by ubiquitination leading to progesterone receptor degradation and epigenetic silencing of target promoters. Cancer Res. 2011, 71, 3422–3431. [Google Scholar] [CrossRef]
- Kuukasjärvi, T.; Kononen, J.; Helin, H.; Holli, K.; Isola, J. Loss of estrogen receptor in recurrent breast cancer is associated with poor response to endocrine therapy. J. Clin. Oncol. 1996, 14, 2584–2589. [Google Scholar] [CrossRef]
- Horwitz, K.B.; Smith, J.A.; Jewett, P.B.; Horwitz, K.B. Heterogeneity of Progesterone Receptor Content and Remodeling by Tamoxifen Characterize Subpopulations of Cultured Human Breast Cancer Cells: Analysis by Quantitative Dual Parameter Flow Cytometry. Cancer Res. 1992, 52, 593–602. [Google Scholar]
- Annaratone, L.; Simonetti, M.; Wernersson, E.; Marchiò, C.; Garnerone, S.; Scalzo, M.S.; Bienko, M.; Chiarle, R.; Sapino, A.; Crosetto, N. Quantification of HER2 and estrogen receptor heterogeneity in breast cancer by single-molecule RNA fluorescence in situ hybridization. Oncotarget 2017, 8, 18680–18698. [Google Scholar] [CrossRef] [PubMed]
- Lindström, L.S.; Yau, C.; Czene, K.; Thompson, C.K.; Hoadley, K.A.; Van’t Veer, L.J.; Balassanian, R.; Bishop, J.W.; Carpenter, P.M.; Chen, Y.Y.; et al. Intratumor heterogeneity of the estrogen receptor and the long-term risk of fatal Breast cancer. J. Natl. Cancer Inst. 2018, 110, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.W.; Zava, D.T.; Locher, G.W.; Goldhirsch, A.; Hartmann, W.H. Receptor heterogeneity of human breast cancer as measured by multiple intratumoral assays of estrogen and progesterone receptor. Eur. J. Cancer Clin. Oncol. 1984, 20, 375–382. [Google Scholar] [CrossRef]
- Chung, G.G.; Zerkowski, M.P.; Ghosh, S.; Camp, R.L.; Rimm, D.L. Quantitative analysis of estrogen receptor heterogeneity in breast cancer. Lab. Investig. 2007, 87, 662–669. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Allott, E.H.; Geradts, J.; Sun, X.; Cohen, S.M.; Zirpoli, G.R.; Khoury, T.; Bshara, W.; Chen, M.; Sherman, M.E.; Palmer, J.R.; et al. Intratumoral heterogeneity as a source of discordance in breast cancer biomarker classification. Breast Cancer Res. 2016, 18, 1–11. [Google Scholar] [CrossRef]
- Zhang, L.; Riethdorf, S.; Wu, G.; Wang, T.; Yang, K.; Peng, G.; Liu, J.; Pantel, K. Meta-analysis of the prognostic value of circulating tumor cells in breast cancer. Clin. Cancer Res. 2012, 18, 5701–5710. [Google Scholar] [CrossRef]
- Babayan, A.; Hannemann, J.; Spötter, J.; Müller, V.; Pantel, K.; Joosse, S.A. Heterogeneity of Estrogen Receptor Expression in Circulating Tumor Cells from Metastatic Breast Cancer Patients. PLoS ONE 2013, 8, e75038. [Google Scholar] [CrossRef]
- Lindstrom, L.S.; Karlsson, E.; Wilking, U.M.; Johansson, U.; Hartman, J.; Lidbrink, E.K.; Hatschek, T.; Skoog, L.; Bergh, J.; Lindström, L.S.; et al. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J. Clin. Oncol. 2012, 30, 2601–2608. [Google Scholar] [CrossRef]
- Arnedos, M.; Nerurkar, A.; Osin, P.; A’Hern, R.; Smith, I.E.; Dowsett, M. Discordance between core needle biopsy (CNB) and excisional biopsy (EB) for estrogen receptor (ER), progesterone receptor (PgR) and HER2 status in early breast cancer (EBC). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2009, 20, 1948–1952. [Google Scholar] [CrossRef]
- Jensen, J.D.; Knoop, A.; Ewertz, M.; Laenkholm, A.-V.V. ER, HER2, and TOP2A expression in primary tumor, synchronous axillary nodes, and asynchronous metastases in breast cancer. Breast Cancer Res. Treat. 2012, 132, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Aitken, S.J.; Thomas, J.S.; Langdon, S.P.; Harrison, D.J.; Faratian, D. Quantitative analysis of changes in ER, PR and HER2 expression in primary breast cancer and paired nodal metastases. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2010, 21, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.H.; Hammond, M.E.H.; Dowsett, M.; McKernin, S.E.; Carey, L.A.; Fitzgibbons, P.L.; Hayes, D.F.; Lakhani, S.R.; Chavez-MacGregor, M.; Perlmutter, J.; et al. Estrogen and progesterone receptor testing in breast cancer: ASCO/CAP guideline update. J. Clin. Oncol. 2020, 38, 1346–1366. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.; Laudico, A.; Carnate, J.; Lim, F.; Fernandez, A.; Rivera, R.; Mapua, C.; Love, R. Breast cancer hormone receptor assay results of core needle biopsy and modified radical mastectomy specimens from the same patients. Clin. Breast Cancer 2010, 10, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Viale, G.; Regan, M.M.; Maiorano, E.; Mastropasqua, M.G.; Dell’Orto, P.; Rasmussen, B.B.; Raffoul, J.; Neven, P.; Orosz, Z.; Braye, S.; et al. Prognostic and predictive value of centrally reviewed expression of estrogen and progesterone receptors in a randomized trial comparing letrozole and tamoxifen adjuvant therapy for postmenopausal early breast cancer: BIG 1-98. J. Clin. Oncol. 2007, 25, 3846–3852. [Google Scholar] [CrossRef]
- Zeng, J.; Piscuoglio, S.; Aggarwal, G.; Magda, J.; Friedlander, M.A.; Murray, M.; Akram, M.; Reis-Filho, J.S.; Weigelt, B.; Edelweiss, M. Hormone receptor and HER2 assessment in breast carcinoma metastatic to bone: A comparison between FNA cell blocks and decalcified core needle biopsies. Cancer Cytopathol. 2020, 128, 133–145. [Google Scholar] [CrossRef]
- Wu, Y.T.; Li, X.; Lu, L.J.; Gan, L.; Dai, W.; Shi, Y.L.; Adhikari, V.P.; Wu, K.N.; Kong, L.Q. Effect of neoadjuvant chemotherapy on the expression of hormone receptors and Ki67 in Chinese breast cancer patients: A retrospective study of 525 patients. J. Biomed. Res. 2018, 32, 191–197. [Google Scholar]
- Hirata, T.; Shimizu, C.; Yonemori, K.; Hirakawa, A.; Kouno, T.; Tamura, K.; Ando, M.; Katsumata, N.; Fujiwara, Y. Change in the hormone receptor status following administration of neoadjuvant chemotherapy and its impact on the long-term outcome in patients with primary breast cancer. Br. J. Cancer 2009, 101, 1529–1536. [Google Scholar] [CrossRef]
- Colleoni, M.; Viale, G.; Zahrieh, D.; Pruneri, G.; Gentilini, O.; Veronesi, P.; Gelber, R.D.; Curigliano, G.; Torrisi, R.; Luini, A.; et al. Chemotherapy is more effective in patients with breast cancer not expressing steroid hormone receptors: A study of preoperative treatment. Clin. Cancer Res. 2004, 10, 6622–6628. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef]
- Masuda, N.; Lee, S.-J.; Ohtani, S.; Im, Y.-H.; Lee, E.-S.; Yokota, I.; Kuroi, K.; Im, S.-A.; Park, B.-W.; Kim, S.-B.; et al. Adjuvant Capecitabine for Breast Cancer after Preoperative Chemotherapy. N. Engl. J. Med. 2017, 376, 2147–2159. [Google Scholar] [CrossRef] [PubMed]
- van de Ven, S.; Smit, V.T.H.B.M.H.B.M.; Dekker, T.J.A.A.; Nortier, J.W.R.R.; Kroep, J.R. Discordances in ER, PR and HER2 receptors after neoadjuvant chemotherapy in breast cancer. Cancer Treat. Rev. 2011, 37, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Tacca, O.; Penault-Llorca, F.; Abrial, C.; Mouret-Reynier, M.; Raoelfils, I.I.I.; Durando, X.; Achard, J.-L.J.; Gimbergues, P.; Curé, H.; Chollet, P.; et al. Changes in and Prognostic Value of Hormone Receptor Status in a Series of Operable Breast Cancer Patients Treated with Neoadjuvant Chemotherapy. Oncologist 2007, 12, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, C.-M.M.; Yu, K.-D.D.; Zhou, R.-J.J.; Shao, Z.-M.M. Prognostic value of a positive-to-negative change in hormone receptor status after neoadjuvant chemotherapy in patients with hormone receptor- positive breast cancer. Ann. Surg. Oncol. 2012, 19, 3002–3011. [Google Scholar] [CrossRef]
- Jin, X.; Jiang, Y.Z.; Chen, S.; Yu, K.D.; Shao, Z.M.; Di, G.H. Prognostic value of receptor conversion after neoadjuvant chemotherapy in breast cancer patients: A prospective observational study. Oncotarget 2015, 6, 9600–9611. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.K.; Lee, M.H.; Park, I.H.; You, J.Y.; Nam, B.-H.; Kim, B.N.; Ro, J.; Lee, K.S.; Jung, S.-Y.; Kwon, Y.M.; et al. Impact of Molecular Subtype Conversion of Breast Cancers after Neoadjuvant Chemotherapy on Clinical Outcome. Cancer Res. Treat. 2016, 48, 133–141. [Google Scholar] [CrossRef]
- Ding, Y.; Ding, K.; Qian, H.; Yu, X.; Zou, D.; Yang, H.; Mo, W.; He, X.; Zhang, F.; Qin, C.; et al. Impact on survival of estrogen receptor, progesterone receptor and Ki-67 expression discordance pre- and post-neoadjuvant chemotherapy in breast cancer. PLoS ONE 2020, 15, e0231895. [Google Scholar] [CrossRef]
- Allevi, G.; Strina, C.; Andreis, D.; Zanoni, V.; Bazzola, L.; Bonardi, S.; Foroni, C.; Milani, M.; Cappelletti, M.R.; Gussago, F.; et al. Increased pathological complete response rate after a long-term neoadjuvant letrozole treatment in postmenopausal oestrogen and/or progesterone receptor-positive breast cancer. Br. J. Cancer 2013, 108, 1587–1592. [Google Scholar] [CrossRef]
- Ellis, M.J.; Tao, Y.; Luo, J.; A’Hern, R.; Evans, D.B.; Bhatnagar, A.S.; Chaudri Ross, H.A.; Von Kameke, A.; Miller, W.R.; Smith, I.; et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J. Natl. Cancer Inst. 2008, 100, 1380–1388. [Google Scholar] [CrossRef]
- Hawkins, R.A.; Tesdale, L.; Anderson, E.D.C.; Levack, P.A.; Chetty, U.; Forrest, A.P.M. Does the oestrogen receptor concentration of a breast cancer change during systemic therapy? Br. J. Cancer 1990, 61, 877–880. [Google Scholar] [CrossRef]
- Miller, W.R.; Dixon, J.M.; Macfarlane, L.; Cameron, D.; Anderson, T.J. Pathological features of breast cancer response following neoadjuvant treatment with either letrozole or tamoxifen. Eur. J. Cancer 2003, 39, 462–468. [Google Scholar] [CrossRef]
- Berry, D.A.; Cronin, K.A.; Plevritis, S.K.; Fryback, D.G.; Clarke, L.; Zelen, M.; Mandelblatt, J.S.; Yakovlev, A.Y.; Habbema, J.D.F.; Feuer, E.J. Effect of Screening and Adjuvant Therapy on Mortality from Breast Cancer. N. Engl. J. Med. 2005, 353, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Abe, O.; Abe, R.; Enomoto, K.; Kikuchi, K.; Koyama, H.; Masuda, H.; Nomura, Y.; Sakai, K.; Sugimachi, K.; Tominaga, T.; et al. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar]
- Simmons, C.; Miller, N.; Geddie, W.; Gianfelice, D.; Oldfield, M.; Dranitsaris, G.; Clemons, M.J. Does confirmatory tumor biopsy alter the management of breast cancer patients with distant metastases? Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2009, 20, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Amir, E.; Miller, N.; Geddie, W.; Freedman, O.; Kassam, F.; Simmons, C.; Oldfield, M.; Dranitsaris, G.; Tomlinson, G.; Laupacis, A.; et al. Prospective study evaluating the impact of tissue confirmation of metastatic disease in patients with breast cancer. J. Clin. Oncol. 2012, 30, 587–592. [Google Scholar] [CrossRef]
- Dieci, M.V.; Barbieri, E.; Piacentini, F.; Ficarra, G.; Bettelli, S.; Dominici, M.; Conte, P.F.; Guarneri, V. Discordance in receptor status between primary and recurrent breast cancer has a prognostic impact: A single-institution analysis. Ann. Oncol. 2013, 24, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Lower, E.E.; Glass, E.L.; Bradley, D.A.; Blau, R.; Heffelfinger, S. Impact of metastatic estrogen receptor and progesterone receptor status on survival. Breast Cancer Res. Treat. 2005, 90, 65–70. [Google Scholar] [CrossRef]
- Stueber, T.N.; Weiss, C.R.; Woeckel, A.; Haeusler, S. Influences of adjuvant treatments in hormone receptor positive breast cancer on receptor conversion in recurrent breast cancer. Arch. Gynecol. Obstet. 2019, 299, 533–541. [Google Scholar] [CrossRef]
- Ongaro, E.; Gerratana, L.; Cinausero, M.; Pelizzari, G.; Poletto, E.; Giangreco, M.; Andreetta, C.; Pizzolitto, S.; Di Loreto, C.; Minisini, A.M.; et al. Comparison of primary breast cancer and paired metastases: Biomarkers discordance influence on outcome and therapy. Future Oncol. 2018, 14, 849–859. [Google Scholar] [CrossRef]
- Liedtke, C.; Broglio, K.; Moulder, S.; Hsu, L.; Kau, S.W.; Symmans, W.F.; Albarracin, C.; Meric-Bernstam, F.; Woodward, W.; Theriault, R.L.; et al. Prognostic impact of discordance between triple-receptor measurements in primary and recurrent breast cancer. Ann. Oncol. 2009, 20, 1953–1958. [Google Scholar] [CrossRef]
- Thompson, A.M.; Jordan, L.B.; Quinlan, P.; Anderson, E.; Skene, A.; Dewar, J.A.; Purdie, C.A. Prospective comparison of switches in biomarker status between primary and recurrent breast cancer: The Breast Recurrence In Tissues Study (BRITS). Breast Cancer Res. 2010, 12, R92. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, C.; Mu, Z.; Rossi, G.; Schiewer, M.J.; Nguyen, T.; Austin, L.; Capoluongo, E.; Knudsen, K.; Cristofanilli, M.; Fortina, P. Detection of activating estrogen receptor gene (ESR1) mutations in single circulating tumor cells. Clin. Cancer Res. 2017, 23, 6086–6093. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.M.; Kurland, B.F.; Schubert, E.K.; Link, J.M.; Gadi, V.K.; Specht, J.M.; Eary, J.F.; Porter, P.; Shankar, L.K.; Mankoff, D.A.; et al. A phase 2 study of 16α-[18F]-fluoro-17β-estradiol positron emission tomography (FES-PET) as a marker of hormone sensitivity in metastatic breast cancer (MBC). Mol. Imaging Biol. 2014, 16, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ayres, K.L.; Goldman, D.A.; Dickler, M.N.; Bardia, A.; Mayer, I.A.; Winer, E.; Fredrickson, J.; Arteaga, C.L.; Baselga, J.; et al. 18F-fluoroestradiol PET/CT measurement of estrogen receptor suppression during a phase I trial of the novel estrogen receptor-targeted therapeutic GDC-0810: Using an imaging biomarker to guide drug dosage in subsequent trials. Clin. Cancer Res. 2017, 23, 3053–3060. [Google Scholar] [CrossRef]
- Mortimer, J.E.; Dehdashti, F.; Siegel, B.A.; Trinkaus, K.; Katzenellenbogen, J.A.; Welch, M.J. Metabolic Flare: Indicator of Hormone Responsiveness in Advanced Breast Cancer. J. Clin. Oncol. 2001, 19, 2797–2803. [Google Scholar] [CrossRef]
- Linden, H.M.; Stekhova, S.A.; Link, J.M.; Gralow, J.R.; Livingston, R.B.; Ellis, G.K.; Petra, P.H.; Peterson, L.M.; Schubert, E.K.; Dunnwald, L.K.; et al. Quantitative fluoroestradiol positron emission tomography imaging predicts response to endocrine treatment in breast cancer. J. Clin. Oncol. 2006, 24, 2793–2799. [Google Scholar] [CrossRef]



| Author | Methods | Site | Previous Treatments | ERα (%) | PgR (%) | HR Status (%) |
|---|---|---|---|---|---|---|
| Hawkins R.A. et al. (1990) [111] | Prospective N = 62 pts | Surgical sample | Various | n.s | - | - |
| Miller W.R. et al. (2003) [112] | Retrospective N = 48 pts | Surgical sample | Letrozole vs tamoxifen | 48 vs 83 | 87 vs 60 | - |
| Tacca O. et al. (2007) [104] | Retrospective N = 459 pts | Surgical sample | Various | - | - | 23 |
| Hirata T. et al. (2009) [99] | Retrospective N = 368 pts | Surgical sample | Various | 14.9 | 29.1 | 16 |
| Aitken S.J.et al. (2010) [93] | Retrospective N = 385 pts | Synchronous LN | None | 28.3 | 23.4 | - |
| Van de VenS. et al. (2011) [103] | Metanalysis N = 32 studies | Surgical sample | Various | 2.5–17 | 5.9–19 | 8–33 |
| Jensen J.D. et al. (2012) [92] | Prospective N = 128 pts | Synchronous LN | None | 4 | - | - |
| Chen S. et al. (2012) [105] | Retrospective N = 224 pts | Surgical sample | CEF (29.0%), NE (35.7%), PC (29.9%), or TE (5.4%) 1 | 16 (+ to −) | 22.2 (+ to −) | 15.2 (+ to −) |
| Jin X. et al. (2015) [106] | Prospective N = 423 pts | Surgical sample | Various | - | - | 18.4 |
| Lim S.K. et al. (2016) [107] | Retrospective N = 322 pts | Surgical sample | Various | 40.7 | 62.1 | 17.9 |
| Ding Y. et al. (2020) [108] | Retrospective N = 482 pts | Surgical sample | Anthracyclines and/or taxanes | 10.4 | 17 | 27.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zattarin, E.; Leporati, R.; Ligorio, F.; Lobefaro, R.; Vingiani, A.; Pruneri, G.; Vernieri, C. Hormone Receptor Loss in Breast Cancer: Molecular Mechanisms, Clinical Settings, and Therapeutic Implications. Cells 2020, 9, 2644. https://doi.org/10.3390/cells9122644
Zattarin E, Leporati R, Ligorio F, Lobefaro R, Vingiani A, Pruneri G, Vernieri C. Hormone Receptor Loss in Breast Cancer: Molecular Mechanisms, Clinical Settings, and Therapeutic Implications. Cells. 2020; 9(12):2644. https://doi.org/10.3390/cells9122644
Chicago/Turabian StyleZattarin, Emma, Rita Leporati, Francesca Ligorio, Riccardo Lobefaro, Andrea Vingiani, Giancarlo Pruneri, and Claudio Vernieri. 2020. "Hormone Receptor Loss in Breast Cancer: Molecular Mechanisms, Clinical Settings, and Therapeutic Implications" Cells 9, no. 12: 2644. https://doi.org/10.3390/cells9122644
APA StyleZattarin, E., Leporati, R., Ligorio, F., Lobefaro, R., Vingiani, A., Pruneri, G., & Vernieri, C. (2020). Hormone Receptor Loss in Breast Cancer: Molecular Mechanisms, Clinical Settings, and Therapeutic Implications. Cells, 9(12), 2644. https://doi.org/10.3390/cells9122644