Notoginsenoside R1 Ameliorates Diabetic Retinopathy through PINK1-Dependent Activation of Mitophagy

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Reagents and Methods
2.1. Materials
2.2. Cell Culture and Drug
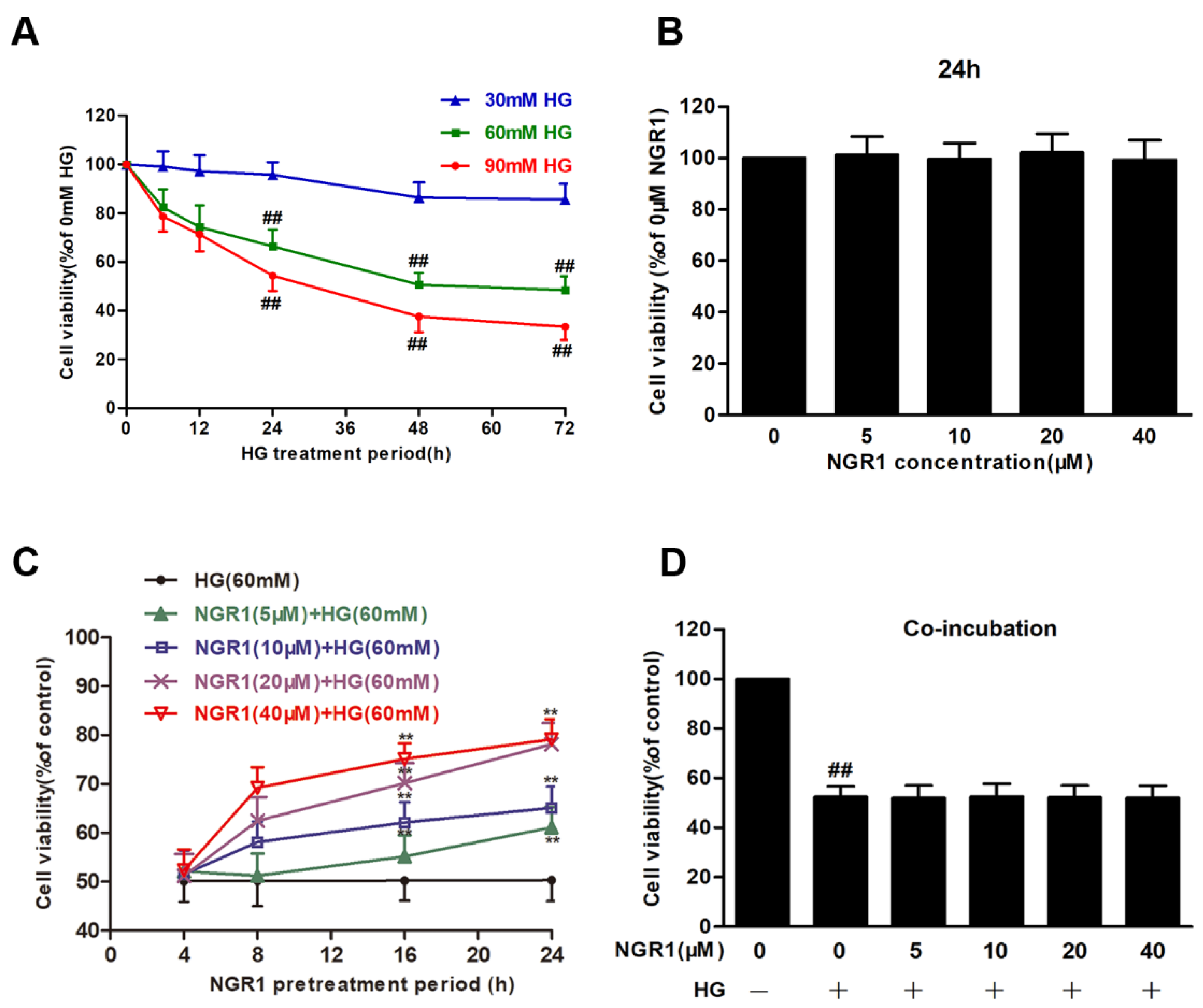
2.3. Analysis of Cell Viability
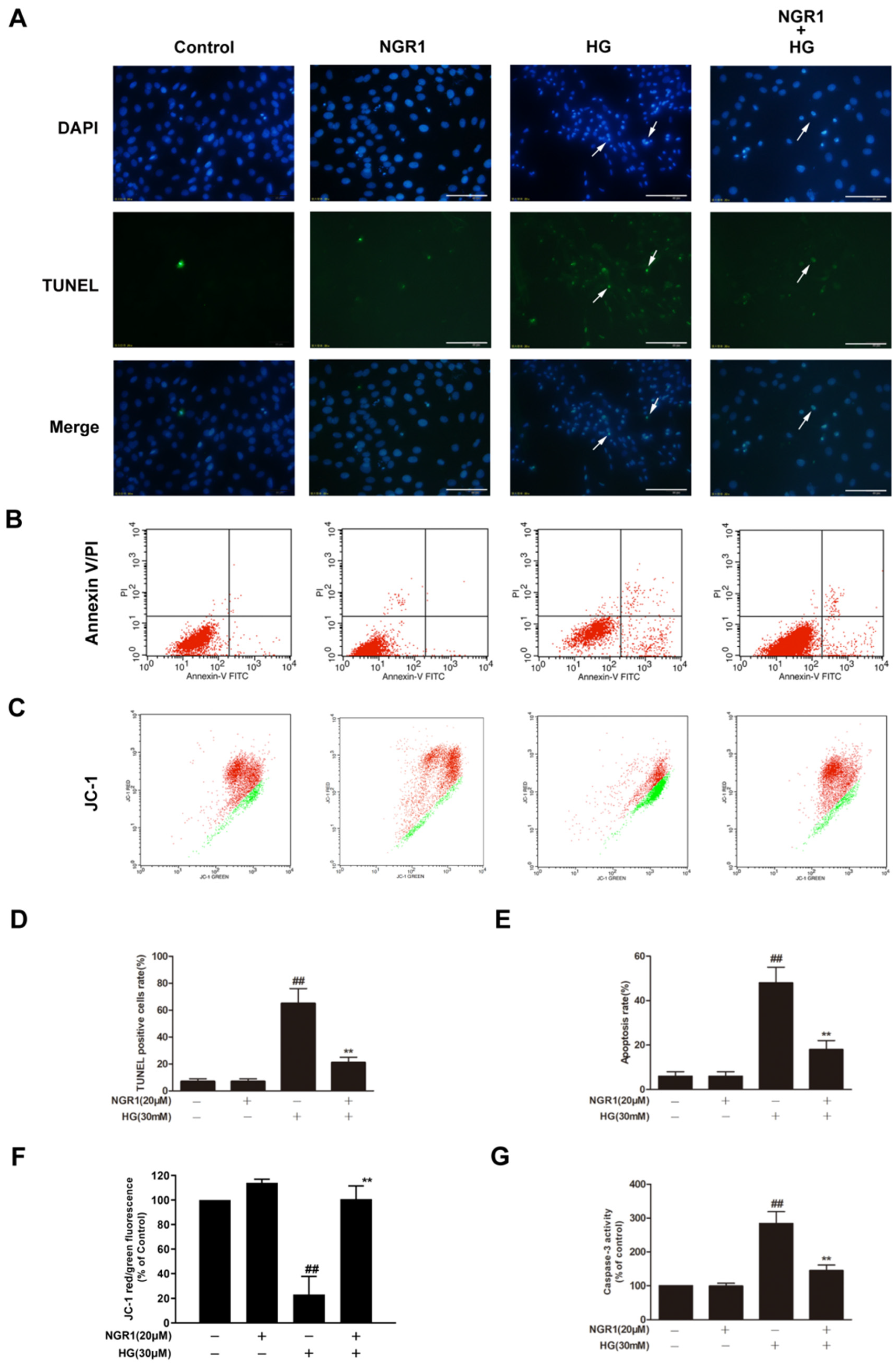
2.4. Measurement of Mitochondrial Membrane Potential
2.5. Detection of the Apoptosis Rate
2.6. Evaluation of DNA Fragmentation
2.7. Detection of Caspase-3 Activity
2.8. Transient Transfection
2.9. Animals
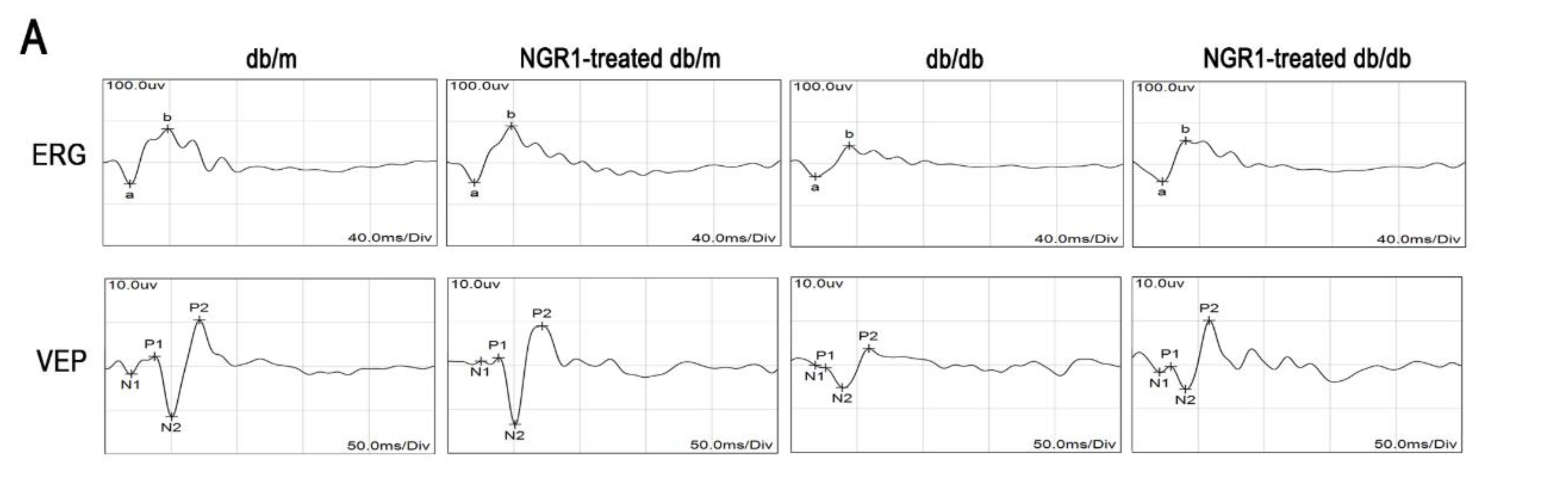
2.10. Electroretinogram and Visual Evoked Potential
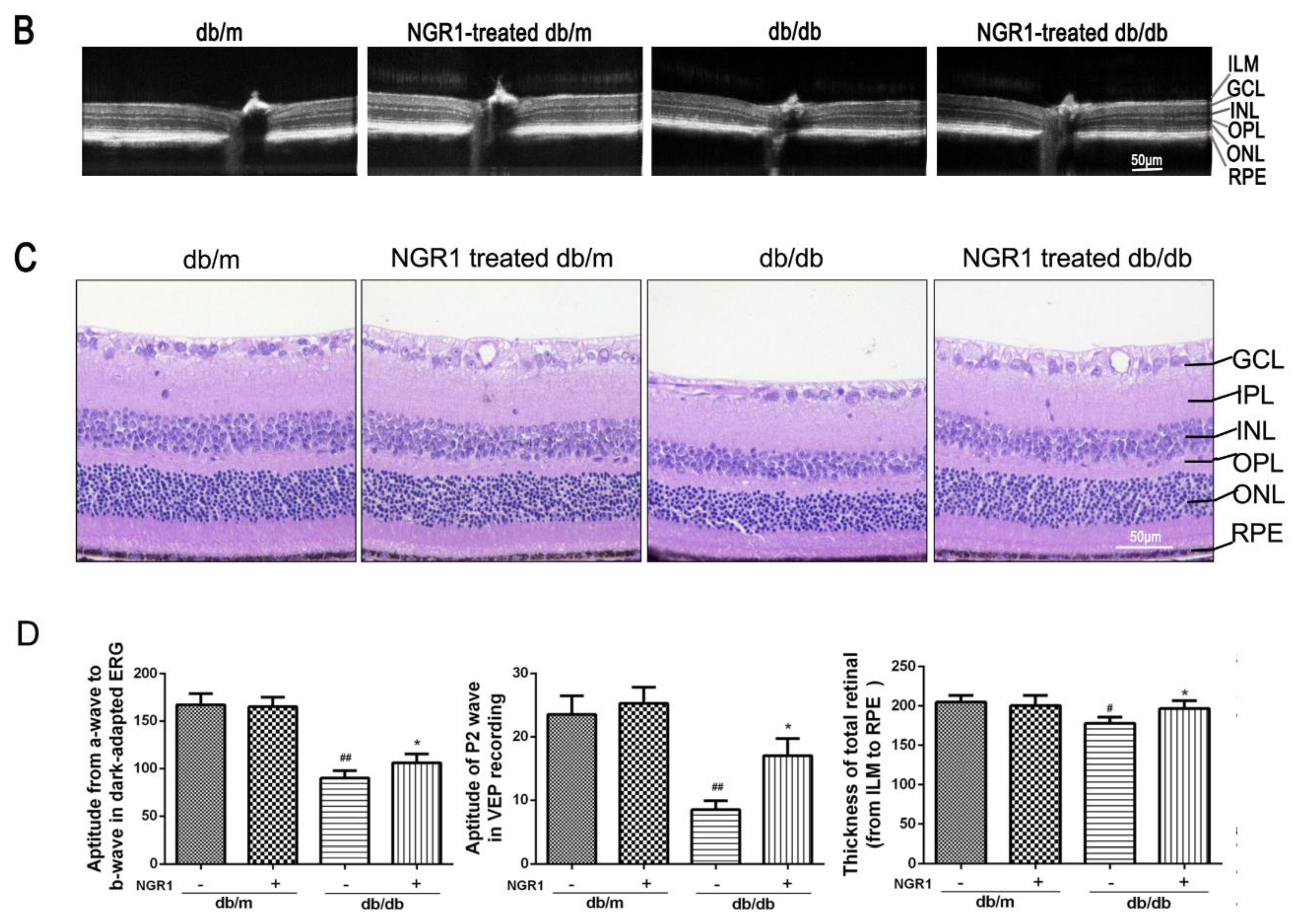
2.11. Optical Coherence Tomography
2.12. Transmission Electron Microscopy Analysis
2.13. HE Staining
2.14. Determination of Mitochondrial ROS
2.15. ELISA
2.16. Western Blotting
2.17. Statistical Analysis
3. Results
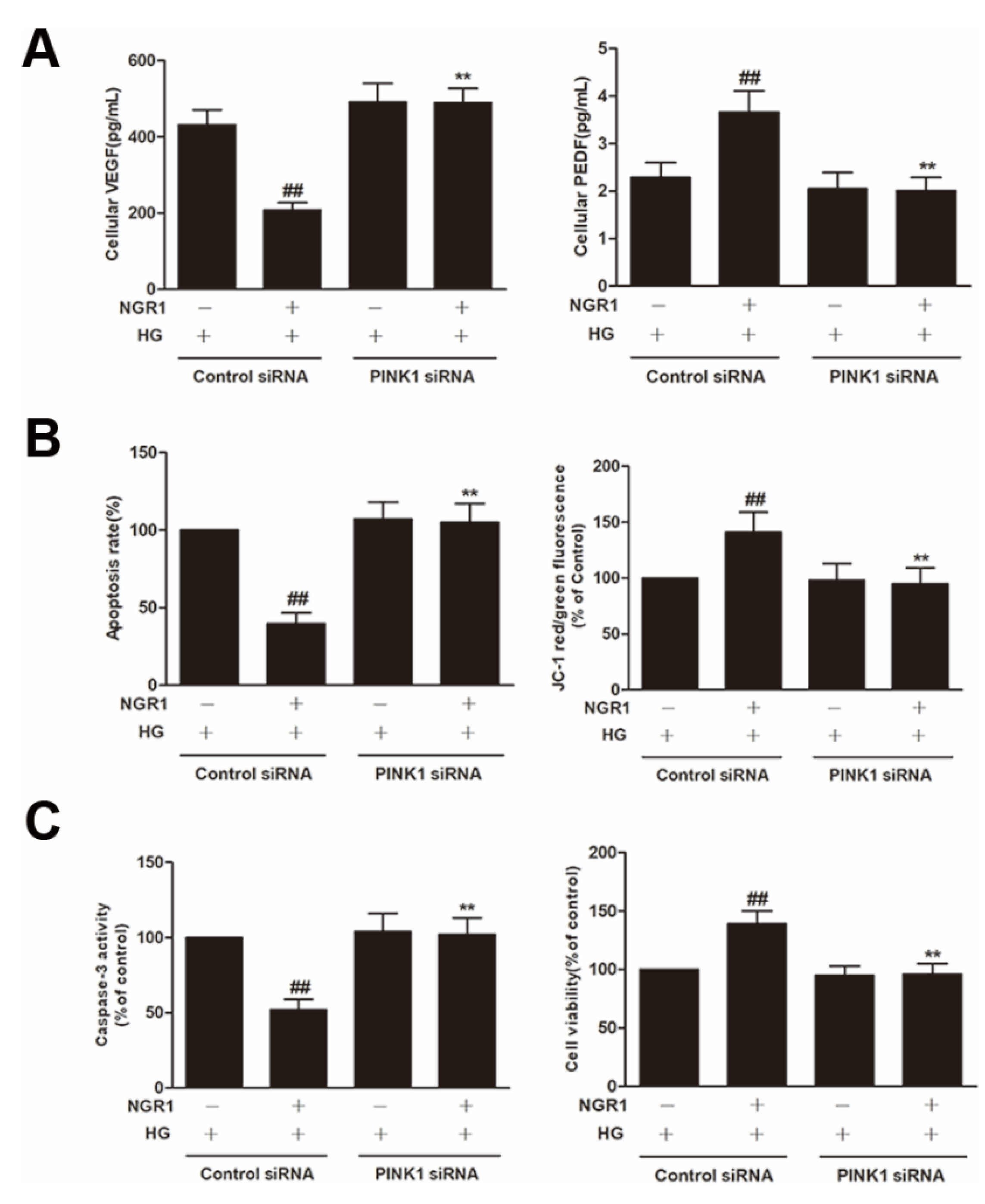
3.1. NGR1 Pre-Treatment Exerted a Positive Effect on HG-Induced Cell Death in rMC-1 Cells
3.2. NGR1 Inhibited HG-Induced Apoptosis in rMC-1 Cells
3.3. NGR1 Significantly Attenuated DR in db/db Mice
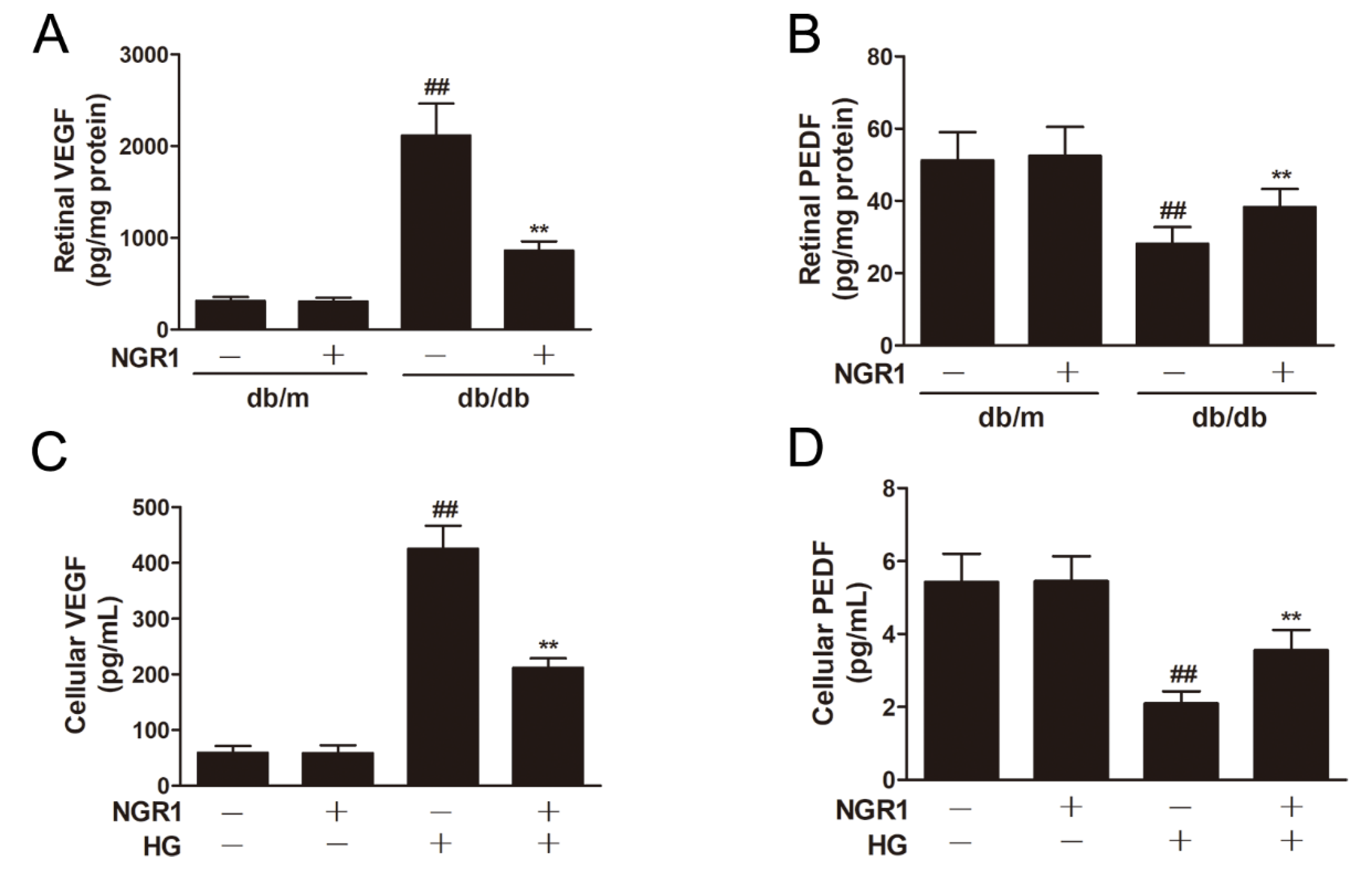
3.4. NGR1 Reversed the Imbalance between VEGF and PEDF In Vivo and In Vitro
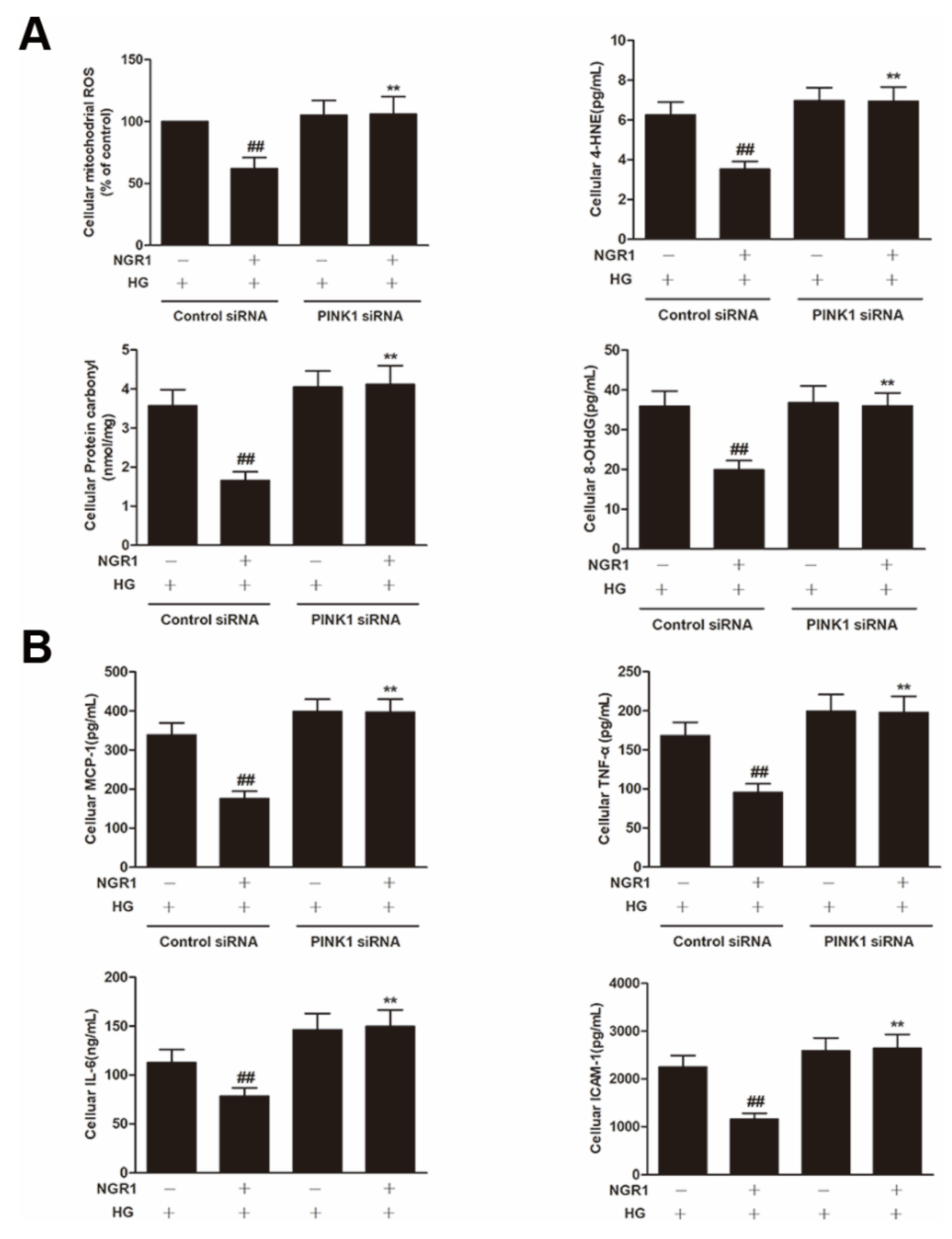
3.5. NGR1 Inhibited Oxidative Stress In Vivo and In Vitro
3.6. NGR1 Inhibited Inflammation In Vivo and In Vitro
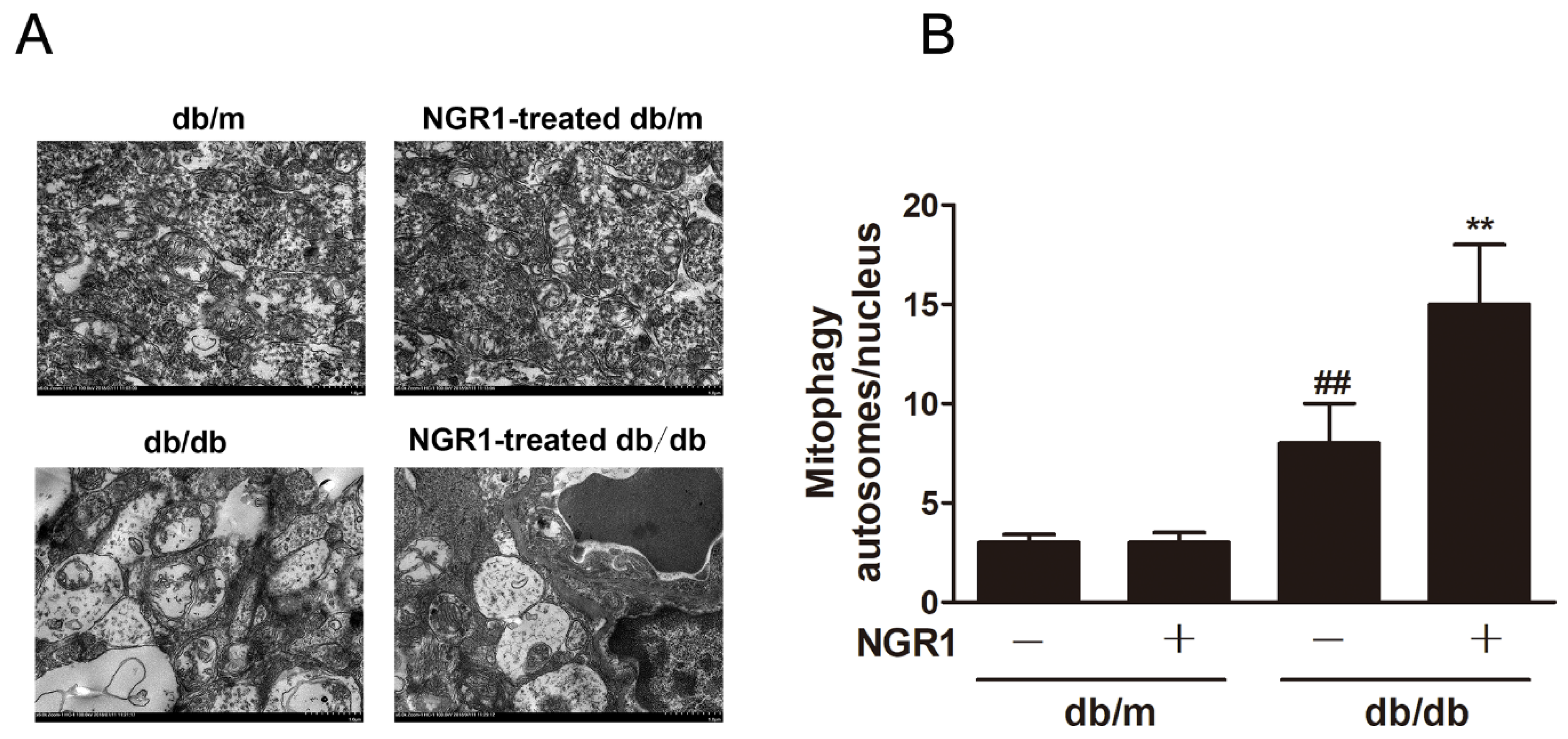
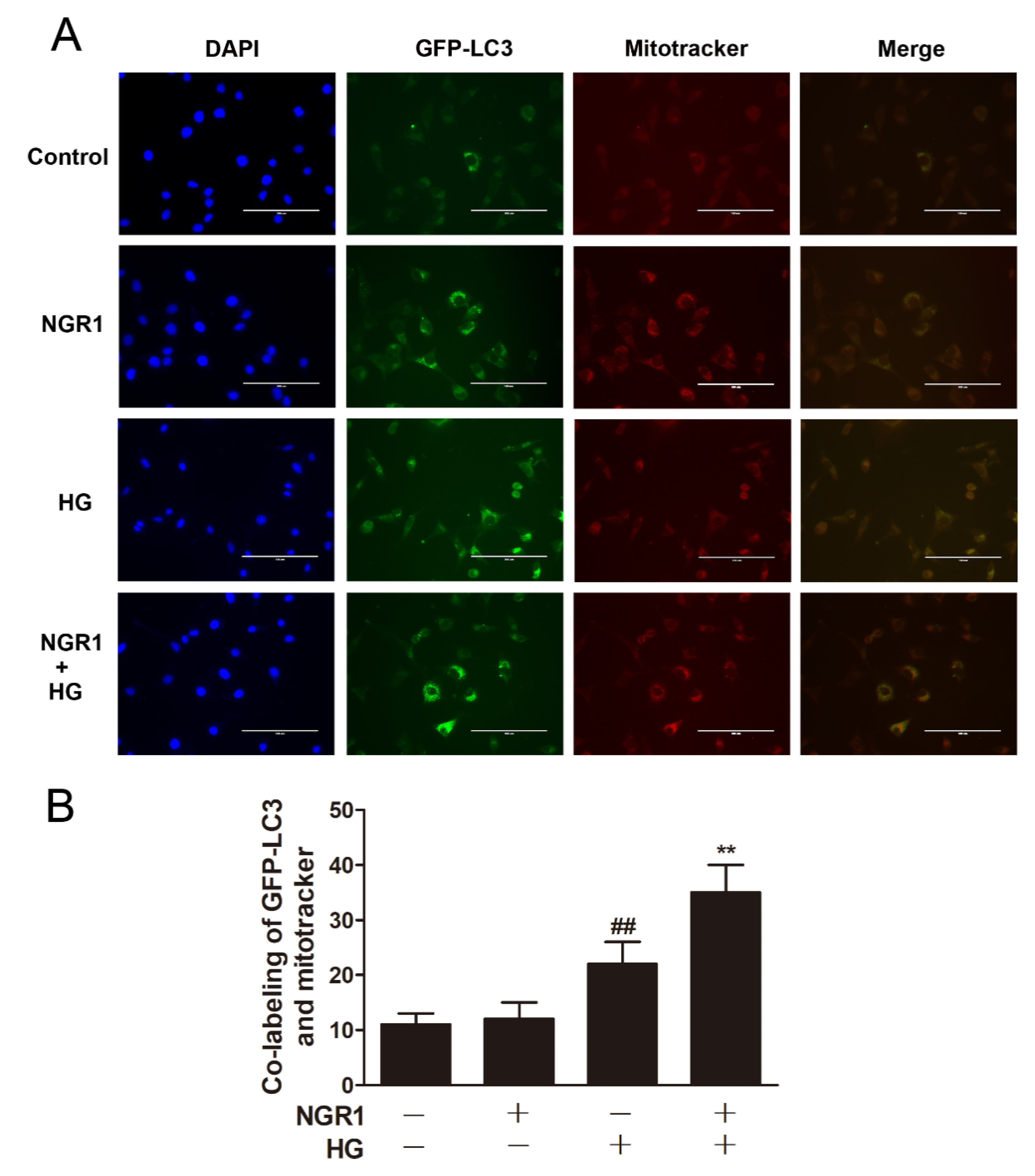
3.7. NGR1 Enhanced Mitophagy In Vivo and In Vitro
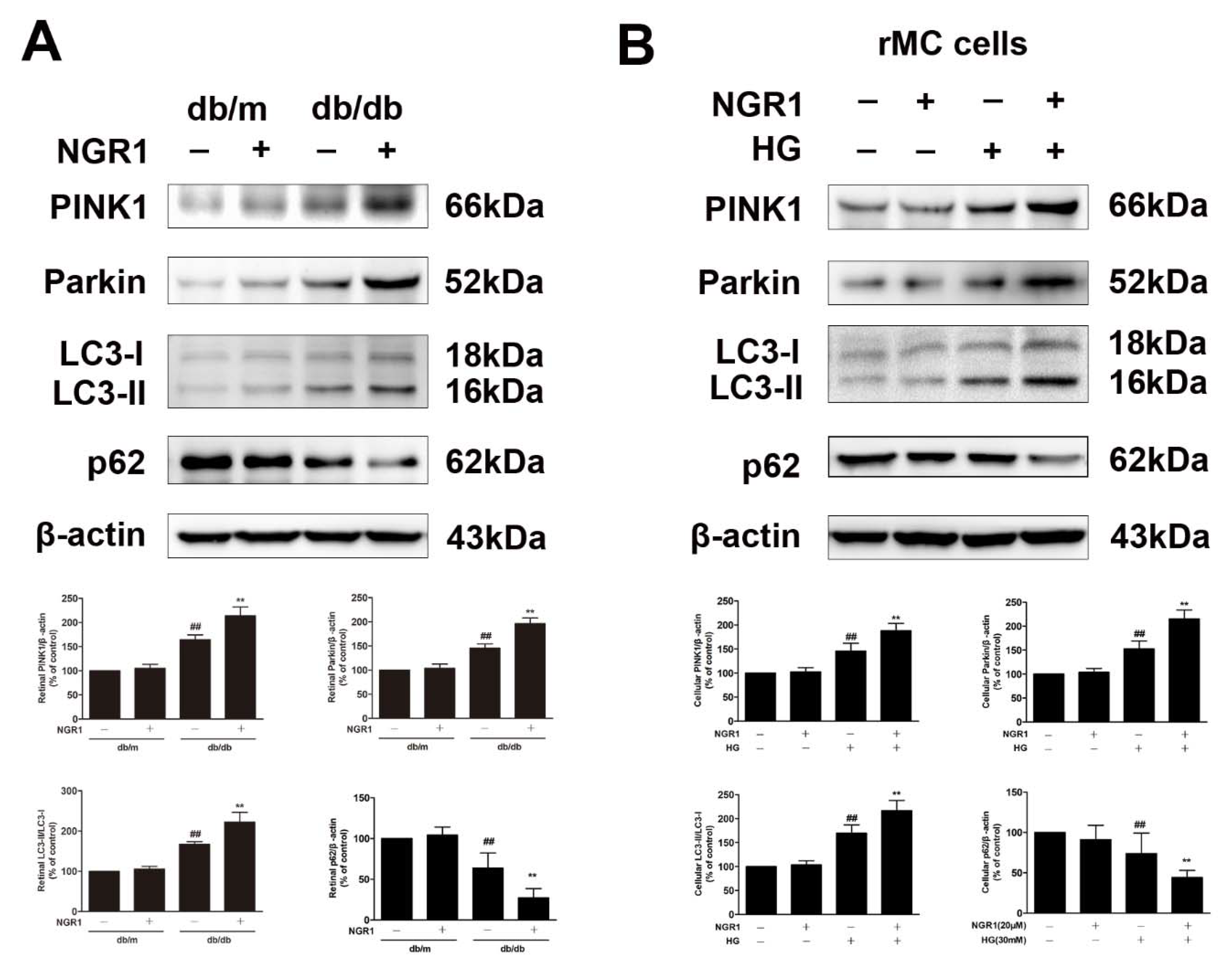
3.8. NGR1 Enhanced Mitophagy by Activating PINK1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gulshan, V.; Peng, L.; Coram, M.; Stumpe, M.C.; Wu, D.; Narayanaswamy, A.; Venugopalan, S.; Widner, K.; Madams, T.; Cuadros, J. Development and Validation of a Deep Learning Algorithm for Detection of Diabetic Retinopathy in Retinal Fundus Photographs. JAMA 2016, 316, 2402. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Kern, T.S.; Song, B.; Stuebe, C. Mechanistic Insights into Pathological Changes in the Diabetic Retina: Implications for Targeting Diabetic Retinopathy. Am. J. Pathol. 2016, 187, 9. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, B.A.; Feenstra, D.J.; Mohr, S. Müller cells and diabetic retinopathy. Vis. Res. 2017, 139, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Abcouwer, S.F. Müller Cell–Microglia Cross Talk Drives Neuroinflammation in Diabetic Retinopathy. Diabetes 2017, 66, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Xin, X.; Jee, K.; Babapoorfarrokhran, S.; Kashiwabuchi, F.; Ma, T.; Bhutto, I.; Hassan, S.J.; Daoud, Y.; Baranano, D. VEGF Secreted by Hypoxic Müller Cells Induces MMP-2 Expression and Activity in Endothelial Cells to Promote Retinal Neovascularization in Proliferative Diabetic Retinopathy. Diabetes 2013, 62, 3863–3873. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.M.; Yafai, Y.; Wiedemann, P.; Kuhrt, H.; Wang, Y.S.; Reichenbach, A.; Eichler, W. Hypoxia-induced upregulation of pigment epithelium-derived factor by retinal glial (Müller) cells. J. Neurosci. Res. 2012, 90, 257. [Google Scholar] [CrossRef] [PubMed]
- Kang, A.Y.; Park, S.K.; Park, S.Y.; Lee, H.J.; Han, Y.; Lee, S.R.; Suh, S.H.; Kim, D.K.; Park, M.K. Therapeutic target achievement in type 2 diabetic patients after hyperglycemia, hypertension, dyslipidemia management. Diabetes Metab. J. 2011, 35, 264. [Google Scholar] [CrossRef] [PubMed]
- Olsen, T.W. Anti-VEGF Pharmacotherapy as an Alternative to Panretinal Laser Photocoagulation for Proliferative Diabetic Retinopathy. JAMA 2015, 314, 2135–2136. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kotwani, A. Chinese herbal drugs for the treatment of diabetic retinopathy. J. Pharm. Pharmacol. 2017, 69, 223. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Guo, Y.; Li, X.; Li, X.; Li, Z.; Xue, M.; Ou, Z.; Liu, M.; Yang, M.; Liu, S.; et al. An Aqueous Extract of Radix Astragali, Angelica sinensis, and Panax notoginseng Is Effective in Preventing Diabetic Retinopathy. Evid.-Based Complement. Altern. Med. 2013, 2013, 578165. [Google Scholar] [CrossRef] [PubMed]
- Fengmei, L.; Lie, W.; Jiaxing, T.; Ming, J.; Shuiping, Z.; Min, Z.; Lijuan, W.; Yanlin, Z.; Yuliang, W.; Mingchang, Z. The effectiveness and safety of a danshen-containing Chinese herbal medicine for diabetic retinopathy: a randomized, double-blind, placebo-controlled multicenter clinical trial. J. Ethnopharmacol. 2015, 164, 71–77. [Google Scholar]
- Zhang, H.W.; Zhang, H.; Grant, S.J.; Wan, X.; Li, G. Single herbal medicine for diabetic retinopathy. Cochrane Database Syst. Rev. 2018, 12, Cd007939. [Google Scholar] [CrossRef] [PubMed]
- Tien, T.; Zhang, J.; Muto, T.; Kim, D.; Sarthy, V.P.; Roy, S. High Glucose Induces Mitochondrial Dysfunction in Retinal Müller Cells: Implications for Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2915–2921. [Google Scholar] [CrossRef] [PubMed]
- Toda, C.; Kim, J.D.; Impellizzeri, D.; Cuzzocrea, S.; Liu, Z.W.; Diano, S. UCP2 Regulates Mitochondrial Fission and Ventromedial Nucleus Control of Glucose Responsiveness. Cell 2016, 164, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Dehdashtian, E.; Mehrzadi, S.; Yousefi, B.; Hosseinzadeh, A.; Reiter, R.J.; Safa, M.; Ghaznavi, H.; Naseripour, M. Diabetic retinopathy pathogenesis and the ameliorating effects of melatonin; involvement of autophagy, inflammation and oxidative stress. Life Sci. 2018, 193, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Zhenyukh, O.; Civantos, E.; Ruizortega, M.; Sánchez, M.S.; Vázquez, C.; Peiró, C.; Egido, J.; Mas, S. High concentration of branched-chain amino acids promotes oxidative stress, inflammation and migration of human peripheral blood mononuclear cells via mTORC1 activation. Free Radic. Biol. Med. 2017, 104, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M. Therapeutic targets for altering mitochondrial dysfunction associated with diabetic retinopathy. Expert Opin. Targets 2018, 22, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Masser, D.R.; Otalora, L.; Clark, N.W.; Kinter, M.T.; Elliott, M.H.; Freeman, W.M. Functional changes in the neural retina occur in the absence of mitochondrial dysfunction in a rodent model of diabetic retinopathy. J. Neurochem. 2017, 143, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.D.; Tewari, S.; Goldberg, A.F.X.; Kowluru, A.R. Mitochondria biogenesis and the development of diabetic retinopathy. Free Radic. Biol. Med. 2011, 51, 1849. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Devi, T.S.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP regulates mitophagy in retinal Müller cells under high-glucose conditions: Implications for diabetic retinopathy. Cell Death Dis. 2017, 8, e2777. [Google Scholar] [CrossRef] [PubMed]
- De, I.C.; Costa, A.C.; Celardo, I.; Tufi, R.; Dinsdale, D.; Loh, S.H.; Martins, L.M. Drosophila ref(2)P is required for the parkin-mediated suppression of mitochondrial dysfunction in pink1 mutants. Cell Death Dis. 2013, 4, e873. [Google Scholar]
- Ashrafi, G.; Schlehe, J.S.; Lavoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 2014, 206, 655. [Google Scholar] [CrossRef] [PubMed]
- Sabanayagam, C. Epidemiology of diabetic retinopathy, diabetic macular edema and vision loss due to diabetes. Diabetes Res. Clin. Pract. 2016, 120, S14. [Google Scholar] [CrossRef]
- Tout, S.; Chan-Ling, T.; Holl?Nder, H.; Stone, J. The role of Müller cells in the formation of the blood-retinal barrier. Neuroscience 1993, 55, 291–301. [Google Scholar] [CrossRef]
- Abukawa, H.; Tomi, M.; Kiyokawa, J.; Hori, S.; Kondo, T.; Terasaki, T.; Hosoya, K. Modulation of retinal capillary endothelial cells by Müller glial cell-derived factors. Mol. Vis. 2009, 15, 451–457. [Google Scholar] [PubMed]
- Zhong, Y.; Li, J.; Chen, Y.; Wang, J.J.; Ratan, R.; Zhang, S.X. Activation of Endoplasmic Reticulum Stress by Hyperglycemia Is Essential for Müller Cell–Derived Inflammatory Cytokine Production in Diabetes. Diabetes 2012, 61, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.W.; Antonetti, D.A.; Barber, A.J.; LaNoue, K.F.; Levison, S.W. Diabetic Retinopathy: More than Meets the Eye. Surv. Ophthalmol. 2002, 47, S253–S262. [Google Scholar] [CrossRef]
- Zhou, T.; Che, D.; Lan, Y.; Fang, Z.; Xie, J.; Gong, H.; Li, C.; Feng, J.; Hong, H.; Qi, W.; et al. Mesenchymal marker expression is elevated in Muller cells exposed to high glucose and in animal models of diabetic retinopathy. Oncotarget 2017, 8, 4582–4594. [Google Scholar] [PubMed]
- Cheung, A.K.; Fung, M.K.; Lo, A.C.; Lam, T.T.; So, K.F.; Chung, S.S.; Chung, S.K. Aldose reductase deficiency prevents diabetes-induced blood-retinal barrier breakdown, apoptosis, and glial reactivation in the retina of db/db mice. Diabetes 2005, 54, 3119–3125. [Google Scholar] [CrossRef] [PubMed]
- Na, Z.; Meng, L.; Zhang, J.; Yue, C.; Li, J.; An, J.; Zhang, J.; Pathology, D.O.; University, N.M. The activation and apoptosis of astrocytes in the hippocampus of the diabetic model rats. Chin. J. Neuroanat. 2016, 32, 13–17. [Google Scholar]
- Bringmann, A.; Reichenbach, A. Role of Muller cells in retinal degenerations. Front. Biosci. 2001, 6, E72–E92. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, A.; Bringmann, A. Müller Cells in the Healthy and Diseased Retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar]
- Xiao-Lu, L.I.; Ya-Ling, M.A. Dynamic expressions of VEGF and PEDF in retina of rats with diabetic retinopathy. Recent Adv. Ophthalmol. 2013, 26, 573–576. [Google Scholar]
- Yang, L.; Cai-Hui, H.E.; Liu, W.X.; Guo, S.Q.; Liu, D.W.; Epidemiology, D.O.; University, H.M. Relationship between polymorphism of VEGF and PEDF with proliferative diabetic retinopathy. Chronic Pathematol. J. 2013, 14, 8–12. [Google Scholar]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2011, 331, 456. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Takamatsu, H.; Liu, S.; Kataoka, K.; Huh, N.H.; Sakaguchi, M. NRF2 Regulates PINK1 Expression under Oxidative Stress Conditions. PLoS ONE 2015, 10, e0142438. [Google Scholar] [CrossRef] [PubMed]
- Soutar, M.P.M.; Kempthorne, L.; Miyakawa, S.; Annuario, E.; Melandri, D.; Harley, J.; O’Sullivan, G.A.; Wray, S.; Hancock, D.C.; Cookson, M.R.; et al. AKT signalling selectively regulates PINK1 mitophagy in SHSY5Y cells and human iPSC-derived neurons. Sci. Rep. 2018, 8, 8855. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Meng, X.; Luo, Y.; Wu, Y.; Ye, T.; Zhou, P.; Ding, S.; Wang, M.; Lu, S.; Zhu, L.; et al. Notoginsenoside R1 ameliorates diabetic encephalopathy by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Oncotarget 2018, 9, 9344–9363. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, P.; Xie, W.; Meng, X.; Zhai, Y.; Dong, X.; Zhang, X.; Sun, G.; Sun, X. Notoginsenoside R1 Ameliorates Diabetic Retinopathy through PINK1-Dependent Activation of Mitophagy. Cells 2019, 8, 213. https://doi.org/10.3390/cells8030213
Zhou P, Xie W, Meng X, Zhai Y, Dong X, Zhang X, Sun G, Sun X. Notoginsenoside R1 Ameliorates Diabetic Retinopathy through PINK1-Dependent Activation of Mitophagy. Cells. 2019; 8(3):213. https://doi.org/10.3390/cells8030213
Chicago/Turabian StyleZhou, Ping, Weijie Xie, Xiangbao Meng, Yadong Zhai, Xi Dong, Xuelian Zhang, Guibo Sun, and Xiaobo Sun. 2019. "Notoginsenoside R1 Ameliorates Diabetic Retinopathy through PINK1-Dependent Activation of Mitophagy" Cells 8, no. 3: 213. https://doi.org/10.3390/cells8030213
APA StyleZhou, P., Xie, W., Meng, X., Zhai, Y., Dong, X., Zhang, X., Sun, G., & Sun, X. (2019). Notoginsenoside R1 Ameliorates Diabetic Retinopathy through PINK1-Dependent Activation of Mitophagy. Cells, 8(3), 213. https://doi.org/10.3390/cells8030213