Perilipin 5 Protects against Cellular Oxidative Stress by Enhancing Mitochondrial Function in HepG2 Cells


Abstract
1. Introduction
2. Results
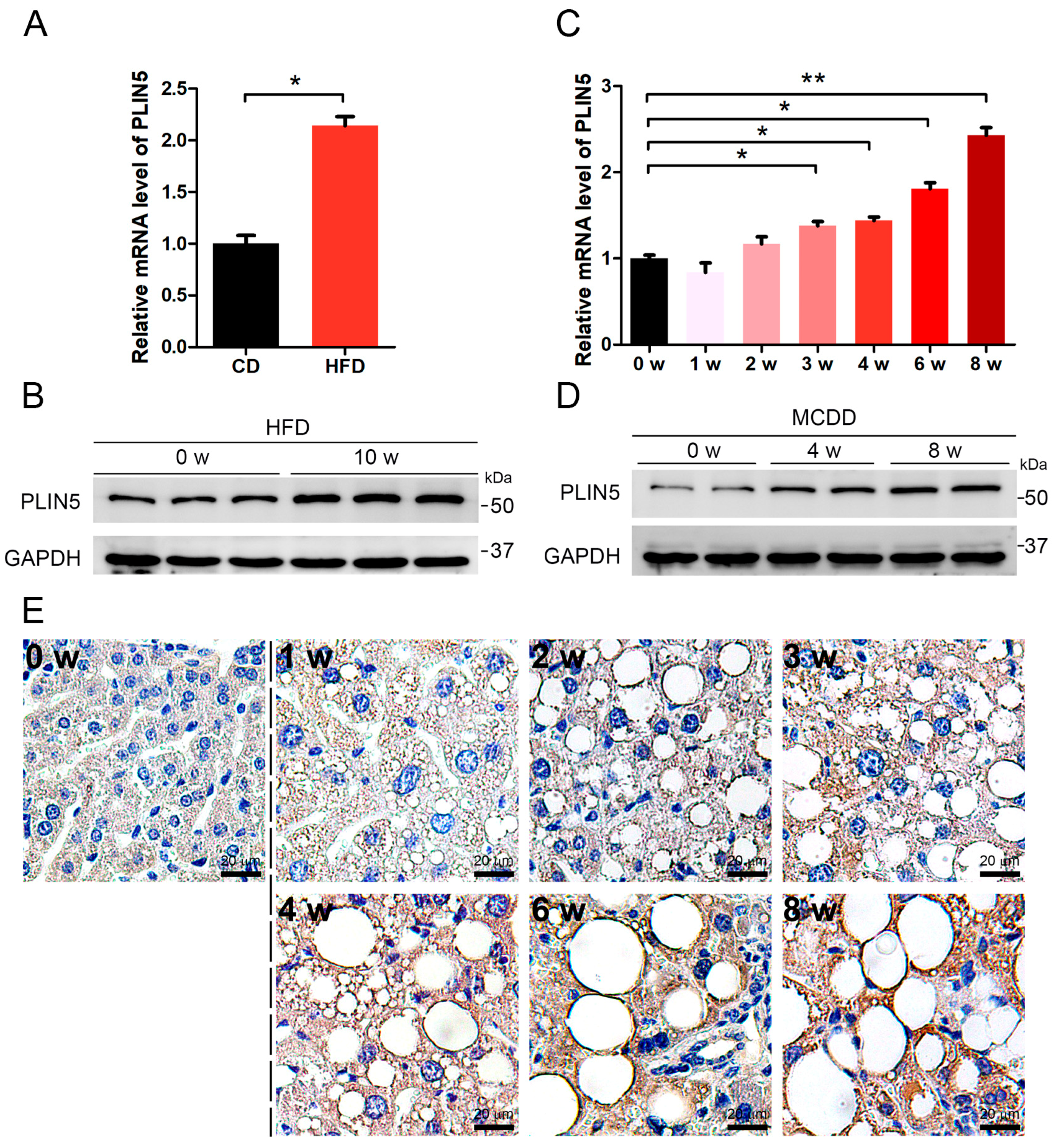
2.1. PLIN5 Was Up-Regulated in Liver Tissues of NAFLD Mice
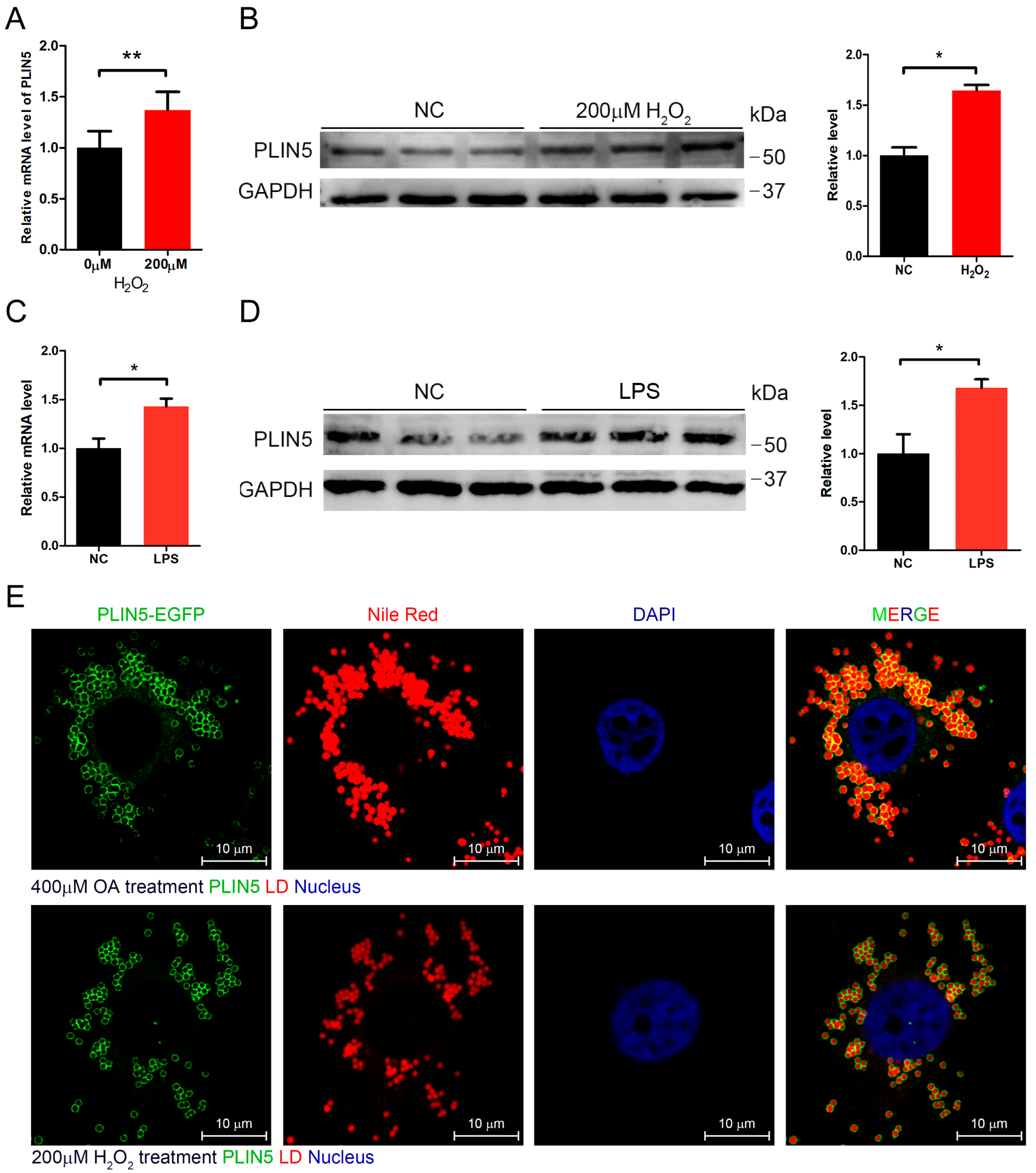
2.2. Hydrogen Peroxide or LPS Treatment Enhanced Expression of PLIN5
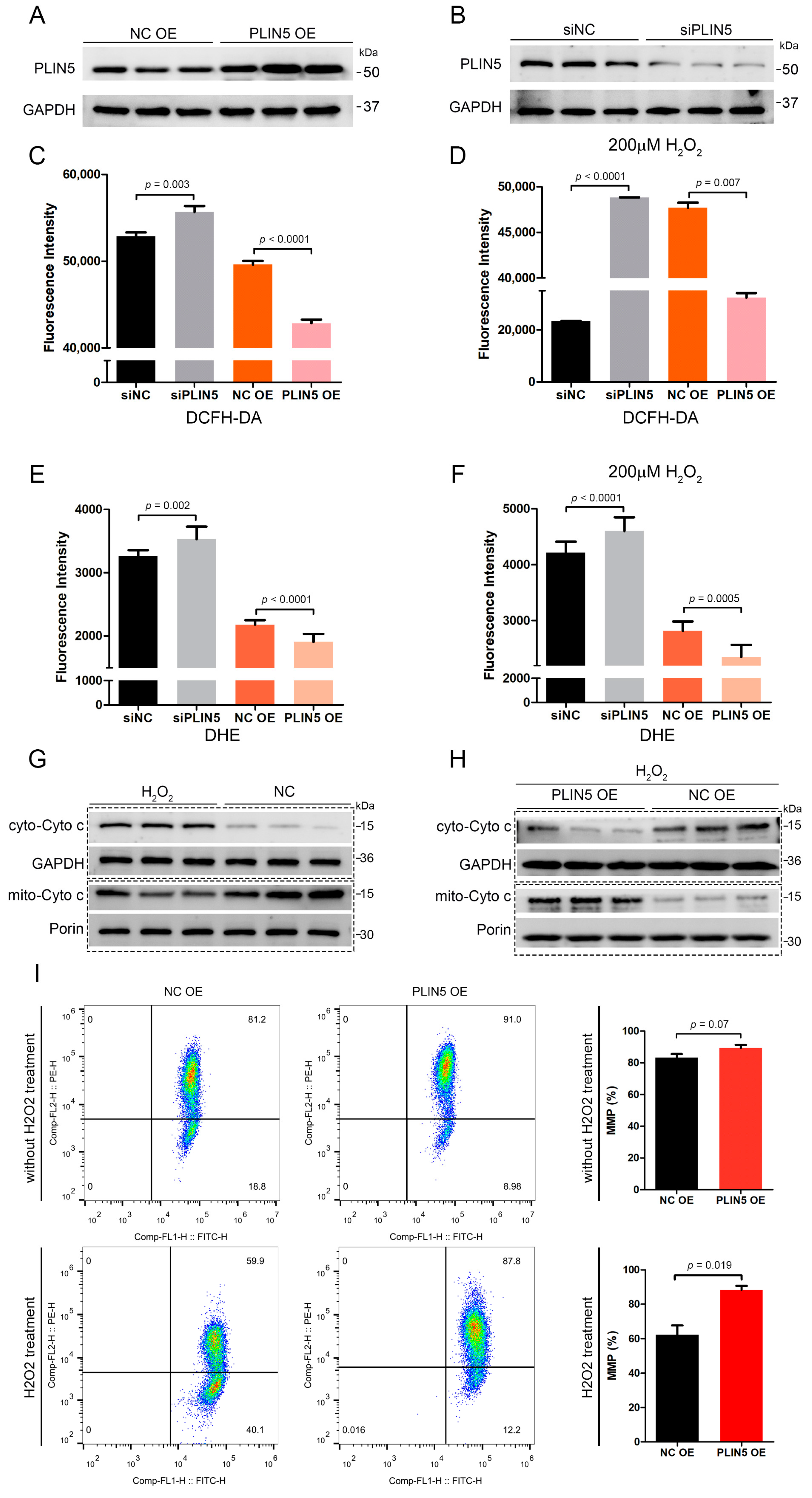
2.3. PLIN5 Regulated Cellular ROS Levels
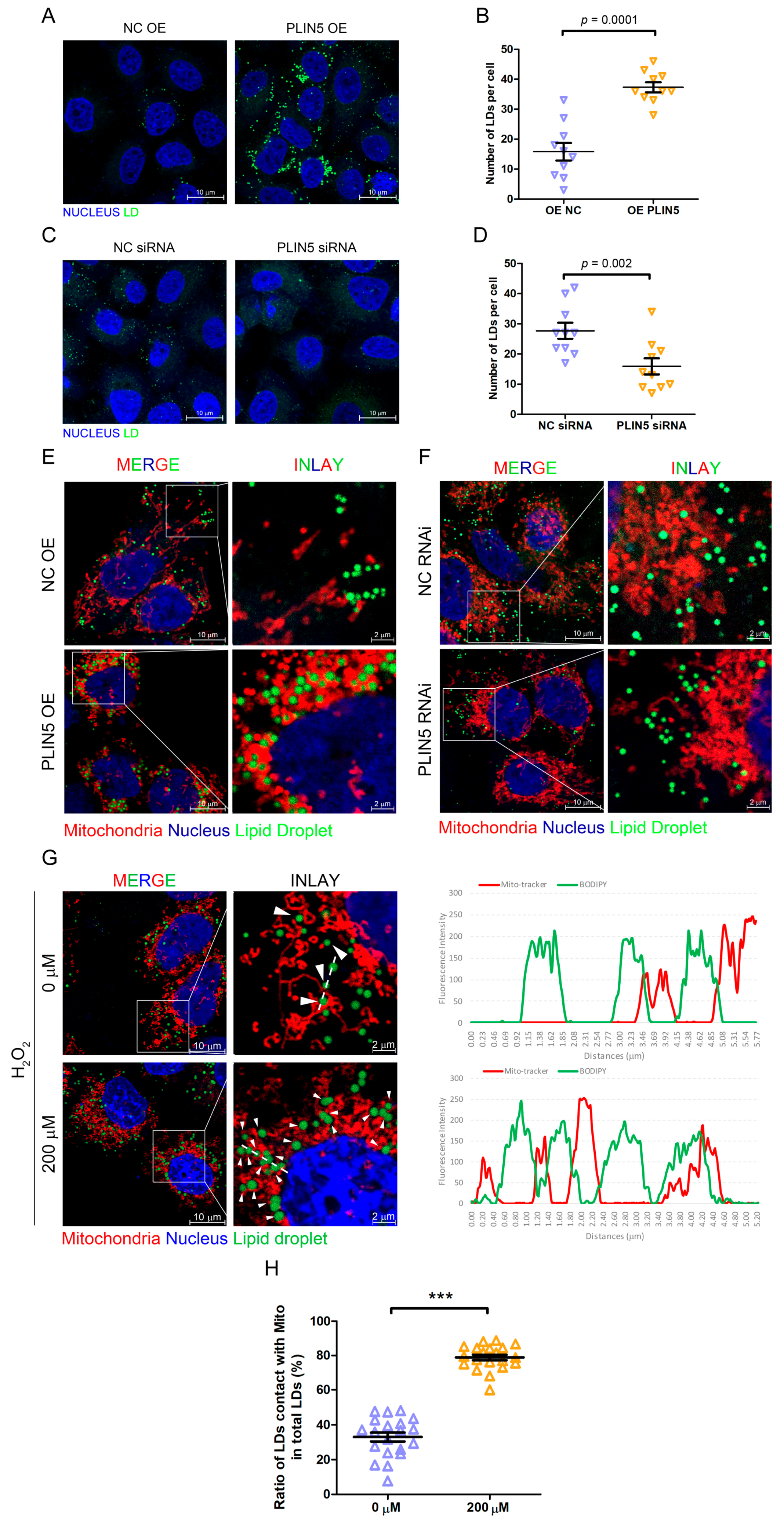
2.4. PLIN5 Promoted LD Formation and Contact with Mitochondria
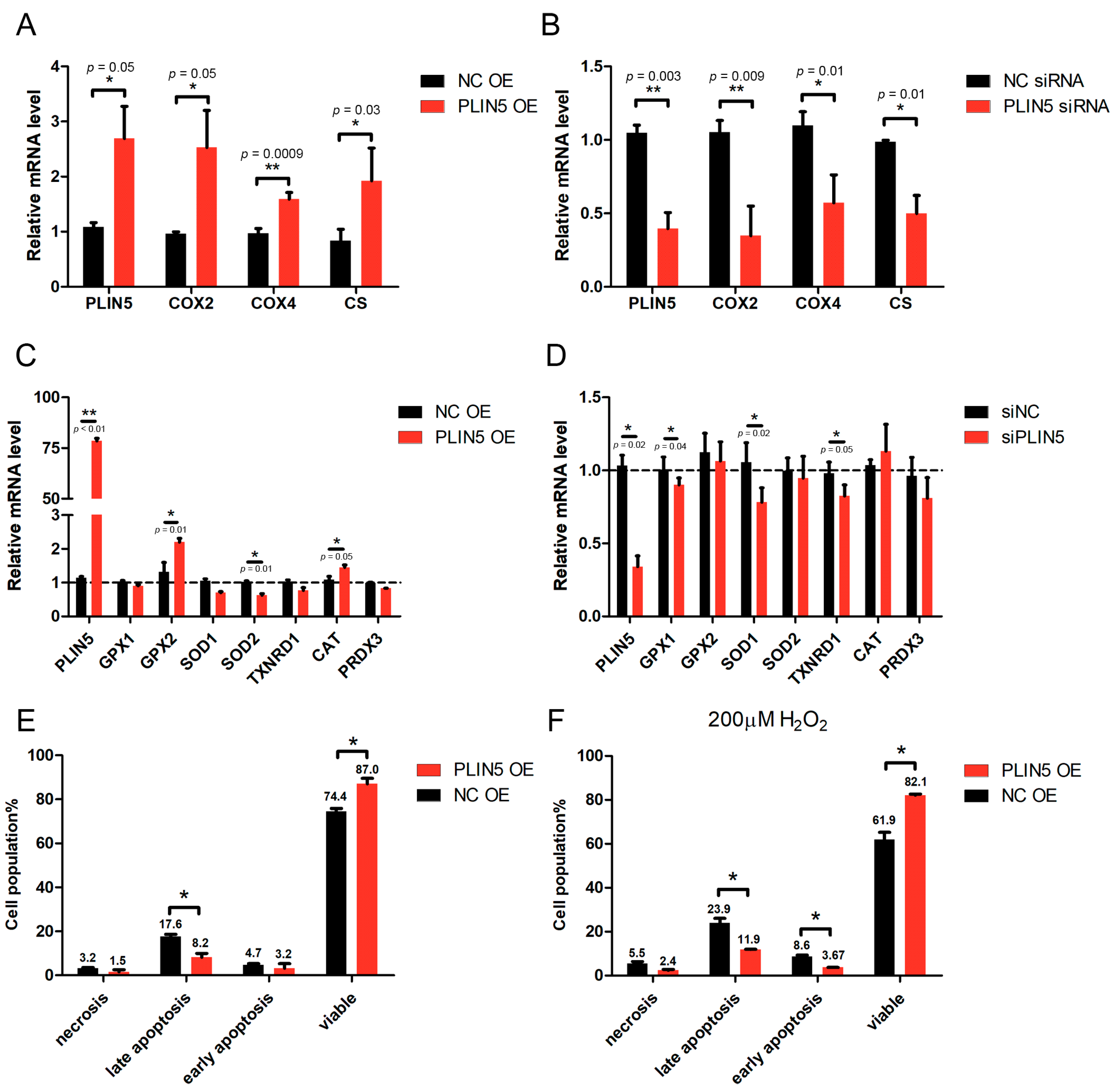
2.5. PLIN5 Regulated the Expression Levels of Mitochondrial Function-Related Genes
2.6. PLIN5 Reduced Apoptotic Rates of HepG2 Cells
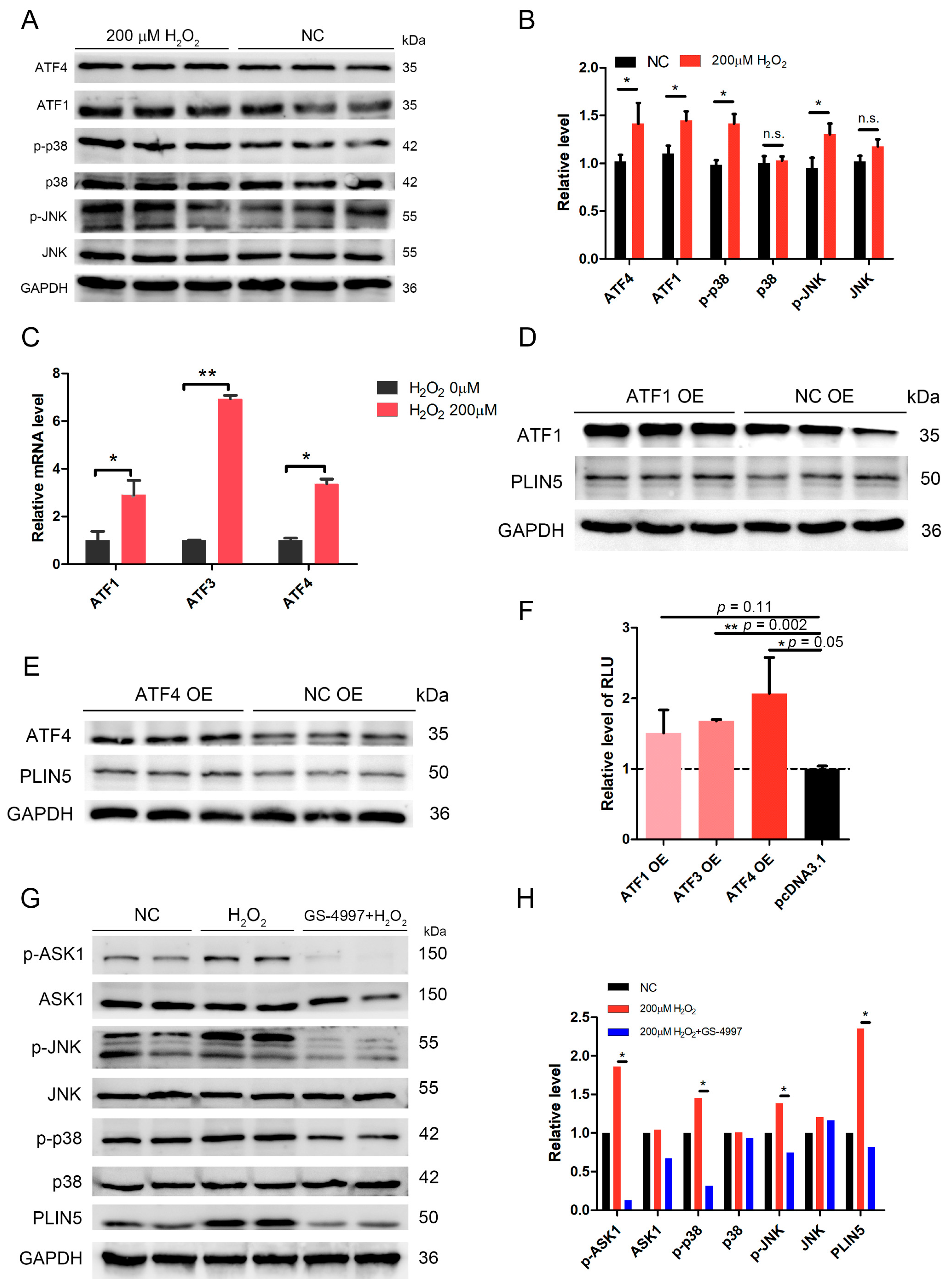
2.7. The Expression of PLIN5 Was Regulated by the JNK-p38-ATF Pathway
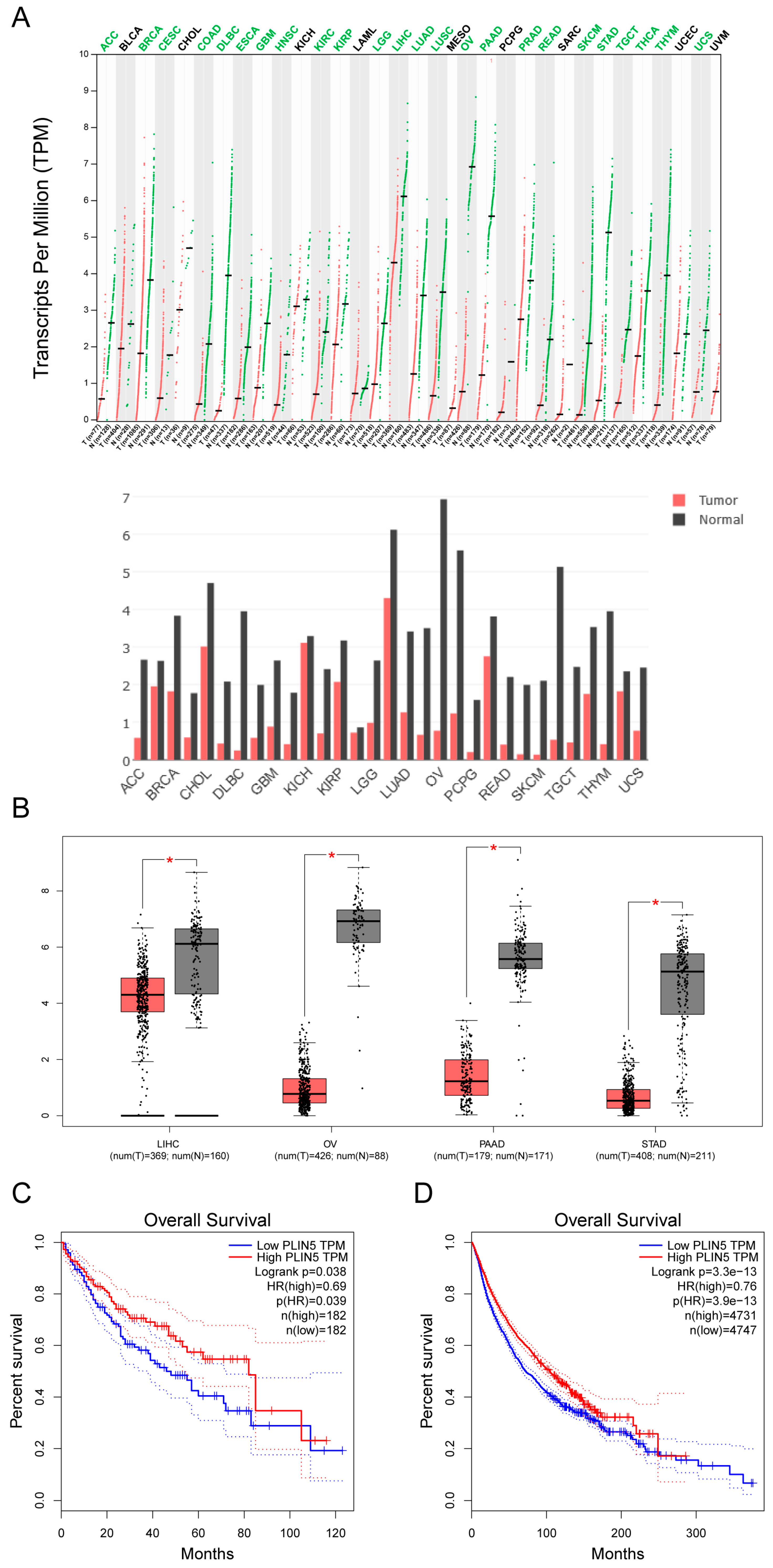
2.8. Low Expression of PLIN5 Is Associated with Poor Prognosis
3. Discussion and Conclusions
4. Materials and Methods
4.1. Animals
4.2. Cell Culture
4.3. Antibodies
4.4. Transfection Assay
4.5. Plasmid DNA Construction
4.6. Hydrogen Peroxide and LPS Treatment
4.7. Lipid Droplets Marking and Observation
4.8. Western Blot
4.9. Real-Time PCR
4.10. Apoptosis and Mitochondrial Membrane Potential Analysis
4.11. Bioinformatics and Data Analysis
4.12. Dual-Luciferase Reporter Assay
4.13. Immunohistochemistry Assay
4.14. Isolation of Cytosolic and Mitochondrial Fractions
4.15. Fluorescence Image Analysis
4.16. Survival Analysis and Normal/Cancer Gene Expression Comparison Analysis
4.17. Statistical Analyses
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | protein kinase AMP-activated catalytic subunit |
| ASK | apoptosis signal-regulating kinase |
| ATF | activating transcription factor |
| BODIPY | 4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene |
| CAT | catalase |
| COX | cytochrome c oxidase subunit IV |
| CS | citrate synthase |
| Cyto c | cytochrome c |
| DCFH-DA | 2,7-Dichlorodihydrofluorescein diacetate |
| GEPIA | gene expression profiling interactive analysis |
| GPX | glutathione peroxidase |
| HFD | high-fat diet |
| JC-1 | 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine |
| JNK | jnk c-Jun N-terminal kinase |
| LD | lipid droplet |
| LIHC | liver hepatocellular carcinoma |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MCDD | methionine-choline-deficient diet |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic hepatitis |
| P38 | mitogen-activated protein kinase 14 |
| PAT | perilipin, adipophilin, and TIP47 |
| PLIN5 | perilipin 5 |
| PNPLA2/ATGL | patatin like phospholipase domain containing 2 |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
References
- Anderson, N.; Borlak, J. Molecular Mechanisms and Therapeutic Targets in Steatosis and Steatohepatitis. Pharmacol. Rev. 2008, 60, 311–357. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Di, R.M.; Nicoletti, F.; Malaguarnera, L. Molecular mechanisms involved in NAFLD progression. J. Mol. Med. 2009, 87, 679. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.S.; Oben, J. Nonalcoholic fatty liver disease and lipids. Curr. Opin. Lipidol. 2012, 23, 345. [Google Scholar] [CrossRef] [PubMed]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quiñones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.A.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef]
- Welte, M.A. How Brain Fat Conquers Stress. Cell 2015, 163, 269–270. [Google Scholar] [CrossRef]
- Valm, A.M.; Cohen, S.; Legant, W.R.; Melunis, J.; Hershberg, U.; Wait, E.; Cohen, A.R.; Davidson, M.W.; Betzig, E.; Lippincott-Schwartz, J. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 2017, 546, 162–167. [Google Scholar] [CrossRef]
- Romanauska, A.; Kohler, A. The Inner Nuclear Membrane is a Metabolically Active Territory that Generates Nuclear Lipid Droplets. Cell 2018, 174, 700–715. [Google Scholar] [CrossRef]
- Soltysik, K.; Ohsaki, Y.; Tatematsu, T.; Cheng, J.L.; Fujimoto, T. Nuclear lipid droplets derive from a lipoprotein precursor and regulate phosphatidylcholine synthesis. Nat. Commun. 2019, 10, 473. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Tan, Y.; Chen, L.; Liu, Y.; Ren, Z. Reactive Oxygen Species Induces Lipid Droplet Accumulation in HepG2 Cells by Increasing Perilipin 2 Expression. Int. J. Mol. Sci. 2018, 19, 3445. [Google Scholar] [CrossRef] [PubMed]
- Dalen, K.T.; Dahl, T.; Holter, E.; Arntsen, B.; Londos, C.; Sztalryd, C.; Nebb, H.I. LSDP5 is a PAT protein specifically expressed in fatty acid oxidizing tissues. BBA-Mol. Cell Biol. Lipids 2007, 1771, 210–227. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.R.; Watt, M.J. Unraveling the roles of PLIN5: Linking cell biology to physiology. Trends Endocrinol. Metab. 2015, 26, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sztalryd, C. Oxidative tissue: Perilipin 5 links storage with the furnace. Trends Endocrinol. Metab. 2011, 22, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Whytock, K.L.; Shepherd, S.O.; Wagenmakers, A.J.M.; Strauss, J.A. Hormone-sensitive lipase preferentially redistributes to lipid droplets associated with perilipin-5 in human skeletal muscle during moderate-intensity exercise. J. Physiol. 2018, 596, 2077–2090. [Google Scholar] [CrossRef] [PubMed]
- Langhi, C.; Marquart, T.J.; Allen, R.M.; Baldan, A. Perilipin-5 is regulated by statins and controls triglyceride contents in the hepatocyte. J. Hepatol. 2014, 61, 358–365. [Google Scholar] [CrossRef]
- Harris, L.A.; Skinner, J.R.; Shew, T.M.; Pietka, T.A.; Abumrad, N.A.; Wolins, N.E. Perilipin 5-Driven Lipid Droplet Accumulation in Skeletal Muscle Stimulates the Expression of Fibroblast Growth Factor 21. Diabetes 2015, 64, 2757–2768. [Google Scholar] [CrossRef]
- Bosma, M.; Sparks, L.M.; Hooiveld, G.J.; Jorgensen, J.A.; Houten, S.M.; Schrauwen, P.; Kersten, S.; Hesselink, M.K.C. Overexpression of PLIN5 in skeletal muscle promotes oxidative gene expression and intramyocellular lipid content without compromising insulin sensitivity. BBA-Mol. Cell Biol. Lipids 2013, 1831, 844–852. [Google Scholar] [CrossRef]
- Bosma, M.; Minnaard, R.; Sparks, L.M.; Schaart, G.; Losen, M.; de Baets, M.H.; Duimel, H.; Kersten, S.; Bickel, P.E.; Schrauwen, P.; et al. The lipid droplet coat protein perilipin 5 also localizes to muscle mitochondria. Histochem. Cell Biol. 2012, 137, 205–216. [Google Scholar] [CrossRef]
- Wang, H.; Sreenevasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acin-Perez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869–885. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Louie, S.M.; Daniele, J.R.; Tran, Q.; Dillin, A.; Zoncu, R.; Nomura, D.K.; Olzmann, J.A. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell 2017, 42, 9–21.e5. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Herms, A.; Bosch, M.; Ariotti, N.; Reddy, B.J.N.; Fajardo, A.; Fernandez-Vidal, A.; Alvarez-Guaita, A.; Fernandez-Rojo, M.A.; Rentero, C.; Tebar, F.; et al. Cell-to-Cell Heterogeneity in Lipid Droplets Suggests a Mechanism to Reduce Lipotoxicity. Curr. Biol. 2013, 23, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Bischof, J.; Salzmann, M.; Streubel, M.K.; Hasek, J.; Geltinger, F.; Duschl, J.; Bresgen, N.; Briza, P.; Haskova, D.; Lejskovas, R.; et al. Clearing the outer mitochondrial membrane from harmful proteins via lipid droplets. Cell Death Discov. 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, A.R.; Sztalryd, C. Perilipin 5, a lipid droplet protein adapted to mitochondrial energy utilization. Curr. Opin. Lipidol. 2014, 25, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Kennedy, J.A. Role of Superoxide Radical in Mitochondrial Dehydrogenase Reactions. Biochem. Biophys. Res. Commun. 1974, 60, 1044–1050. [Google Scholar] [CrossRef]
- Loschen, G.; Azzi, A.; Richter, C.; Flohe, L. Superoxide Radicals as Precursors of Mitochondrial Hydrogen-Peroxide. FEBS Lett. 1974, 42, 68–72. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Ngoei, K.R.; Zhao, T.T.; Yeap, Y.Y.; Ng, D.C. c-Jun N-terminal kinase (JNK) signaling: Recent advances and challenges. Biochim. Biophys. Acta 2010, 1804, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Chen, F. JNK-induced apoptosis, compensatory growth, and cancer stem cells. Cancer Res. 2012, 72, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. p38(MAPK): Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A. Expanding Roles for Lipid Droplets. Curr. Biol. 2015, 25, R470–R481. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Targeting endoplasmic reticulum stress in metabolic disease. Expert Opin. Ther. Targets 2013, 17, 437–448. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Song, Y.; Zhang, L.J.; Gu, Y.; Li, F.F.; Pan, S.Y.; Jiang, L.N.; Liu, F.; Ye, J.; Li, Q. LSDP5 enhances triglyceride storage in hepatocytes by influencing lipolysis and fatty acid beta-oxidation of lipid droplets. PLoS ONE 2012, 7, e36712. [Google Scholar] [CrossRef]
- Yuki, O.; Jinglei, C.; Michitaka, S.; Akikazu, F.; Toyoshi, F. Lipid droplets are arrested in the ER membrane by tight binding of lipidated apolipoprotein B-100. J. Cell Sci. 2008, 121, 2415–2422. [Google Scholar]
- Jacquier, N.; Choudhary, V.; Mari, M.; Toulmay, A.; Reggiori, F.; Schneiter, R. Lipid droplets are functionally connected to the endoplasmic reticulum in Saccharomyces cerevisiae. J. Cell Sci. 2011, 124, 2424–2437. [Google Scholar] [CrossRef]
- Alexandra, G.; Laura, B.; Gabriel, M.; Charlotta, F.; Fatima-Zahra, I.; Francesco, M.; Raul, G.; Júlia, M.; Eduard, S.; Pedro, C. The seipin complex Fld1/Ldb16 stabilizes ER-lipid droplet contact sites. J. Cell Biol. 2015, 211, 829–844. [Google Scholar]
- Wang, H.; Becuwe, M.; Housden, B.E.; Chitraju, C.; Porras, A.J.; Graham, M.M.; Liu, X.N.; Thiam, A.R.; Savage, D.B.; Agarwal, A.K. Seipin is required for converting nascent to mature lipid droplets. eLife 2016, 5, e16582. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, J.; Esposti, M.D.; Gilmore, A.P. Bcl-2 proteins and mitochondria-Specificity in membrane targeting for death. BBA-Mol. Cell Res. 2011, 1813, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Happo, L.; Strasser, A.; Cory, S. BH3-only proteins in apoptosis at a glance. J. Cell Sci. 2012, 125, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Oltvai, Z.N.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 Heterodimerizes in-Vivo with a Conserved Homolog, Bax, That Accelerates Programmed Cell-Death. Cell 1993, 74, 609–619. [Google Scholar] [CrossRef]
- Westphal, D.; Dewson, G.; Czabotar, P.E.; Kluck, R.M. Molecular biology of Bax and Bak activation and action. BBA-Mol. Cell Res. 2011, 1813, 521–531. [Google Scholar] [CrossRef]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef]
- Sun, P.; Zeng, Q.; Cheng, D.; Zhang, K.; Zheng, J.; Liu, Y.; Yuan, Y.F.; Tang, Y.D. Caspase Recruitment Domain Protein 6 protects against hepatic steatosis and insulin resistance by suppressing Ask1. Hepatology 2018, 68, 2212–2229. [Google Scholar] [CrossRef]
- Ye, P.; Xiang, M.; Liao, H.; Liu, J.; Luo, H.; Wang, Y.; Huang, L.; Chen, M.; Xia, J. Dual-specificity Phosphatase 9 Protects Against Non-alcoholic Fatty Liver Disease in Mice via ASK1 Suppression. Hepatology 2018, 69, 76–93. [Google Scholar] [CrossRef]
- Lawan, A.; Zhang, L.; Gatzke, F.; Min, K.; Jurczak, M.J.; Al-Mutairi, M.; Richter, P.; Camporez, J.P.; Couvillon, A.; Pesta, D. Hepatic mitogen-activated protein kinase phosphatase 1 selectively regulates glucose metabolism and energy homeostasis. Mol. Cell. Biol. 2015, 35, 26. [Google Scholar] [CrossRef]
- Cingolani, F.; Czaja, M.J. Oxidized Albumin—A Trojan Horse for p38 MAPK-Mediated Inflammation in Decompensated Cirrhosis. Hepatology 2018, 68, 1678–1680. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.Y.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER Stress Cooperates with Hypernutrition to Trigger TNF-Dependent Spontaneous HCC Development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Ohki, T.; Tateishi, R.; Shiina, S.; Goto, E.; Sato, T.; Nakagawa, H.; Masuzaki, R.; Goto, T.; Hamamura, K.; Kanai, F.; et al. Visceral fat accumulation is an independent risk factor for hepatocellular carcinoma recurrence after curative treatment in patients with suspected NASH. Gut 2009, 58, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Huff, T.B.; Cheng, J.X. Coherent anti-Stokes Raman scattering imaging of lipids in cancer metastasis. BMC Cancer 2009, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.V.; Taylor, H.B. Lipid-rich carcinoma of the breast. A clinicopathologic analysis of 13 examples. Cancer 1974, 33, 812–819. [Google Scholar] [CrossRef]
- de los Monteros, A.E.; Hellmen, E.; Ramirez, G.A.; Herraez, P.; Rodriguez, F.; Ordas, J.; Millán, Y.; Lara, A.; de las Mulas, J.M. Lipid-rich Carcinomas of the Mammary Gland in Seven Dogs: Clinicopathologic and Immunohistochemical Features. Vet. Pathol. 2003, 40, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Jin, Y.; Zhou, Y.; Jin, J.; Ma, Z.; Ren, Z. Deep sequencing of transcriptome profiling of GSTM2 knock-down in swine testis cells. Sci. Rep. 2016, 6, 38254. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Primer Sequence 5′-3′ |
|---|---|
| PLIN5 (Perilipin 5) | Forward: AAGGCCCTGAAGTGGGTTC |
| Reverse: GCATGTGGTCTATCAGCTCCA | |
| CS | Forward: GCTCCTGTTTCCATGGGTCA |
| Reverse: TGCCAAAGCATGTCCAGCTA | |
| COX2 | Forward: GCTGTCCCCACATTAGGCTT |
| Reverse: ACCGTAGTATACCCCCGGTC | |
| COX4 | Forward: CCCGGCATTTTACGACGTTC |
| Reverse: AAAAATGTACACCTGCCGCC | |
| ATF1 | Forward: TTCGGATCTACCTGGGAGGG |
| Reverse: CTGATAAAGATGATACCTGTTGAGC | |
| ATF3 | Forward: GACCAACCATGCCTTGAGGA |
| Reverse: GGATGGCAAACCTCAGCTCT | |
| ATF4 | Forward: TAAGCCATGGCGTGAGTACC |
| Reverse: GCGCTCGTTAAATCGCTTCC | |
| GAPDH | Forward: CTGGGCTACACTGAGCACC |
| Reverse: AAGTGGTCGTTGAGGGCAATG | |
| CAT | Forward: TGGGATCTCGTTGGAAATAACAC |
| Reverse: TCAGGACGTAGGCTCCAGAAG | |
| GPX1 | Forward: CAGTCGGTGTATGCCTTCTCG |
| Reverse: GAGGGACGCCACATTCTCG | |
| GPX2 | Forward: GAATGGGCAGAACGAGCATC |
| Reverse: CCGGCCCTATGAGGAACTTC | |
| SOD1 | Forward: GGTGGGCCAAAGGATGAAGAG |
| Reverse: CCACAAGCCAAACGACTTCC | |
| SOD2 | Forward: TTTCAATAAGGAACGGGGACAC |
| Reverse: GTGCTCCCACACATCAATCC | |
| TXNRD1 | Forward: ATGGGCAATTTATTGGTCCTCAC |
| Reverse: CCCAAGTAACGTGGTCTTTCAC | |
| PRDX3 | Forward: ACTGTGAAGTTGTCGCAGTCT |
| Reverse: CACACCGTAGTCTCGGGAAA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, Y.; Jin, Y.; Wang, Q.; Huang, J.; Wu, X.; Ren, Z. Perilipin 5 Protects against Cellular Oxidative Stress by Enhancing Mitochondrial Function in HepG2 Cells. Cells 2019, 8, 1241. https://doi.org/10.3390/cells8101241
Tan Y, Jin Y, Wang Q, Huang J, Wu X, Ren Z. Perilipin 5 Protects against Cellular Oxidative Stress by Enhancing Mitochondrial Function in HepG2 Cells. Cells. 2019; 8(10):1241. https://doi.org/10.3390/cells8101241
Chicago/Turabian StyleTan, Yanjie, Yi Jin, Qian Wang, Jin Huang, Xiang Wu, and Zhuqing Ren. 2019. "Perilipin 5 Protects against Cellular Oxidative Stress by Enhancing Mitochondrial Function in HepG2 Cells" Cells 8, no. 10: 1241. https://doi.org/10.3390/cells8101241
APA StyleTan, Y., Jin, Y., Wang, Q., Huang, J., Wu, X., & Ren, Z. (2019). Perilipin 5 Protects against Cellular Oxidative Stress by Enhancing Mitochondrial Function in HepG2 Cells. Cells, 8(10), 1241. https://doi.org/10.3390/cells8101241