Neuronal Ceroid Lipofuscinoses: Connecting Calcium Signalling through Calmodulin

Abstract
1. Introduction
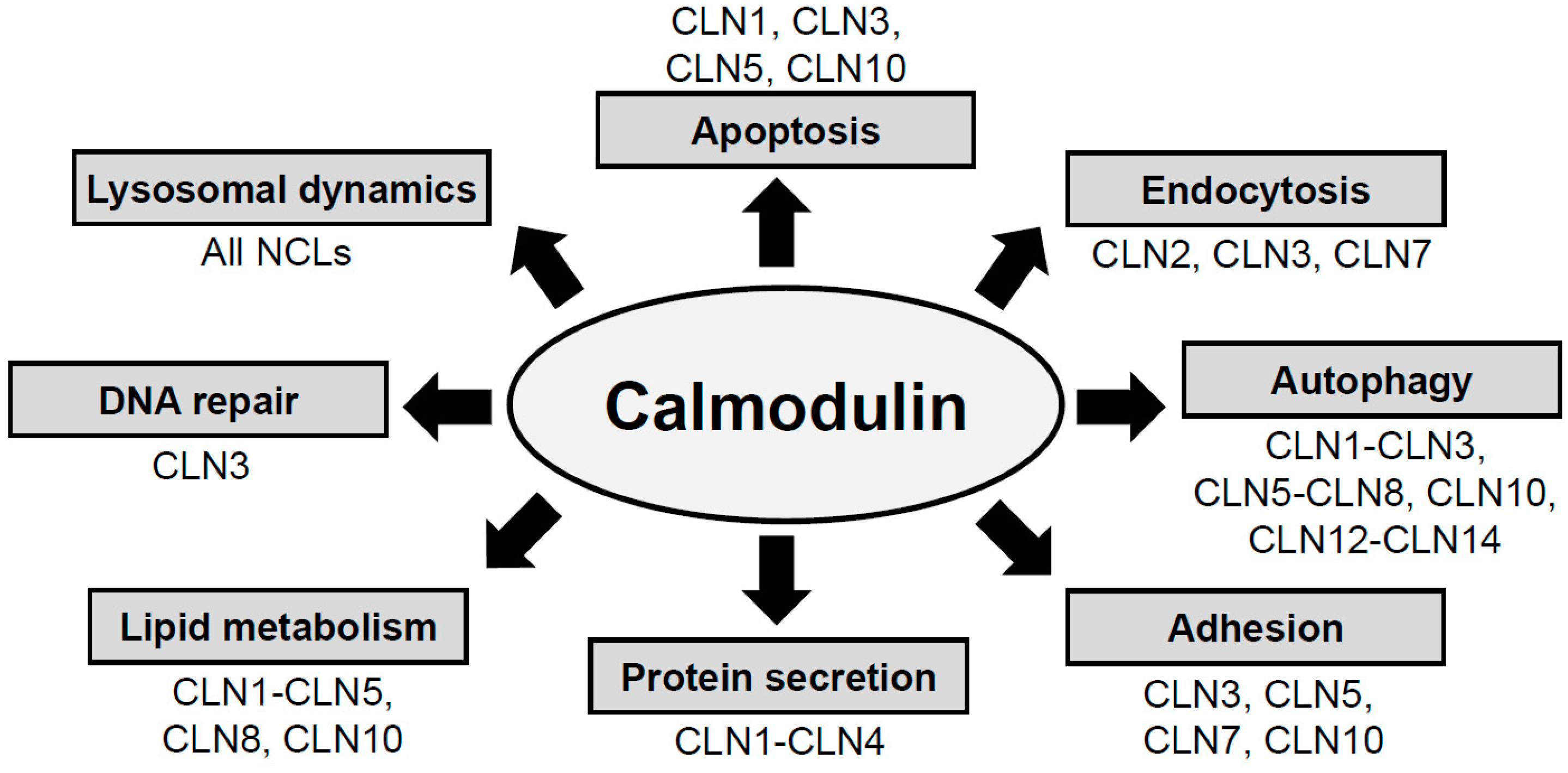
2. NCL Proteins Influence Cellular Pathways That Are Regulated by CaM
3. NCL Proteins Have Putative Binding Domains for CaM
4. Clinical Relevance for the Presence of CaMBDs in NCL Proteins
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CaM | Calmodulin |
| CaMKII | Calcium/CaM-dependent protein kinase II |
| CaMBDs | CaM-binding domains |
| CaMBPs | CaM-binding proteins |
| CLN | ceroid lipofuscinosis neuronal |
| CTS | cathepsin |
| NCL | Neuronal ceroid lipofuscinosis |
References
- Batten, F.E. Cerebral degeneration with symmetrical changes in the maculae in two members of a family. Trans. Ophthalmol. Soc. UK 1903, 23, 386–390. [Google Scholar]
- Dolisca, S.B.; Mehta, M.; Pearce, D.A.; Mink, J.W.; Maria, B.L. Batten disease: Clinical aspects, molecular mechanisms, translational science, and future directions. J. Child Neurol. 2013, 28, 1074–1100. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.; Kohlschütter, A.; Mink, J.; Simonati, A.; Williams, R. NCL diseases–clinical perspectives. Biochim. Biophys. Acta 2013, 1832, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Cerliponase alfa: First global approval. Drugs 2017, 77, 1247–1249. [Google Scholar] [CrossRef] [PubMed]
- Kyttälä, A.; Lahtinen, U.; Braulke, T.; Hofmann, S.L. Functional biology of the neuronal ceroid lipofuscinoses (NCL) proteins. Biochim. Biophys. Acta 2006, 1762, 920–933. [Google Scholar] [CrossRef] [PubMed]
- Danyukova, T.; Ariunbat, K.; Thelen, M.; Brocke-Ahmadinejad, N.; Mole, S.E.; Storch, S. Loss of CLN7 results in depletion of soluble lysosomal proteins and impaired mTOR reactivation. Hum. Mol. Genet. 2018, 27, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Staropoli, J.F.; Karaa, A.; Lim, E.T.; Kirby, A.; Elbalalesy, N.; Romansky, S.G.; Leydiker, K.B.; Coppel, S.H.; Barone, R.; Xin, W.; et al. A homozygous mutation in KCTD7 links neuronal ceroid lipofuscinosis to the ubiquitin-proteasome system. Hum. Mol. Genet. 2012, 91, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Kohan, R.; Cismondi, I.A.; Dodelson Kremer, R.; Muller, V.J.; Guelbert, N.; Tapia Anzolini, V.; Fietz, M.J.; Oller Ramírez, A.M.; Noher Halac, I. An integrated strategy for the diagnosis of neuronal ceroid lipofuscinosis types 1 (CLN1) and 2 (CLN2) in eleven Latin American patients. Clin. Genet. 2009, 76, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Holopainen, J.M.; Saarikoski, J.; Kinnunen, P.K.; Järvelä, I. Elevated lysosomal pH in neuronal ceroid lipofuscinoses (NCLs). Eur. J. Biochem. 2001, 268, 5851–5856. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Wirak, G.S.; Zhang, Y.Q.; Dai, F.; Ginsberg, S.D.; Dolzhanskaya, N.; Staropoli, J.F.; Nijssen, P.C.; Lam, T.T.; Roth, A.F.; et al. Neuronal ceroid lipofuscinosis with DNAJC5/CSPα mutation has PPT1 pathology and exhibit aberrant protein palmitoylation. Acta Neuropathol. 2016, 131, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Persaud-Sawin, D.A.; Mousallem, T.; Wang, C.; Zucker, A.; Kominami, E.; Boustany, R.M. Neuronal ceroid lipofuscinosis: A common pathway? Pediatr. Res. 2007, 61, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.E.; Aberg, L.; Autti, T.; Goebel, H.H.; Kohlschütter, A.; Lönnqvist, T. Diagnosis of the neuronal ceroid lipofuscinoses: An update. Biochim. Biophys. Acta 2006, 1762, 865–872. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, J.E.; Guidotti, G. Batten disease and the control of the Fo subunit c pore by cGMP and calcium. Eur. J. Paediatr. Neurol. 2001, 5, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.E.; Kielian, T. Astrocytes in juvenile neuronal ceroid lipofuscinosis (CLN3) display metabolic and calcium signaling abnormalities. J. Neurochem. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Ahtiainen, L.; Kolikova, J.; Mutka, A.L.; Luiro, K.; Gentile, M.; Ikonen, E.; Khiroug, L.; Jalanko, A.; Kopra, O. Palmitoyl protein thioesterase 1 (Ppt1)-deficient mouse neurons show alterations in cholesterol metabolism and calcium homeostasis prior to synaptic dysfunction. Neurobiol. Dis. 2007, 28, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Polisetty, R.V.; Gautam, P.; Sharma, R.; Harsha, H.C.; Nair, S.C.; Gupta, M.K.; Uppin, M.S.; Challa, S.; Puligopu, A.K.; Ankathi, P.; et al. LC-MS/MS analysis of differentially expressed glioblastoma membrane proteome reveals altered calcium signaling and other protein groups of regulatory functions. Mol. Cell. Proteomics. 2012, 11, M111.013565. [Google Scholar] [CrossRef] [PubMed]
- Kolikova, J.; Afzalov, R.; Surin, A.; Lehesjoki, A.E.; Khiroug, L. Deficient mitochondrial Ca(2+) buffering in the Cln8(mnd) mouse model of neuronal ceroid lipofuscinosis. Cell Calcium 2011, 50, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.; Zhao, X.; Hu, J.; Wang, Z.G.; Zhao, X.T. HCCS1 overexpression induces apoptosis via cathepsin D and intracellular calcium, and HCCS1 disruption in mice causes placental abnormality. Cell Death Differ. 2008, 15, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Warnock, A.; Tan, L.; Li, C.; an Haack, K.; Narayan, S.B.; Bennett, M.J. Amlodipine prevents apoptotic cell death by correction of elevated intracellular calcium in a primary neuronal model of Batten disease (CLN3 disease). Biochem. Biophys. Res. Commun. 2013, 436, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J.; Myre, M.A.; Cotman, S.L. Loss of Cln3 function in the social amoeba Dictyostelium discoideum causes pleiotropic effects that are rescued by human CLN3. PLoS ONE 2014, 9, e110544. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J.; Myre, M.A.; Cotman, S.L. Aberrant adhesion impacts early development in a Dictyostelium model for juvenile neuronal ceroid lipofuscinosis. Cell Adh. Migr. 2017, 11, 399–418. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.J.; Falk, M.J.; Bennett, M.J. Flunarizine rescues reduced lifespan in CLN3 triple knock-out Caenorhabditis elegans model of batten disease. J. Inherit Metab. Dis. 2017, 40, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 1995, 80, 259–268. [Google Scholar] [CrossRef]
- Cheung, W.Y. Calmodulin plays a pivotal role in cellular regulation. Science 1980, 207, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.R.; Friedberg, F. Sequence motifs for calmodulin recognition. FASEB J. 1997, 11, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Kim, J.; Truong, K.; Sherman, M.; Yuan, T.; Ikura, M. Calmodulin target database. J. Struct. Funct. Genomics. 2000, 1, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Chafouleas, J.G.; Bolton, W.E.; Means, A.R. Potentiation of bleomycin lethality by anticalmodulin drugs: A role for calmodulin in DNA repair. Science 1984, 224, 1346–1348. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, M.W.; Villalobo, A. The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim. Biophys. Acta 2014, 1843, 398–435. [Google Scholar] [CrossRef] [PubMed]
- Luiro, K.; Yliannala, K.; Ahtiainen, L.; Maunu, H.; Järvelä, I.; Kyttälä, A.; Jalanko, A. Interconnections of CLN3, Hook1 and Rab proteins link Batten disease to defects in the endocytic pathway. Hum. Mol. Genet. 2004, 13, 3017–3027. [Google Scholar] [CrossRef] [PubMed]
- Donsante, A.; Boulis, N.M. Progress in gene and cell therapies for the neuronal ceroid lipofuscinoses. Expert Opin. Biol. Ther. 2018, 18, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.N.; Barry, L.A.; Tyynelä, J.; Cooper, J.D. NCL disease mechanisms. Biochim. Biophys. Acta. 2013, 1832, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.H.; Pearce, D.A. What have microarrays told us about the neuronal ceroid lipofuscinoses? Curr. Genom. 2005, 6, 257–268. [Google Scholar] [CrossRef]
- Kopra, O.; Vesa, J.; von Schantz, C.; Manninen, T.; Minye, H.; Fabritius, A.L.; Rapola, J.; Diggelen, O.P.; Saarela, J.; Jalanko, A.; et al. A mouse model for Finnish variant late infantile neuronal ceroid lipofuscinosis, CLN5, reveals neuropathology associated with early aging. Hum. Mol. Genet. 2004, 13, 2893–2906. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H.; Eshak, K.; Myre, M.A. Calmodulin binding proteins and Alzheimer’s disease. J. Alzheimers Dis. 2015, 46, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, Y.H.; Patel, V.M.; Berman, D.E.; Kothiya, M.J.; Neufeld, J.L.; Vardarajan, B.; Tang, M.; Reyes-Dumeyer, D.; Lantigua, R.; Medrano, M.; et al. An Alzheimer’s disease-linked loss-of-function CLN5 variant impairs cathepsin D maturation, consistent with a retromer trafficking defect. Mol. Cell Biol. 2018, 38, E00011-18. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H.; Myre, M.A. Calmodulin-binding domains in Alzheimer’s disease proteins: Extending the calcium hypothesis. Biochem. Biophys. Res. Commun. 2004, 320, 1051–1054. [Google Scholar] [CrossRef] [PubMed]
- Mruk, K.; Farley, B.M.; Ritacco, A.W.; Kobertz, W.R. Calmodulation meta-analysis: Predicting calmodulin binding via canonical motif clustering. J. Gen. Physiol. 2014, 144, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Canobbio, I.; Catricalà, S.; Balduini, C.; Torti, M. Calmodulin regulates the non-amyloidogenic metabolism of amyloid precursor protein in platelets. Biochim. Biophys. Acta 2011, 1813, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Salazar, D.A.; Butler, V.J.; Argouarch, A.R.; Hsu, T.Y.; Mason, A.; Nakamura, A.; McCurdy, H.; Cox, D.; Ng, R.; Pan, G.; et al. The progranulin cleavage products, granulins, exacerbate TDP-43 toxicity and increase TDP-43 levels. J. Neurosci. 2015, 35, 9315–9328. [Google Scholar] [CrossRef] [PubMed]
- Steenhuis, P.; Froemming, J.; Reinheckel, T.; Storch, S. Proteolytic cleavage of the disease-related lysosomal membrane glycoprotein CLN7. Biochim. Biophys. Acta 2012, 1822, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Stankowski, J.N.; Chew, J.; Cook, C.N.; Lam, Y.W.; Almeida, S.; Carlomagno, Y.; Lau, K.F.; Prudencio, M.; Gao, F.B.; et al. The lysosomal protein cathepsin L is a progranulin protease. Mol. Neurodegener. 2017, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Sevenich, L.; Pennacchio, L.A.; Peters, C.; Reinheckel, T. Human cathepsin L rescues the neurodegeneration and lethality in cathepsin B/L double-deficient mice. Biol. Chem. 2006, 387, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Tang, W.; Sondheimer, N.; Avadhani, N.G. Role of calcineurin, hnRNPA2 and Akt in mitochondrial respiratory stress-mediated transcription activation of nuclear gene targets. Biochim. Biophys. Acta 2010, 1797, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.E. The genetic spectrum of human neuronal ceroid-lipofuscinoses. Brain Pathol. 2004, 14, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Dudek, N.L.; Dai, Y.; Muma, N.A. Protective effects of interrupting the binding of calmodulin to mutant huntingtin. J. Neuropathol. Exp. Neurol. 2008, 67, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Dudek, N.L.; Dai, Y.; Muma, N.A. Neuroprotective effects of calmodulin peptide 76-121aa: Disruption of calmodulin binding to mutant huntingtin. Brain Pathol. 2010, 20, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.; kleine Holthaus, S.M.; Tammen, I.; Tear, G.; Russell, C. Use of model organisms for the study of neuronal ceroid lipofuscinosis. Biochim. Biophys. Acta 2013, 1832, 1842–1865. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Calcium-dependent calmodulin-binding motifs | |
| 1–10 Subclasses | |
| 1–10 | (FILVW)xxxxxxxx(FILVW) |
| 1–5–10 | (FILVW)xxx(FAILVW)xxxx(FILVW) |
| Basic 1–5–10 | (RK)(RK)(RK)(FAILVW)xxx(FILV)xxxx(FILVW) |
| 1–14 Subclasses | |
| 1–14 | (FILVW)xxxxxxxxxxxx(FILVW) |
| 1–8–14 | (FILVW)xxxxxx(FAILVW)xxxxx(FILVW) |
| Basic 1–8–14 | (RK)(RK)(RK)(FILVW)xxxxxx(FAILVW)xxxxx(FILVW) |
| 1–5–8–14 | (FILVW)xxx(FAILVW)xx(FAILVW)xxxxx(FILVW) |
| 1–16 Subclasses | |
| 1–16 | (FILVW)xxxxxxxxxxxxxx(FILVW) |
| Calcium-independent calmodulin-binding motifs | |
| IQ Subclasses | |
| IQ | (FILVW)Qxxx(RK)Gxxx(RK)xx(FILVWY) |
| IQ-like | (FILVW)Qxxx(RK)xxxxxxxx |
| NCL Protein | Putative CaMBD | Motif |
|---|---|---|
| CLN1 Region 1 (159–183) | ICDFIRKTLNAGAYSKVVQERLVQA | 1–14 |
| ICDFIRKTLNAGAYSKVVQERLVQA | 1–14 | |
| ICDFIRKTLNAGAYSKVVQERLVQA | 1–14 | |
| CLN1 Region 2 (207–232) | QERGINESYKKNLMALKKFVMVKFLN | 1–16 |
| QERGINESYKKNLMALKKFVMVKFLN | 1–10 | |
| CLN2 (332–358) | LSSAYIQRVNTELMKAAARGLTLLFAS | 1–16 |
| LSSAYIQRVNTELMKAAARGLTLLFAS | 1–16 | |
| CLN3 (318–352) | YRWYQMLYQAGVFASRSSLRCCRIRFTWALALLQ | 1–5–10 |
| YRWYQMLYQAGVFASRSSLRCCRIRFTWALALLQ | 1–16 | |
| YRWYQMLYQAGVFASRSSLRCCRIRFTWALALLQ | 1–14 | |
| YRWYQMLYQAGVFASRSSLRCCRIRFTWALALLQ | 1–10 | |
| YRWYQMLYQAGVFASRSSLRCCRIRFTWALALLQ | 1–8–14 | |
| YRWYQMLYQAGVFASRSSLRCCRIRFTWALALLQ | 1–5–10 | |
| CLN4 (65–87) | AILTDATKRNIYDKYGSLGLYVA | 1–5–10 |
| AILTDATKRNIYDKYGSLGLYVA | 1–16 | |
| AILTDATKRNIYDKYGSLGLYVA | 1–10 | |
| CLN5 (57–84) | QGAEMRRGAGAARGRASWCWALALLWL | 1–5–10 |
| CLN6 (245–259) | VLHQKRKRLFLDSNG | 1–10 |
| VLHQKRKRLFLDSNG | 1–10 | |
| CLN8 (45–72) | LSSSLNATYRSLVAREKVFWDLAATRA | 1–16 |
| CLN10 Region 1 (166–189) | ASALGGVKVERQVFGEATKQPGIT | 1–10 |
| ASALGGVKVERQVFGEATKQPGIT | 1–10 | |
| CLN10 Region 2 (250–275) | DSKYYKGSLSYLNVTRKAYWQVHLD | 1–14 |
| DSKYYKGSLSYLNVTRKAYWQVHLD | 1–16 | |
| CLN12 (152–179) | EEAVSVGQKRVLRYYLFQGQRYIWIETQ | 1–8–14 |
| EEAVSVGQKRVLRYYLFQGQRYIWIETQ | 1–14 | |
| EEAVSVGQKRVLRYYLFQGQRYIWIETQ | 1–14 | |
| EEAVSVGQKRVLRYYLFQGQRYIWIETQ | 1–10 | |
| CLN13 (39–80) | LLAPTRFALEMFNRGRAAGTRAVLGLVRGRVRRAGQGSLYSL | 1–16 |
| LLAPTRFALEMFNRGRAAGTRAVLGLVRGRVRRAGQGSLYSL | 1–16 | |
| LLAPTRFALEMFNRGRAAGTRAVLGLVRGRVRRAGQGSLYSL | 1–16 | |
| LLAPTRFALEMFNRGRAAGTRAVLGLVRGRVRRAGQGSLYSL | 1–16 | |
| CLN14 (148–166) | HLERIVEIARLRAVQRKARFAKLKVCVFKEEMP | IQ-Like |
| HLERIVEIARLRAVQRKARFAKLKVCVFKEEMP | 1–5–10 | |
| HLERIVEIARLRAVQRKARFAKLKVCVFKEEMP | 1–5–10 | |
| HLERIVEIARLRAVQRKARFAKLKVCVFKEEMP | 1–16 | |
| HLERIVEIARLRAVQRKARFAKLKVCVFKEEMP | 1–16 | |
| HLERIVEIARLRAVQRKARFAKLKVCVFKEEMP | 1–10 | |
| HLERIVEIARLRAVQRKARFAKLKVCVFKEEMP | 1–5–10 | |
| HLERIVEIARLRAVQRKARFAKLKVCVFKEEMP | 1–8–14 | |
| CTSL (226–247) | VDIPKQEKALMKAVATVGPISV | 1–10 |
| VDIPKQEKALMKAVATVGPISV | 1–14 |
| NCL Type | NCL Protein | CaMBD (aa) | NCL Patient Mutations Present within CaMBD | Mutations Last Updated |
|---|---|---|---|---|
| Infantile | CLN1 | 159–183 | p.Gln177Glu, p.Val181Met, p.Val181Leu | 28 November 2017 |
| Infantile | CLN1 | 207–232 | p.Leu222Pro, p.Val228Gly | 28 November 2017 |
| Late Infantile | CLN2 | 332–358 | p.Glu343Lys, p.Arg339Trp, p.Leu355Pro, p.Thr353Pro, p.Arg339Gln, p.Arg350Trp | 13 November 2017 |
| Juvenile | CLN3 | 318–352 | p.Arg334Cys, p.Val330Phe, p.Arg334Trp, p.Val330Ile | 28 November 2017 |
| Adult | CLN4 | 65–87 | None documented | 26 February 2018 |
| Variant Late Infantile | CLN5 | 57–84 | p.Trp75Arg | 26 February 2018 |
| Variant Late Infantile | CLN6 | 245–260 | p.Arg252His, p.Gly259Val, p.Gly259Ser, p.Asp256Glu | 26 February 2018 |
| Variant Late Infantile | CLN7 | N/A | - | 26 February 2018 |
| Variant Late Infantile | CLN8 | 45–72 | p.Arg70His | 26 February 2018 |
| Congenital | CLN10 | 166–189 | None documented | 26 February 2018 |
| Congenital | CLN10 | 250–275 | None documented | 26 February 2018 |
| Adult | CLN11 | N/A | - | 26 February 2018 |
| Juvenile | CLN12 | 152–179 | None documented | 26 February 2018 |
| Adult | CLN13 | 39–80 | None documented | 4 December 2017 |
| Infantile | CLN14 | 148–166 | None documented | 4 December 2017 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathavarajah, S.; O’Day, D.H.; Huber, R.J. Neuronal Ceroid Lipofuscinoses: Connecting Calcium Signalling through Calmodulin. Cells 2018, 7, 188. https://doi.org/10.3390/cells7110188
Mathavarajah S, O’Day DH, Huber RJ. Neuronal Ceroid Lipofuscinoses: Connecting Calcium Signalling through Calmodulin. Cells. 2018; 7(11):188. https://doi.org/10.3390/cells7110188
Chicago/Turabian StyleMathavarajah, Sabateeshan, Danton H. O’Day, and Robert J. Huber. 2018. "Neuronal Ceroid Lipofuscinoses: Connecting Calcium Signalling through Calmodulin" Cells 7, no. 11: 188. https://doi.org/10.3390/cells7110188
APA StyleMathavarajah, S., O’Day, D. H., & Huber, R. J. (2018). Neuronal Ceroid Lipofuscinoses: Connecting Calcium Signalling through Calmodulin. Cells, 7(11), 188. https://doi.org/10.3390/cells7110188