
Stage-Specific Serum Proteomic Signatures Reveal Early Biomarkers and Molecular Pathways in Huntington’s Disease Progression


Abstract
1. Introduction
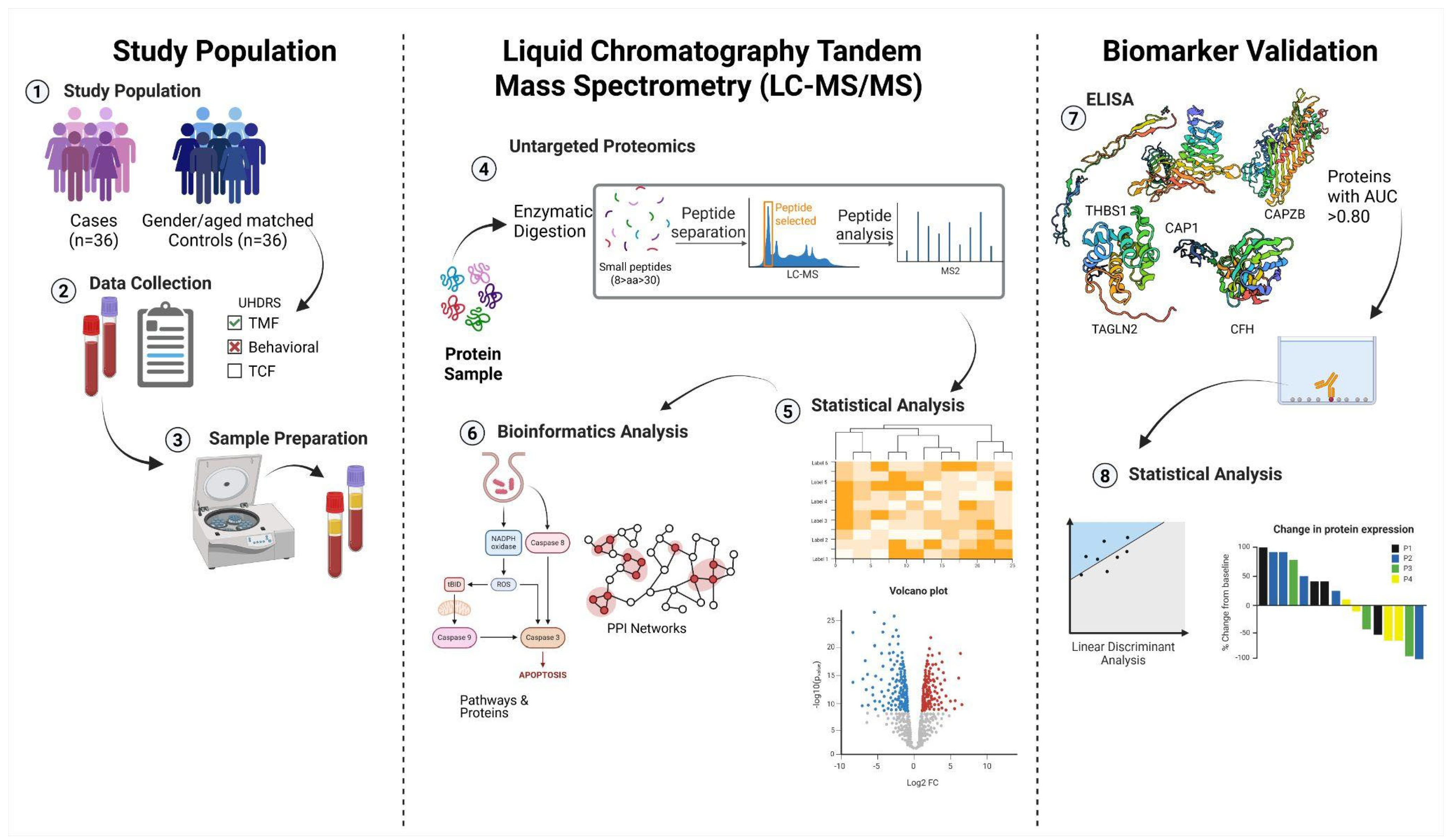
2. Materials and Methods
2.1. HD Patients and Controls
2.2. Data Collection
- (i)
- Demographics: Sex, date of birth, birthplace and city of residence, birthplace of parents, family status (single, married, divorced or widowed) and occupation.
- (ii)
- Medical and family history of HD: Presence and age of HD symptoms and family members who have HD.
- (iii)
- Additional information: Height and weight prior to symptom onset in early symptomatic and symptomatic advanced stages.
- (iv)
- Medical history, CAG repeats, treatments and other comorbidities obtained from the patient’s medical records or via self-reporting for controls.
- (v)
- Lifestyle: Dietary intake, MD adherence, physical activity and smoking status.
2.3. Huntington’s Disease Assessment
2.4. Blood Collection and Serum Extraction
2.5. Sample Preparation
2.5.1. High-Abundance Protein Depletion
2.5.2. Protein Digestion
2.5.3. Nano-Liquid Chromatography and Mass Spectrometry (LC-MS/MS) Analysis
2.5.4. Proteomic Data Analysis
2.5.5. Quantification of CFH, CAPZB, CAP1, THBS1 and TAGLN2 in Serum via ELISA
2.6. Statistical Analysis
2.7. Bioinformatics Analysis Pipeline for Proteomic Data
2.7.1. Identification and Determination of the DEPs in Stage-Specific HD
2.7.2. Pathway and Gene Ontology Enrichment Analysis of Differentially Expressed Proteins in Stage-Specific HD
2.7.3. Identification of Shared and Exclusive Proteins Across HD Stages
2.7.4. Filtering and Evaluation of Biomarker Candidates for Huntington’s Disease Progression
2.7.5. ELISA-Based Validation of Stage-Specific Biomarker Candidates in Huntington’s Disease
2.7.6. Analysis of Protein–Protein Interaction Networks Across Huntington’s Disease Stages
3. Results
3.1. Demographics and Anthropometric Characteristics of Cohort
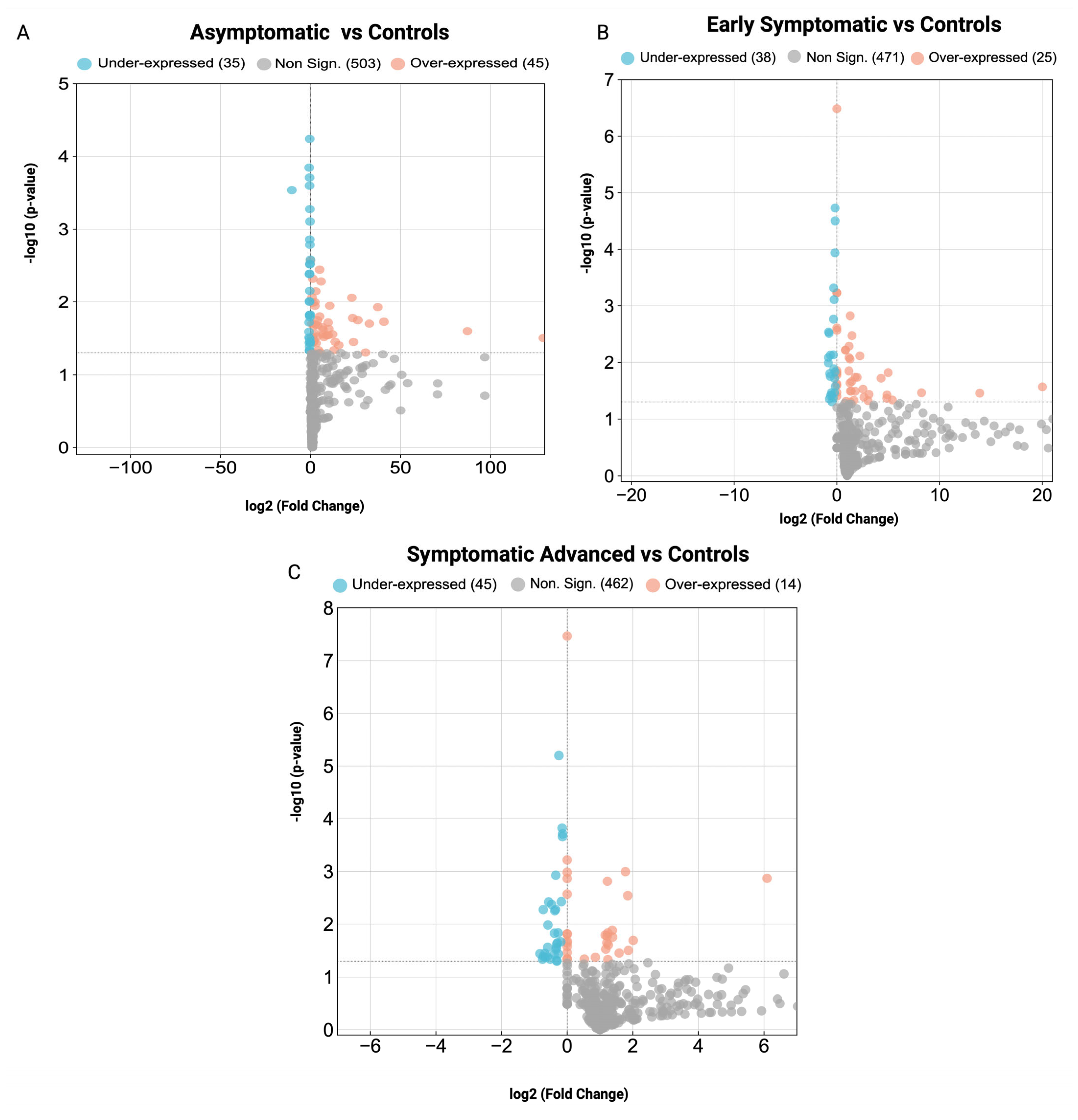
3.2. Key Proteins and Identification of Differentially Expressed Proteins in Huntington’s Disease Pathogenesis
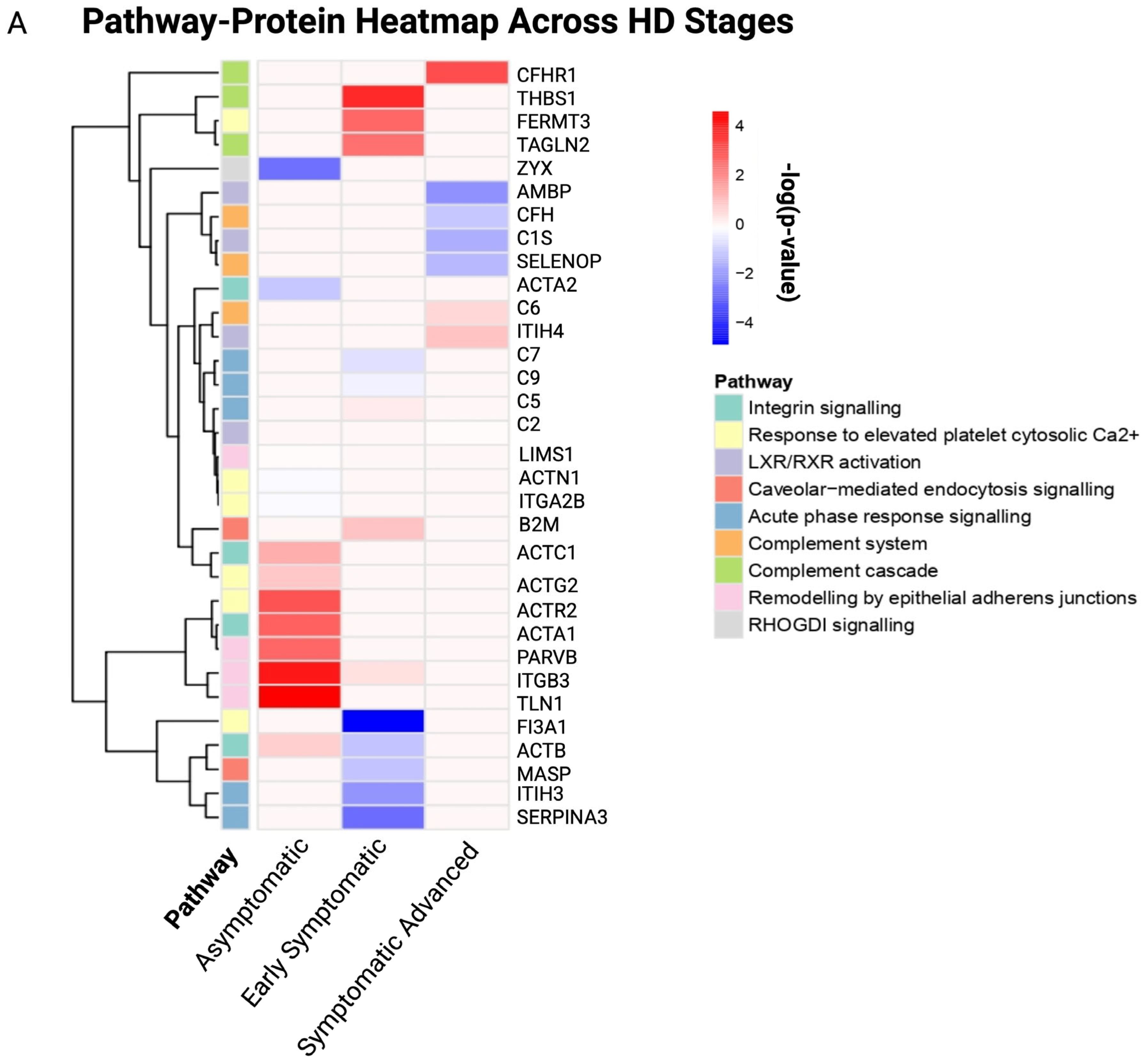
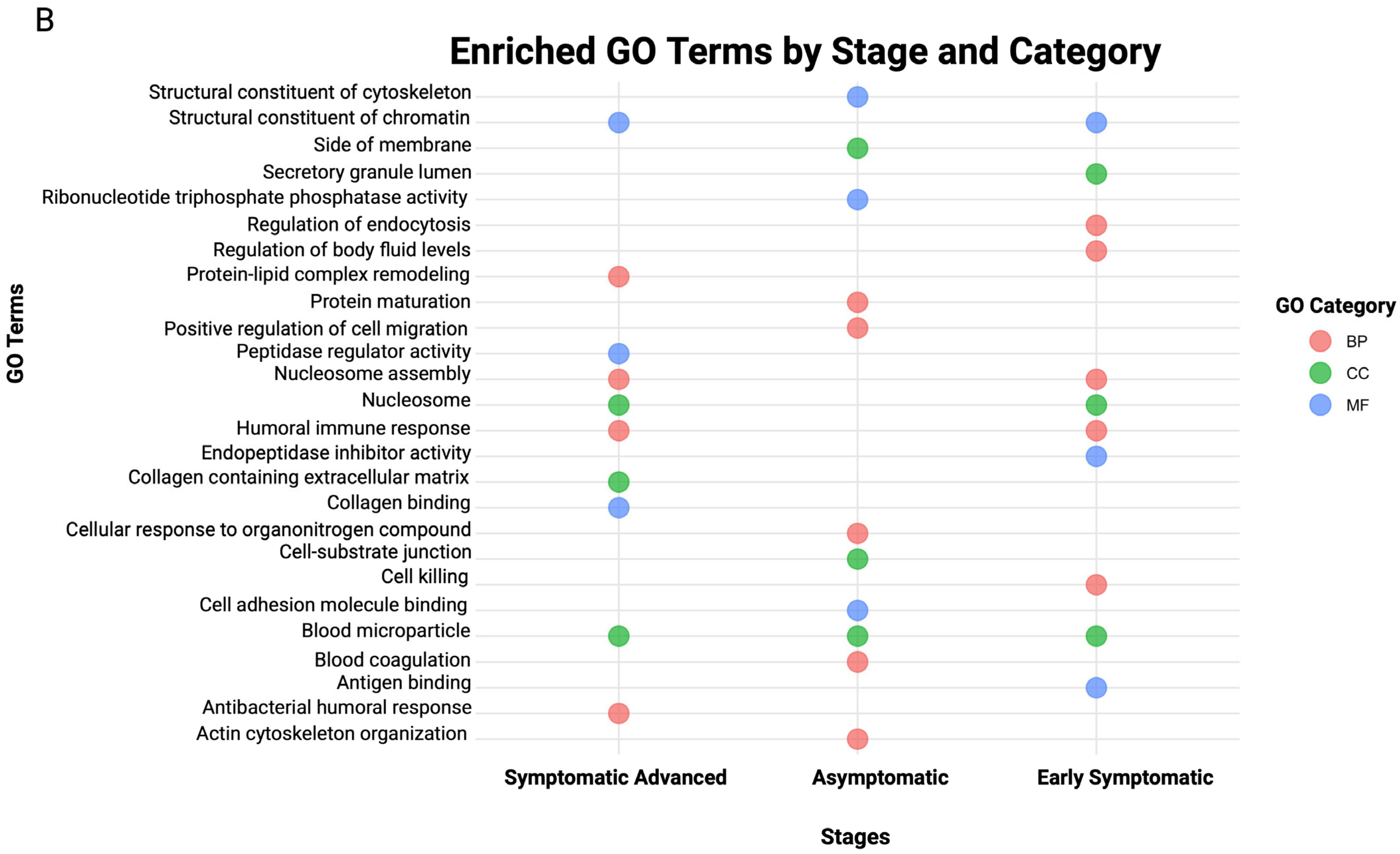
3.3. Biological Pathways and GO Enrichment Terms in Stage-Specific Huntington’s Disease
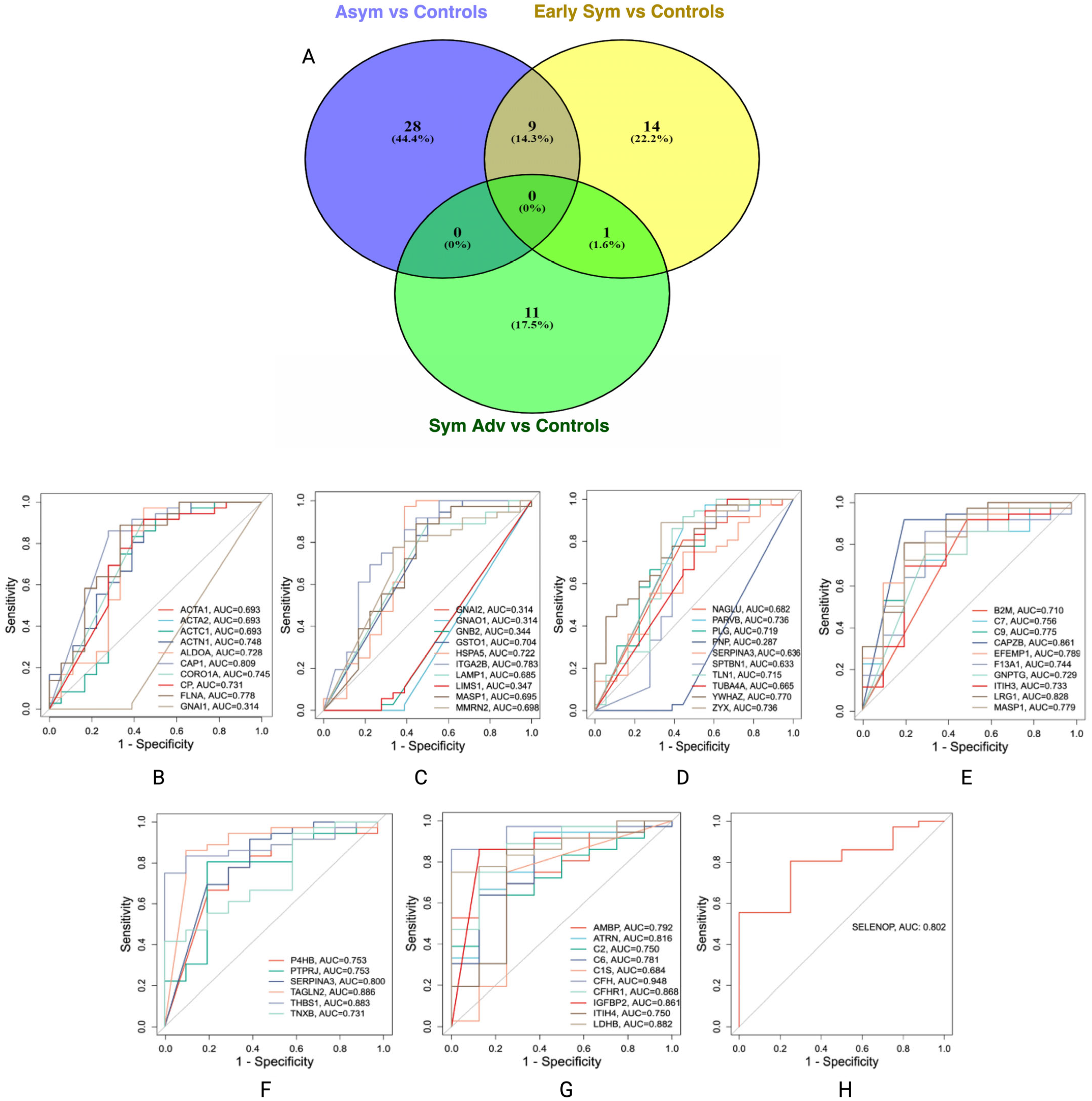
3.4. Potential Protein Biomarker Candidates for Stage-Specific Huntington’s Disease
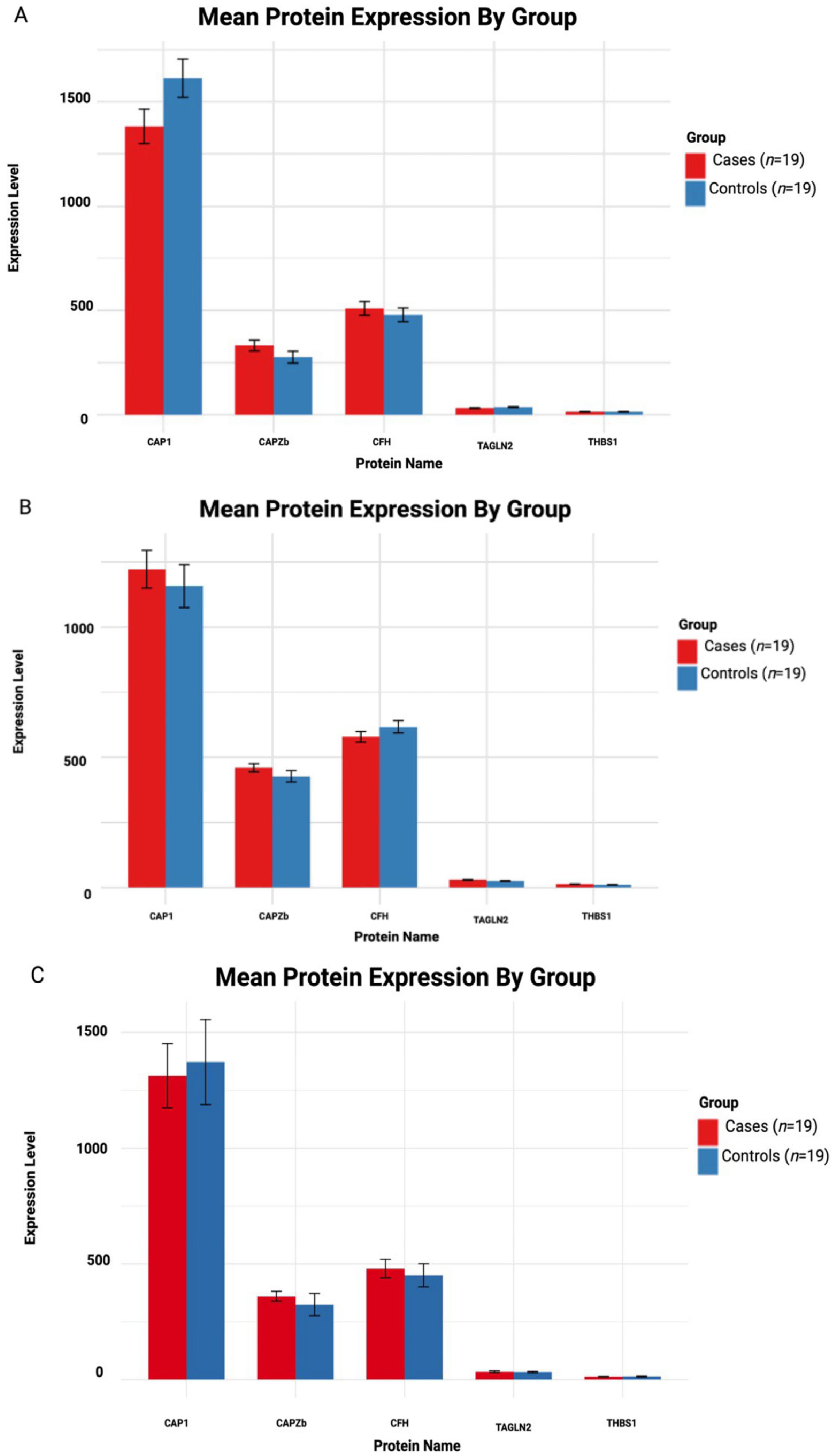
3.5. Independent Validation of Proteomic Candidates in Huntington’s Disease
3.5.1. Asymptomatic HD vs. Controls
3.5.2. Early Symptomatic HD vs. Controls
3.5.3. Symptomatic Advanced HD vs. Controls

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group Name | Protein | Estimate | SE | adj. p-Value | Mean Cases | Mean Control | Cohen’s d |
|---|---|---|---|---|---|---|---|
| Asymptomatic HD vs. Controls | CAP1 | −230.76 | 61.45 | 0.0027 * | 1382.10 | 1612.86 | −5.31 |
| CAPZB | 55.81 | 61.45 | 0.381 | 331.94 | 276.12 | 1.280 | |
| CFH | 30.27 | 61.45 | 0.631 | 509.47 | 479.19 | 0.699 | |
| TAGLN2 | −4.81 | 61.45 | 0.938 | 31.43 | 36.24 | −0.111 | |
| THBS1 | −0.056 | 61.45 | 0.992 | 14.56 | 14.61 | −0.001 | |
| Symptomatic HD vs. Controls | CAP1 | 65.08 | 58.11 | 0.281 | 1222.53 | 1157.44 | 1.580 |
| CAPZB | 33.20 | 58.11 | 0.576 | 460.65 | 427.45 | 0.808 | |
| CFH | −38.53 | 58.11 | 0.518 | 579.30 | 617.84 | −0.938 | |
| TAGLN2 | 4.15 | 58.11 | 0.944 | 29.15 | 25.00 | 0.101 | |
| THBS1 | 2.74 | 58.11 | 0.963 | 13.72 | 10.98 | 0.066 | |
| Symptomatic Advanced HD vs. Controls | CAP1 | −59.04 | 112.30 | 0.617 | 1314.06 | 1373.11 | −0.743 |
| CAPZB | 36.05 | 112.30 | 0.759 | 360.14 | 324.09 | 0.454 | |
| CFH | 28.79 | 112.30 | 0.800 | 479.70 | 450.90 | 0.362 | |
| TAGLN2 | 1.89 | 112.30 | 0.987 | 33.71 | 31.81 | 0.024 | |
| THBS1 | −0.70 | 112.30 | 0.995 | 12.44 | 13.57 | −0.008 |
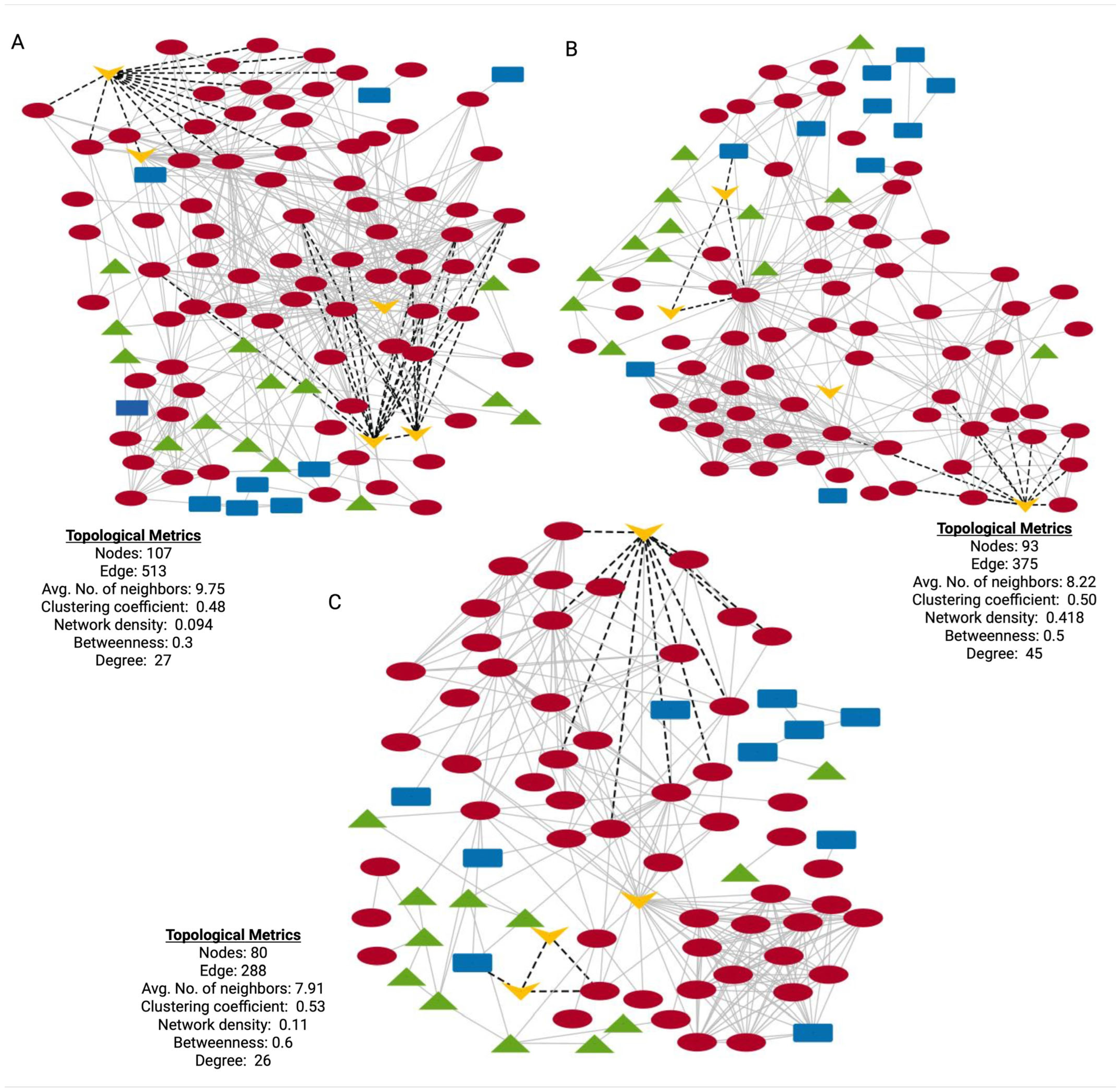
3.6. Protein–Protein Interaction Networks and Functional Context for DEPs of Stage-Specific HD
4. Discussion
- Strengths and Limitations
- Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 24-OHC | 24S-Hydroxycholesterol |
| AD | Alzheimer’s Disease |
| ALS | Amyotrophic Lateral Sclerosis |
| AGC | Automatic Gain Control |
| AOO | Age of Onset |
| AUC | Area Under the Curve |
| BMI | Body Mass Index |
| CAG | Cytosine–Adenine–Guanine |
| CNS | Central Nervous System |
| CSF | Cerebrospinal Fluid |
| DDA | Data-Dependent Acquisition |
| DEP | Differentially Expressed Proteins |
| DTT | DL-dithiothreitol |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| FC | Fold Change |
| FDR | False Discovery Rate |
| GO | Gene Ontology |
| GWAS | Genome-Wide Association Studies |
| HD | Huntington’s Disease |
| HTT | Huntingtin |
| IAA | Iodoacetamide |
| IPA | Ingenuity Pathway Analysis |
| iPSCs | Induced Pluripotent Stem Cells |
| IS | Independence Scale |
| LC-MS/MS | Liquid Chromatography Tandem Mass Spectrometry |
| LMM | Linear Mixed-Effects Model |
| LXR | Liver X Receptor |
| MSN | Medium Spiny Neurons |
| mHTT | Mutant Huntingtin |
| NCE | Normalized Collision Energy |
| ND | Neurodegenerative Disease |
| NS | Nervous System |
| PD | Parkinson’s Disease |
| PPI | Protein–Protein Interaction Networks |
| SSRI | Selective Serotonin Reuptake Inhibitors |
| TBS | Total Behavioral Score |
| TFC | Total Functional Capacity |
| TMS | Total Motor Score |
| UHDRS | Unified Huntington’s Disease Rating Scale |
References
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid. Med. Cell. Longev. 2016, 90578. [Google Scholar] [CrossRef]
- Roos, R.A. Huntington’s Disease: A Clinical Review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef]
- Demetriou, C.A.; Heraclides, A.; Salafori, C.; Tanteles, G.A.; Christodoulou, K.; Christou, Y.; Zamba-Papanicolaou, E. Epidemiology of Huntington Disease in Cyprus: A 20-Year Retrospective Study. Clin. Genet. 2018, 3, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Al-Amrani, S.; Al-Jabri, Z.; Al-Zaabi, A.; Alshekaili, J.; Al-Khabori, M. Proteomics: Concepts and Applications in Human Medicine. World J. Biol. Chem. 2021, 12, 57–69. [Google Scholar] [CrossRef]
- Gregorich, Z.R.; Ge, Y. Top-down Proteomics in Health and Disease: Challenges and Opportunities. Proteomics 2014, 14, 1195–1210. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Biswas, A. Molecular Diagnostics of Neurodegenerative Disorders. Front. Mol. Biosci. 2015, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mees, I.; Li, S.; Tran, H.; Ang, C.S.; Williamson, N.A.; Hannan, A.J.; Renoir, T. Phosphoproteomic Dysregulation in Huntington’s Disease Mice Is Rescued by Environmental Enrichment. Brain Commun. 2022, 4, fcac305. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Miller, B.R.; Rebec, G.V.; Clemmer, D.E. Protein Expression in the Striatum and Cortex Regions of the Brain for a Mouse Model of Huntington’s Disease. J. Proteome Res. 2007, 6, 3134–3142. [Google Scholar] [CrossRef]
- Perluigi, M.; Poon, H.F.; Maragos, W.; Pierce, W.M.; Klein, J.B.; Calabrese, V.; Cini, C.; De Marco, C.; Butterfield, D.A. Proteomic Analysis of Protein Expression and Oxidative Modification in R6/2 Transgenic Mice: A Model of Huntington Disease. Mol. Cell. Proteom. 2005, 4, 1849–1861. [Google Scholar] [CrossRef]
- Deschepper, M.; Hoogendoorn, B.; Brooks, S.; Dunnett, S.B.; Jones, L. Proteomic Changes in the Brains of Huntington’s Disease Mouse Models Reflect Pathology and Implicate Mitochondrial Changes. Brain Res. Bull. 2012, 88, 210–222. [Google Scholar] [CrossRef]
- Cozzolino, F.; Landolfi, A.; Iacobucci, I.; Monaco, V.; Caterino, M.; Celentano, S.; Zuccato, C.; Cattaneo, E.; Monti, M. New Label-Free Methods for Protein Relative Quantification Applied to the Investigation of an Animal Model of Huntington Disease. PLoS ONE 2020, 15, e0238037. [Google Scholar] [CrossRef]
- Vodicka, P.; Mo, S.; Tousley, A.; Green, K.M.; Sapp, E.; Iuliano, M.; Sadri-Vakili, G.; Shaffer, S.A.; Aronin, N.; DiFiglia, M.; et al. Mass Spectrometry Analysis of Wild-Type and Knock-in Q140/Q140 Huntington’s Disease Mouse Brains Reveals Changes in Glycerophospholipids Including Alterations in Phosphatidic Acid and Lyso-Phosphatidic Acid. J. Huntingt. Dis. 2015, 4, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Sap, K.A.; Guler, A.T.; Bezstarosti, K.; Bury, A.E.; Juenemann, K.; Demmers, J.A.A.; Reits, E.A. Global Proteome and Ubiquitinome Changes in the Soluble and Insoluble Fractions of Q175 Huntington Mice Brains. Mol. Cell. Proteom. 2019, 18, 1705–1720. [Google Scholar] [CrossRef]
- Ratovitski, T.; Chighladze, E.; Arbez, N.; Boronina, T.; Herbrich, S.; Cole, R.N.; Ross, C.A. Huntingtin Protein Interactions Altered by Polyglutamine Expansion as Determined by Quantitative Proteomic Analysis. Cell Cycle 2012, 11, 2006–2021. [Google Scholar] [CrossRef]
- Liu, L.; Huang, J.S.; Han, C.; Zhang, G.X.; Xu, X.Y.; Shen, Y.; Li, J.; Jiang, H.Y.; Lin, Z.C.; Xiong, N.; et al. Induced Pluripotent Stem Cells in Huntington’s Disease: Disease Modeling and the Potential for Cell-Based Therapy. Mol. Neurobiol. 2016, 53, 6698–6708. [Google Scholar] [CrossRef]
- Zhang, N.; Bailus, B.J.; Ring, K.L.; Ellerby, L.M. IPSC-Based Drug Screening for Huntingtons Disease. Brain Res. 2016, 1638, 42–56. [Google Scholar] [CrossRef]
- Kaye, J.; Reisine, T.; Finkbeiner, S. Huntington’s Disease IPSC Models—Using Human Patient Cells to Understand the Pathology Caused by Expanded CAG Repeats. Fac. Rev. 2022, 11. [Google Scholar] [CrossRef]
- Wu, G.H.; Smith-Geater, C.; Galaz-Montoya, J.G.; Gu, Y.; Gupte, S.R.; Aviner, R.; Mitchell, P.G.; Hsu, J.; Miramontes, R.; Wang, K.Q.; et al. CryoET Reveals Organelle Phenotypes in Huntington Disease Patient IPSC-Derived and Mouse Primary Neurons. Nat. Commun. 2023, 14, 692. [Google Scholar] [CrossRef]
- Mehta, S.R.; Tom, C.M.; Wang, Y.; Bresee, C.; Rushton, D.; Mathkar, P.P.; Tang, J.; Mattis, V.B. Human Huntington’s Disease IPSC-Derived Cortical Neurons Display Altered Transcriptomics, Morphology, and Maturation. Cell Rep. 2018, 25, 1081–1096.e6. [Google Scholar] [CrossRef] [PubMed]
- Akimov, S.S.; Jiang, M.; Kedaigle, A.J.; Arbez, N.; Marque, L.O.; Eddings, C.R.; Ranum, P.T.; Whelan, E.; Tang, A.; Wang, R.; et al. Immortalized Striatal Precursor Neurons from Huntington’s Disease Patient-Derived IPS Cells as a Platform for Target Identification and Screening for Experimental Therapeutics. Hum. Mol. Genet. 2021, 30, 2469–2487. [Google Scholar] [CrossRef] [PubMed]
- Chiu, F.L.; Lin, J.T.; Chuang, C.Y.; Chien, T.; Chen, C.M.; Chen, K.H.; Hsiao, H.Y.; Lin, Y.S.; Chern, Y.; Kuo, H.C. Elucidating the Role of the A2A Adenosine Receptor in Neurodegeneration Using Neurons Derived from Huntington’s Disease IPSCs. Hum. Mol. Genet. 2015, 24, 6066–6079. [Google Scholar] [CrossRef]
- Malakhova, A.A.; Grigor’eva, E.V.; Malankhanova, T.B.; Pavlova, S.V.; Valetdinova, K.R.; Abramycheva, N.Y.; Vetchinova, A.S.; Illarioshkin, S.N.; Zakian, S.M. Generation of Induced Pluripotent Stem Cell Line ICGi018-A from Peripheral Blood Mononuclear Cells of a Patient with Huntington’s Disease. Stem Cell Res. 2020, 44, 101743. [Google Scholar] [CrossRef]
- Jeon, I.; Choi, C.; Lee, N.; Im, W.; Kim, M.; Oh, S.H.; Park, I.H.; Kim, H.S.; Song, J. In Vivo Roles of a Patient-Derived Induced Pluripotent Stem Cell Line (HD72-IPSC) in the YAC128 Model of Huntington’s Disease. Int. J. Stem Cells 2014, 7, 43–47. [Google Scholar] [CrossRef]
- Mattis, V.B.; Tom, C.; Akimov, S.; Saeedian, J.; Østergaard, M.E.; Southwell, A.L.; Doty, C.N.; Ornelas, L.; Sahabian, A.; Lenaeus, L.; et al. HD IPSC-Derived Neural Progenitors Accumulate in Culture and Are Susceptible to BDNF Withdrawal Due to Glutamate Toxicity. Hum. Mol. Genet. 2014, 24, 3257–3271. [Google Scholar] [CrossRef]
- Berlet, R.; Galang Cabantan, D.A.; Gonzales-Portillo, D.; Borlongan, C.V. Enriched Environment and Exercise Enhance Stem Cell Therapy for Stroke, Parkinson’s Disease, and Huntington’s Disease. Front. Cell Dev. Biol. 2022, 10, 798826. [Google Scholar] [CrossRef]
- Dalrymple, A.; Wild, E.J.; Joubert, R.; Sathasivam, K.; Björkqvist, M.; Petersén, Å.; Jackson, G.S.; Isaacs, J.D.; Kristiansen, M.; Bates, G.P.; et al. Proteomic Profiling of Plasma in Huntington’s Disease Reveals Neuroinflammatory Activation and Biomarker Candidates. J. Proteome Res. 2007, 6, 2833–2840. [Google Scholar] [CrossRef] [PubMed]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and Oxidative Stress Analysis in Human Brain Samples of Huntington Disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Schönberger, S.J.; Jezdic, D.; Faull, R.L.M.; Cooper, G.J.S. Proteomic Analysis of the Human Brain in Huntington’s Disease Indicates Pathogenesis by Molecular Processes Linked to Other Neurodegenerative Diseases and to Type-2 Diabetes. J. Huntingt. Dis. 2013, 2, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lu, F.F.; Seeman, P.; Liu, F. Quantitative Proteomic Analysis of Human Substantia Nigra in Alzheimer’s Disease, Huntington’s Disease and Multiple Sclerosis. Neurochem. Res. 2012, 37, 2805–2813. [Google Scholar] [CrossRef]
- Fang, Q.; Strand, A.; Law, W.; Faca, V.M.; Fitzgibbon, M.P.; Hamel, N.; Houle, B.; Liu, X.; May, D.H.; Poschmann, G.; et al. Brain-Specific Proteins Decline in the Cerebrospinal Fluid of Humans with Huntington Disease. Mol. Cell. Proteom. 2009, 8, 451–466. [Google Scholar] [CrossRef]
- Cha, J.H.J. Transcriptional Signatures in Huntington’s Disease. Prog. Neurobiol. 2007, 83, 228–248. [Google Scholar] [CrossRef] [PubMed]
- Kieburtz, K.; MacDonald, M.; Shih, C.; Feigin, A.; Steinberg, K.; Bordwell, K.; Zimmerman, C.; Srinidhi, J.; Sotack, J.; Gusella, J.; et al. Trinucleotide Repeat Length and Progression of Illness in Huntington’s Disease. J. Med. Genet. 1994, 31, 872–874. [Google Scholar] [CrossRef]
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D.; Heisterkamp, S.; Van Willigen, B.; Ranke, J.; EISPACK. R Core Team Package “nlme” Title Linear and Nonlinear Mixed Effects Models. 2025. Available online: https://www.google.com.hk/url?sa=t&source=web&rct=j&opi=89978449&url=https://cran.r-project.org/web/packages/nlme/nlme.pdf&ved=2ahUKEwjpg-aSzeGOAxUOkq8BHYwUEjMQFnoECBkQAQ&usg=AOvVaw09hrCNDc-emI7xk8ixqn0Q (accessed on 3 May 2025).
- Gilbert, G.E. Linear Mixed Models: A Practical Guide Using Statistical Software. J. Am. Stat. Assoc. 2008, 103, 427–428. [Google Scholar] [CrossRef]
- Gueorguieva, R.; Krystal, J.H. Move Over ANOVA Progress in Analyzing Repeated-Measures Data and Its Reflection in Papers Published in the Archives of General Psychiatry. Arch. Gen. Psychiatry 2004, 61, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Lenth, V.R.; Banfai, B.; Bolker, B.; Buerkner, P.; Iago, G.-V.; Herve, M.; Jung, M.; Love, J.; Miguez, F.; Piaskowski, J.; et al. October. 2024. Available online: https://bibbase.org/network/publication/lenth-banfai-bolker-buerkner-ginvzquez-herve-jung-love-etal-emmeansestimatedmarginalmeansakaleastsquaresmeans-2024 (accessed on 5 May 2025).
- Korthauer, K.; Kimes, P.K.; Duvallet, C.; Reyes, A.; Subramanian, A.; Teng, M.; Shukla, C.; Alm, E.J.; Hicks, S.C. A Practical Guide to Methods Controlling False Discoveries in Computational Biology. Genome Biol. 2019, 20, 118. [Google Scholar] [CrossRef]
- Wickham, H.; Chang, W.; Henry, L.; Pedersen, T.L.; Takahashi, K.; Wilke, C.; Woo, K.; Yutani, H.; Dunnington, D.; van den Brand, T. Create Elegant Data Visualisations Using the Grammar of Graphics. Version 3.5.1. CRAN. 2024. Available online: https://ggplot2.tidyverse.org (accessed on 5 May 2025).
- Kassambara, A. ggpubr: ‘ggplot2’ Based Publication Ready Plots. R Package Version 0.6.0. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 5 May 2025).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Kolde, R. pheatmap: Pretty Heatmaps, R Package Version 1.0.13. 2025. Available online: https://github.com/raivokolde/pheatmap (accessed on 4 February 2025).
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, J.C.; Bates, D.M. Linear Mixed-Effects Models: Basic Concepts and Examples. In Linear Mixed-Effects Models in S and S-Plus; Springer: New York, NY, USA, 2000; Volume 96, pp. 33–56. ISBN 978-0-387-98957-0. [Google Scholar]
- Piñero, J.; Queralt-Rosinach, N.; Bravo, À.; Deu-Pons, J.; Bauer-Mehren, A.; Baron, M.; Sanz, F.; Furlong, L.I. DisGeNET: A Discovery Platform for the Dynamical Exploration of Human Diseases and Their Genes. Database 2015, 2015, bav028. [Google Scholar] [CrossRef]
- Sollis, E.; Mosaku, A.; Abid, A.; Buniello, A.; Cerezo, M.; Gil, L.; Groza, T.; Güneş, O.; Hall, P.; Hayhurst, J.; et al. The NHGRI-EBI GWAS Catalog: Knowledgebase and Deposition Resource. Nucleic Acids Res. 2023, 51, D977–D985. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Marchina, E.; Misasi, S.; Bozzato, A.; Ferraboli, S.; Agosti, C.; Rozzini, L.; Borsani, G.; Barlati, S.; Padovani, A. Gene Expression Profile in Fibroblasts of Huntington’s Disease Patients and Controls. J. Neurol. Sci. 2014, 337, 42–46. [Google Scholar] [CrossRef]
- Wright, G.E.B.; Caron, N.S.; Ng, B.; Casal, L.; Casazza, W.; Xu, X.; Ooi, J.; Pouladi, M.A.; Mostafavi, S.; Ross, C.J.D.; et al. Gene Expression Profiles Complement the Analysis of Genomic Modifiers of the Clinical Onset of Huntington Disease. Hum. Mol. Genet. 2020, 29, 2788–2802. [Google Scholar] [CrossRef]
- Moss, D.J.H.; Tabrizi, S.J.; Mead, S.; Lo, K.; Pardiñas, A.F.; Holmans, P.; Jones, L.; Langbehn, D.; Coleman, A.; Santos, R.D.; et al. Identification of Genetic Variants Associated with Huntington’s Disease Progression: A Genome-Wide Association Study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tollefsbol, T.O. DNA Methylation Methods: Global DNA Methylation and Methylomic Analyses; Elsevier Inc.: Amsterdam, The Netherlands, 2021; Volume 187, ISBN 2059344573. [Google Scholar]
- Neueder, A.; Kojer, K.; Hering, T.; Lavery, D.J.; Chen, J.; Birth, N.; Hallitsch, J.; Trautmann, S.; Parker, J.; Flower, M.; et al. Abnormal Molecular Signatures of Inflammation, Energy Metabolism, and Vesicle Biology in Human Huntington Disease Peripheral Tissues. Genome Biol. 2022, 23, 189. [Google Scholar] [CrossRef]
- Miller, S.J.; Glatzer, J.C.; Hsieh, Y.-C.; Rothstein, J.D. Cortical Astroglia Undergo Transcriptomic Dysregulation in the G93A SOD1 ALS Mouse Model. J. Neurogenet. 2018, 32, 322–335. [Google Scholar] [CrossRef] [PubMed]
- Seefelder, M.; Kochanek, S. A Meta-Analysis of Transcriptomic Profiles of Huntington’s Disease Patients. PLoS ONE 2021, 16, e0253037. [Google Scholar] [CrossRef]
- Sinha, M.; Mukhopadhyay, S.; Bhattacharyya, N.P. Mechanism(s) of Alteration of Micro Rna Expressions in Huntington’s Disease and Their Possible Contributions to the Observed Cellular and Molecular Dysfunctions in the Disease. Neuromol. Med. 2012, 14, 221–243. [Google Scholar] [CrossRef]
- Mastrokolias, A.; Pool, R.; Mina, E.; Hettne, K.M.; van Duijn, E.; van der Mast, R.C.; van Ommen, G.J.; ‘t Hoen, P.A.C.; Prehn, C.; Adamski, J.; et al. Integration of Targeted Metabolomics and Transcriptomics Identifies Deregulation of Phosphatidylcholine Metabolism in Huntington’s Disease Peripheral Blood Samples. Metabolomics 2016, 12, 137. [Google Scholar] [CrossRef]
- Borovecki, F.; Lovrecic, L.; Zhou, J.; Jeong, H.; Then, F.; Rosas, H.D.; Hersch, S.M.; Hogarth, P.; Bouzou, B.; Jensen, R.V.; et al. Genome-Wide Expression Profiling of Human Blood Reveals Biomarkers for Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2005, 102, 11023–11028. [Google Scholar] [CrossRef]
- Stopa, E.G.; Tanis, K.Q.; Miller, M.C.; Nikonova, E.V.; Podtelezhnikov, A.A.; Finney, E.M.; Stone, D.J.; Camargo, L.M.; Parker, L.; Verma, A.; et al. Comparative Transcriptomics of Choroid Plexus in Alzheimer’s Disease, Frontotemporal Dementia and Huntington’s Disease: Implications for CSF Homeostasis. Fluids Barriers CNS 2018, 15, 18. [Google Scholar] [CrossRef]
- Runne, H.; Kuhn, A.; Wild, E.J.; Pratyaksha, W.; Kristiansen, M.; Isaacs, J.D.; Ré, E.; Delorenzi, M.; Tabrizi, S.J.; Luthi-Carter, R. Analysis of Potential Transcriptomic Biomarkers for Huntington’s Disease in Peripheral Blood. Proc. Natl. Acad. Sci. USA 2007, 104, 14424–14429. [Google Scholar] [CrossRef]
- Rosas, H.D.; Doros, G.; Bhasin, S.; Thomas, B.; Gevorkian, S.; Malarick, K.; Matson, W.; Hersch, S.M. A Systems-Level “Misunderstanding”: The Plasma Metabolome in Huntington’s Disease. Ann. Clin. Transl. Neurol. 2015, 2, 756–768. [Google Scholar] [CrossRef]
- Herman, S.; Niemelä, V.; Emami Khoonsari, P.; Sundblom, J.; Burman, J.; Landtblom, A.M.; Spjuth, O.; Nyholm, D.; Kultima, K. Alterations in the Tyrosine and Phenylalanine Pathways Revealed by Biochemical Profiling in Cerebrospinal Fluid of Huntington’s Disease Subjects. Sci. Rep. 2019, 9, 4129. [Google Scholar] [CrossRef]
- McGarry, A.; Gaughan, J.; Hackmyer, C.; Lovett, J.; Khadeer, M.; Shaikh, H.; Pradhan, B.; Ferraro, T.N.; Wainer, I.W.; Moaddel, R. Cross-Sectional Analysis of Plasma and CSF Metabolomic Markers in Huntington’s Disease for Participants of Varying Functional Disability: A Pilot Study. Sci. Rep. 2020, 10, 20490. [Google Scholar] [CrossRef]
- Cheng, M.L.; Chang, K.H.; Wu, Y.R.; Chen, C.M. Metabolic Disturbances in Plasma as Biomarkers for Huntington’s Disease. J. Nutr. Biochem. 2016, 31, 38–44. [Google Scholar] [CrossRef]
- Graham, S.F.; Pan, X.; Yilmaz, A.; Macias, S.; Robinson, A.; Mann, D.; Green, B.D. Targeted Biochemical Profiling of Brain from Huntington’s Disease Patients Reveals Novel Metabolic Pathways of Interest. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2430–2437. [Google Scholar] [CrossRef]
- Patassini, S.; Begley, P.; Xu, J.; Church, S.J.; Reid, S.J.; Kim, E.H.; Curtis, M.A.; Dragunow, M.; Waldvogel, H.J.; Snell, R.G.; et al. Metabolite Mapping Reveals Severe Widespread Perturbation of Multiple Metabolic Processes in Huntington’s Disease Human Brain. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 1650–1662. [Google Scholar] [CrossRef]
- Ellrichmann, G.; Reick, C.; Saft, C.; Linker, R.A. The Role of the Immune System in Huntington’s Disease. Clin. Dev. Immunol. 2013, 2013, 541259. [Google Scholar] [CrossRef]
- Singhrao, S.K.; Neal, J.W.; Morgan, B.P.; Gasque, P. Increased Complement Biosynthesis By Microglia and Complement Activation on Neurons in Huntington’s Disease. Exp. Neurol. 1999, 9, 362–376. [Google Scholar] [CrossRef]
- Stennett, A.; Friston, K.; Harris, C.L.; Wollman, A.J.M.; Bronowska, A.K.; Madden, K.S. The Case for Complement Component 5 as a Target in Neurodegenerative Disease. Expert. Opin. Ther. Targets 2023, 27, 97–109. [Google Scholar] [CrossRef]
- Dobó, J.; Kocsis, A.; Farkas, B.; Demeter, F.; Cervenak, L.; Gál, P. The Lectin Pathway of the Complement System—Activation, Regulation, Disease Connections and Interplay with Other (Proteolytic) Systems. Int. J. Mol. Sci. 2024, 25, 1566. [Google Scholar] [CrossRef]
- Matsushita, M.; Thiel, S.; Jensenius, J.; Teraim Itaru; Fujita, T. Proteolytic Activities of Two Types of Mannose-Binding Lectin-Associated Protease. J. Immunol. 2000, 5, 2637–2642. [Google Scholar] [CrossRef]
- Massri, M.; Toonen, E.J.M.; Sarg, B.; Kremser, L.; Grasse, M.; Fleischer, V.; Torres-Quesada, O.; Hengst, L.; Skjoedt, M.O.; Bayarri-Olmos, R.; et al. Complement C7 and Clusterin Form a Complex in Circulation. Front. Immunol. 2024, 15, 1330095. [Google Scholar] [CrossRef]
- Franc, V.; Yang, Y.; Heck, A.J.R. Proteoform Profile Mapping of the Human Serum Complement Component C9 Revealing Unexpected New Features of N-, O-, and C-Glycosylation. Anal. Chem. 2017, 89, 3483–3491. [Google Scholar] [CrossRef]
- Wilton, D.K.; Mastro, K.; Heller, M.D.; Gergits, F.W.; Willing, C.R.; Fahey, J.B.; Frouin, A.; Daggett, A.; Gu, X.; Kim, Y.A.; et al. Microglia and Complement Mediate Early Corticostriatal Synapse Loss and Cognitive Dysfunction in Huntington’s Disease. Nat. Med. 2023, 29, 2866–2884. [Google Scholar] [CrossRef]
- Hamilton, J.; Farag, M.; Tabrizi, S.J. Complementary Insights into Corticostriatal Synapse Loss and Cognition in Huntington’s Disease. Cell Rep. Med. 2023, 4, 101314. [Google Scholar] [CrossRef]
- Shen, Q.; Bai, Y.; Chang, K.C.N.; Wang, Y.; Burris, T.P.; Freedman, L.P.; Thompson, C.C.; Nagpal, S. Liver X Receptor-Retinoid X Receptor (LXR-RXR) Heterodimer Cistrome Reveals Coordination of LXR and AP1 Signaling in Keratinocytes. J. Biol. Chem. 2011, 286, 14554–14563. [Google Scholar] [CrossRef]
- Courtney, R.; Landreth, G.E. LXR Regulation of Brain Cholesterol: From Development to Disease. Trends Endocrinol. Metab. 2016, 27, 404–414. [Google Scholar] [CrossRef]
- Paterniti, I.; Campolo, M.; Siracusa, R.; Cordaro, M.; Di Paola, R.; Calabrese, V.; Navarra, M.; Cuzzocrea, S.; Esposito, E. Liver X Receptors Activation, through TO901317 Binding, Reduces Neuroinflammation in Parkinson’s Disease. PLoS ONE 2017, 12, e0174470. [Google Scholar] [CrossRef]
- Alnaaim, S.A.; Al-Kuraishy, H.M.; Alexiou, A.; Papadakis, M.; Saad, H.M.; Batiha, G.E.S. Role of Brain Liver X Receptor in Parkinson’s Disease: Hidden Treasure and Emerging Opportunities. Mol. Neurobiol. 2024, 61, 341–357. [Google Scholar] [CrossRef]
- Zhang, R.; Wuerch, E.; Yong, V.W.; Xue, M. LXR Agonism for CNS Diseases: Promises and Challenges. J. Neuroinflamm. 2024, 21, 97. [Google Scholar] [CrossRef]
- Ban, S.Y.; Nam, Y.; Do, T.T.; Kim, B.H.; Shin, S.J.; Thi Nguyen, M.T.; Kim, J.; Moon, M.; Park, J.T. Liver-X Receptor β-Selective Agonist CE9A215 Regulates Alzheimer’s Disease-Associated Pathology in a 3xTg-AD Mouse Model. Biomed. Pharmacother. 2025, 184, 117895. [Google Scholar] [CrossRef]
- Zelcer, N.; Khanlou, N.; Clare, R.; Jiang, Q.; Reed-Geaghan, E.G.; Landreth, G.E.; Vinters, H.V.; Tontonoz, P. Attenuation of Neuroinflammation and Alzheimer’s Disease Pathology by Liver x Receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 10601–10606. [Google Scholar] [CrossRef]
- Leoni, V.; Caccia, C. The Impairment of Cholesterol Metabolism in Huntington Disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 1095–1105. [Google Scholar] [CrossRef]
- Futter, M.; Diekmann, H.; Schoenmakers, E.; Sadiq, O.; Chatterjee, K.; Rubinsztein, D.C. Wild-Type but Not Mutant Huntingtin Modulates the Transcriptional Activity of Liver X Receptors. J. Med. Genet. 2009, 46, 438–446. [Google Scholar] [CrossRef]
- Gray, S.M.; Dai, J.; Smith, A.C.; Beckley, J.T.; Rahmati, N.; Lewis, M.C.; Quirk, M.C. Changes in 24(S)-Hydroxycholesterol Are Associated with Cognitive Performance in Early Huntington’s Disease: Data from the TRACK and ENROLL HD Cohorts. J. Huntingt. Dis. 2024, 13, 449–465. [Google Scholar] [CrossRef]
- Martí-Martínez, S.; Valor, L.M. A Glimpse of Molecular Biomarkers in Huntington’s Disease. Int. J. Mol. Sci. 2022, 23, 5411. [Google Scholar] [CrossRef]
- Johnson, E.B.; Byrne, L.M.; Gregory, S.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.R.; Roos, R.A.; Zetterberg, H.; Tabrizi, S.J.; et al. Neurofilament Light Protein in Blood Predicts Regional Atrophy in Huntington Disease. Neurology 2018, 90, e717–e723. [Google Scholar] [CrossRef]
- Chang, K.H.; Cheng, M.L.; Lo, C.J.; Fan, C.M.; Wu, Y.R.; Chen, C.M. Alternations of Lipoprotein Profiles in the Plasma as Biomarkers of Huntington’s Disease. Cells 2023, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Bankov, G.; Kim, M.; Wretlind, A.; Lord, J.; Green, R.; Hodges, A.; Hye, A.; Aarsland, D.; Velayudhan, L.; et al. Integrated Lipidomics and Proteomics Network Analysis Highlights Lipid and Immunity Pathways Associated with Alzheimer’s Disease. Transl. Neurodegener. 2020, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Cheyuo, C.; Yang, W.L.; Wang, P. The Critical Role of Adrenomedullin and Its Binding Protein, AMBP-1, in Neuroprotection. Biol. Chem. 2012, 393, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Pihl, R.; Jensen, R.K.; Poulsen, E.C.; Jensen, L.; Hansen, A.G.; Thøgersen, I.B.; Dobó, J.; Gál, P.; Andersen, G.R.; Enghild, J.J.; et al. ITIH4 Acts as a Protease Inhibitor by a Novel Inhibitory Mechanism. Sci. Adv. 2021, 7, eaba7381. [Google Scholar] [CrossRef]
- Tanaka, H.; Shimazawa, M.; Takata, M.; Kaneko, H.; Tsuruma, K.; Ikeda, T.; Warita, H.; Aoki, M.; Yamada, M.; Takahashi, H.; et al. ITIH4 and Gpx3 Are Potential Biomarkers for Amyotrophic Lateral Sclerosis. J. Neurol. 2013, 260, 1782–1797. [Google Scholar] [CrossRef]
- Shi, X.; Ohta, Y.; Liu, X.; Shang, J.; Morihara, R.; Nakano, Y.; Feng, T.; Huang, Y.; Sato, K.; Takemoto, M.; et al. Acute Anti-Inflammatory Markers ITIH4 and AHSG in Mice Brain of a Novel Alzheimer’s Disease Model. J. Alzheimer’s Dis. 2019, 68, 1667–1675. [Google Scholar] [CrossRef]
- Nimmo, J.; Byrne, R.A.J.; Daskoulidou, N.; Watkins, L.M.; Carpanini, S.M.; Zelek, W.M.; Morgan, B.P. The Complement System in Neurodegenerative Diseases. Clin. Sci. 2024, 138, 387–412. [Google Scholar] [CrossRef]
- Zhang, D.F.; Fan, Y.; Xu, M.; Wang, G.; Wang, D.; Li, J.; Kong, L.L.; Zhou, H.; Luo, R.; Bi, R.; et al. Complement C7 Is a Novel Risk Gene for Alzheimer’s Disease in Han Chinese. Natl. Sci. Rev. 2019, 6, 257–274. [Google Scholar] [CrossRef]
- Rust, M.B.; Marcello, E. Disease Association of Cyclase-Associated Protein (CAP): Lessons from Gene-Targeted Mice and Human Genetic Studies. Eur. J. Cell Biol. 2022, 101, 151207. [Google Scholar] [CrossRef]
- Chang, K.H.; Chen, Y.C.; Wu, Y.R.; Lee, W.F.; Chen, C.M. Downregulation of Genes Involved in Metabolism and Oxidative Stress in the Peripheral Leukocytes of Huntington’s Disease Patients. PLoS ONE 2012, 7, e46492. [Google Scholar] [CrossRef] [PubMed]
- Vanderburg, C.R.; Davis, D.A.; Diamond, R.E.; Kao, P.F.; Delalle, I. Capzb2 Protein Expression in the Brains of Patients Diagnosed with Alzheimer’s Disease and Huntington’s Disease. Transl. Neurosci. 2010, 1, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Zanon Agapito-Tenfen, S.; Guerra, M.P.; Nodari, R.O.; Wikmark, O.G. Untargeted Proteomics-Based Approach to Investigate Unintended Changes in Genetically Modified Maize for Environmental Risk Assessment Purpose. Front. Toxicol. 2021, 3, 655968. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shukla, H.D.; Wu, C.; Saxena, S. Challenges and Solutions in Proteomics. Curr. Genom. 2007, 8, 21–28. [Google Scholar] [CrossRef]
| UHDRS Characteristics of Cypriot HD Patients | |||||
|---|---|---|---|---|---|
| Variable | HD (n = 36) | Asymptomatic (n = 18) | Early Symptomatic (n = 10) | Symptomatic Advanced (n = 8) | |
| TMS (0–124) | Mean (SD) | 21.38 (20.19) | 5.4 (12.1) | 29.4 (6.94) | 47.2 (7.20) |
| TBS (0–224) | Mean (SD) | 18.66 (18.06) | 8.0 (10.3) | 22.2 (10.60) | 38.2 (21.9) |
| IS (0–100%) | Mean (SD) | 65 (5.27) | 90 (5) | 58 (8) | 15 (10) |
| TFC (0–13) | Mean (SD) | 7.08 (5.27) | 11 (4.14) | 5.1 (2.13) | 0.75 (0.70) |
| Behavioral Milestones | |||||
| Confused | N (%) | ||||
| No/Yes | 29 (64)/7 (19) | 16 (89)/2 (11) | 9 (90)/1 (10) | 4 (50)/4 (50) | |
| Demented | N (%) | ||||
| No/Yes | 23 (64)/13 (36) | 10 (56)/8 (44) | 6 (60)/4 (40) | 1 (13)/7 (88) | |
| Depressed | N (%) | ||||
| No/Yes | 13 (36)/23 (64) | 10 (56)/8 (44) | 1 (10)/9 (90) | 2 (25)/6 (75) | |
| Requiring SSRI’s | N (%) | ||||
| No/Yes | 13 (36)/23 (64) | 10 (56)/8 (44) | 0/10 (100) | 3 (38)/5 (63) | |
| Group Name | Total Proteins Identified (n = 1638) | Number of Differentially Over-Expressed Proteins (FC > 1.2, p < 0.05) | Number of Differentially Under-Expressed Proteins (FC < −1/1.2, p < 0.05) |
|---|---|---|---|
| Asymptomatic HD vs. Control | 583 | 45 | 35 |
| Early Symptomatic HD vs. Control | 534 | 25 | 38 |
| Symptomatic Advanced HD vs. Control | 521 | 14 | 45 |
| Protein Name | UniProt ID | Biological Function | Previously Reported in HD or Neurodegeneration | Ref. |
|---|---|---|---|---|
| Complement Component 5 (C5) | P01031 | Phagocytosis, innate immune response, inflammation | AD, PD ALS, HD | [94] |
| Complement Component (C7) | Q8TCS7 | Membrane attack complex, innate immune response | AD, ALS, HD | [95] |
| Complement Component (C9) | A0A8Q3SI37 | Membrane attack complex, innate and adaptive immune response | AD, HD | [94] |
| Mannan-Binding Lectin Serine Protease 1 (MASP1) | P48740 | Lectin pathway involvement in complement system, activation of MASP2 and MASP3 | AD, HD | [94] |
| Alpha-1-Microglobulin/Bikunin Precursor (AMBP) | P02760 | Antioxidant, tissue repair, reed cell homeostasis | AD, HD | [90] |
| Inter-α Trypsin Inhibitor Heavy Chain 4 (ITIH4) | Q14624 | Anti-inflammatory, inflammation and host defense | AD, HD | [93] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christodoulou, C.C.; Demetriou, C.A.; Zamba-Papanicolaou, E. Stage-Specific Serum Proteomic Signatures Reveal Early Biomarkers and Molecular Pathways in Huntington’s Disease Progression. Cells 2025, 14, 1195. https://doi.org/10.3390/cells14151195
Christodoulou CC, Demetriou CA, Zamba-Papanicolaou E. Stage-Specific Serum Proteomic Signatures Reveal Early Biomarkers and Molecular Pathways in Huntington’s Disease Progression. Cells. 2025; 14(15):1195. https://doi.org/10.3390/cells14151195
Chicago/Turabian StyleChristodoulou, Christiana C., Christiana A. Demetriou, and Eleni Zamba-Papanicolaou. 2025. "Stage-Specific Serum Proteomic Signatures Reveal Early Biomarkers and Molecular Pathways in Huntington’s Disease Progression" Cells 14, no. 15: 1195. https://doi.org/10.3390/cells14151195
APA StyleChristodoulou, C. C., Demetriou, C. A., & Zamba-Papanicolaou, E. (2025). Stage-Specific Serum Proteomic Signatures Reveal Early Biomarkers and Molecular Pathways in Huntington’s Disease Progression. Cells, 14(15), 1195. https://doi.org/10.3390/cells14151195