
INHIBITION OF THE PROSTAGLANDIN-DEGRADING ENZYME 15-PGDH AMELIORATES MASH-ASSOCIATED APOPTOSIS AND FIBROSIS IN MICE

 ,
, 
Abstract
1. INTRODUCTION
2. MATERIALS and METHODS
2.1. Animal Model
2.2. Metabolic Compartment Analysis
2.3. Plasma and Tissue Collection
2.4. Plasma Treatment and Mass Spectrometry
2.5. Protein Expression
2.6. Morphology
2.7. Liver Fat Content
2.8. Liver Fibrosis
2.9. Liver Cellular Apoptosis
2.10. Statistical Analysis
3. RESULTS
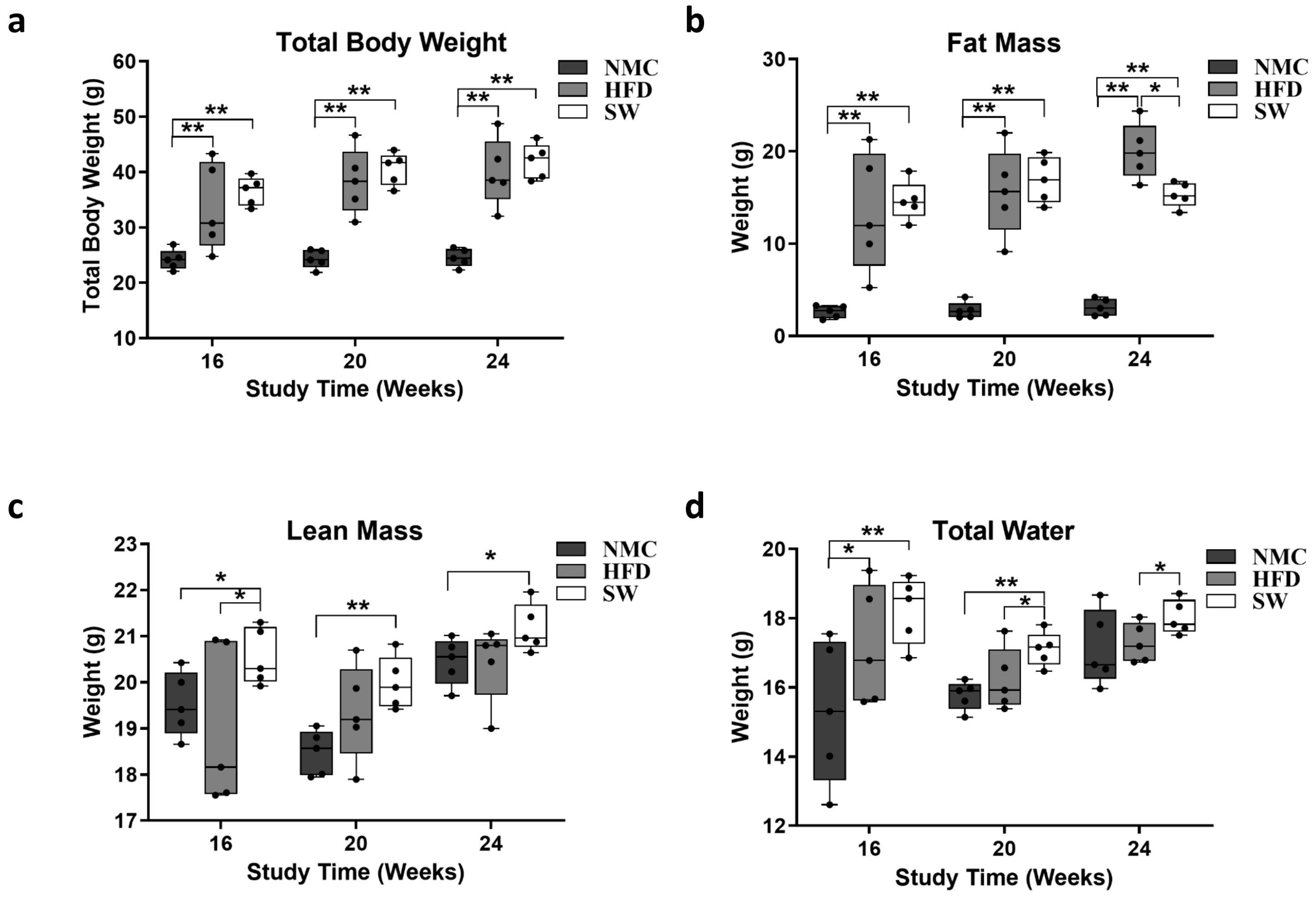
3.1. Total Body Weight and Fat Mass
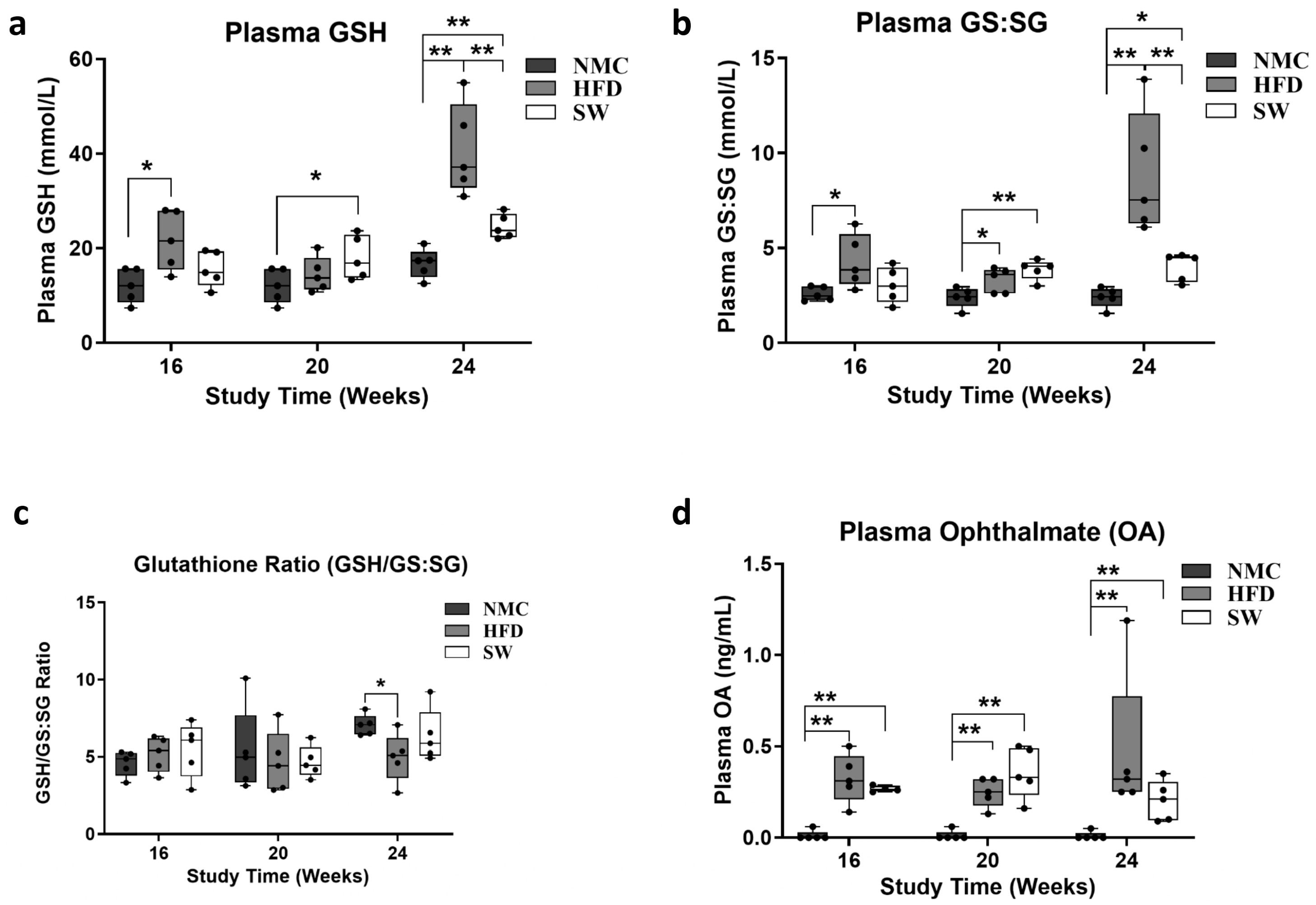
3.2. Oxidative Stress (Glutathione Species and Ophthalmic Acid)
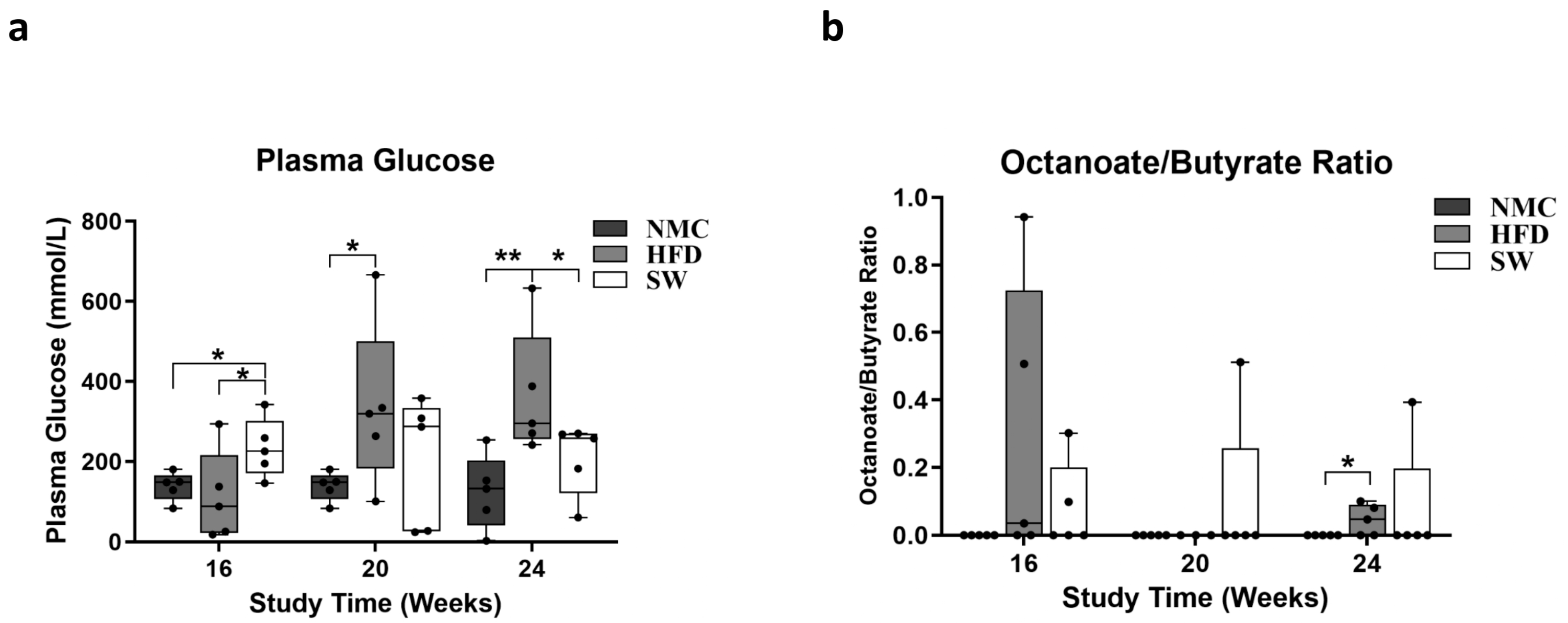
3.3. Insulin Resistance and Lipid β-Oxidation
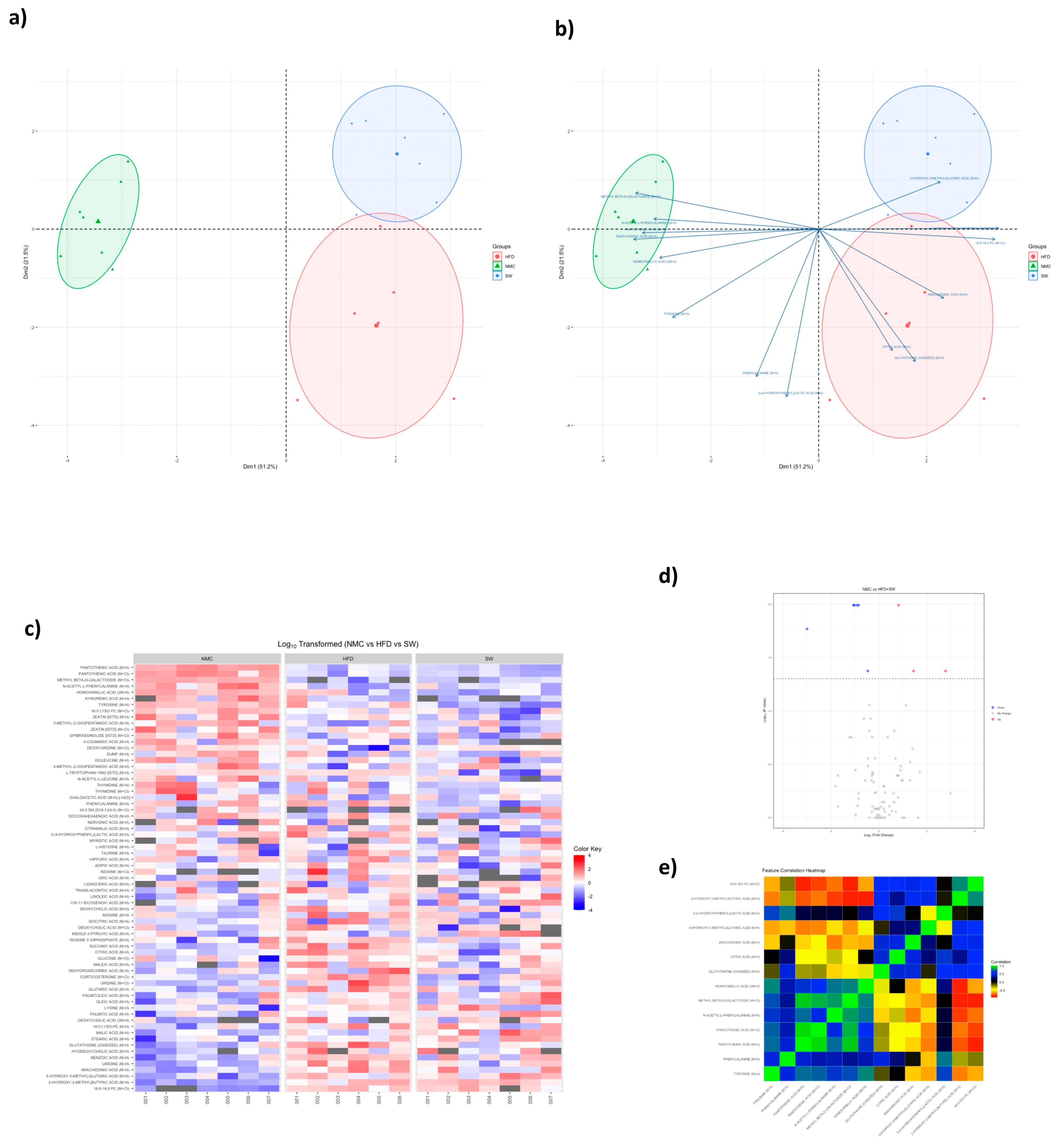
3.4. Non-Targeted Metabolomics
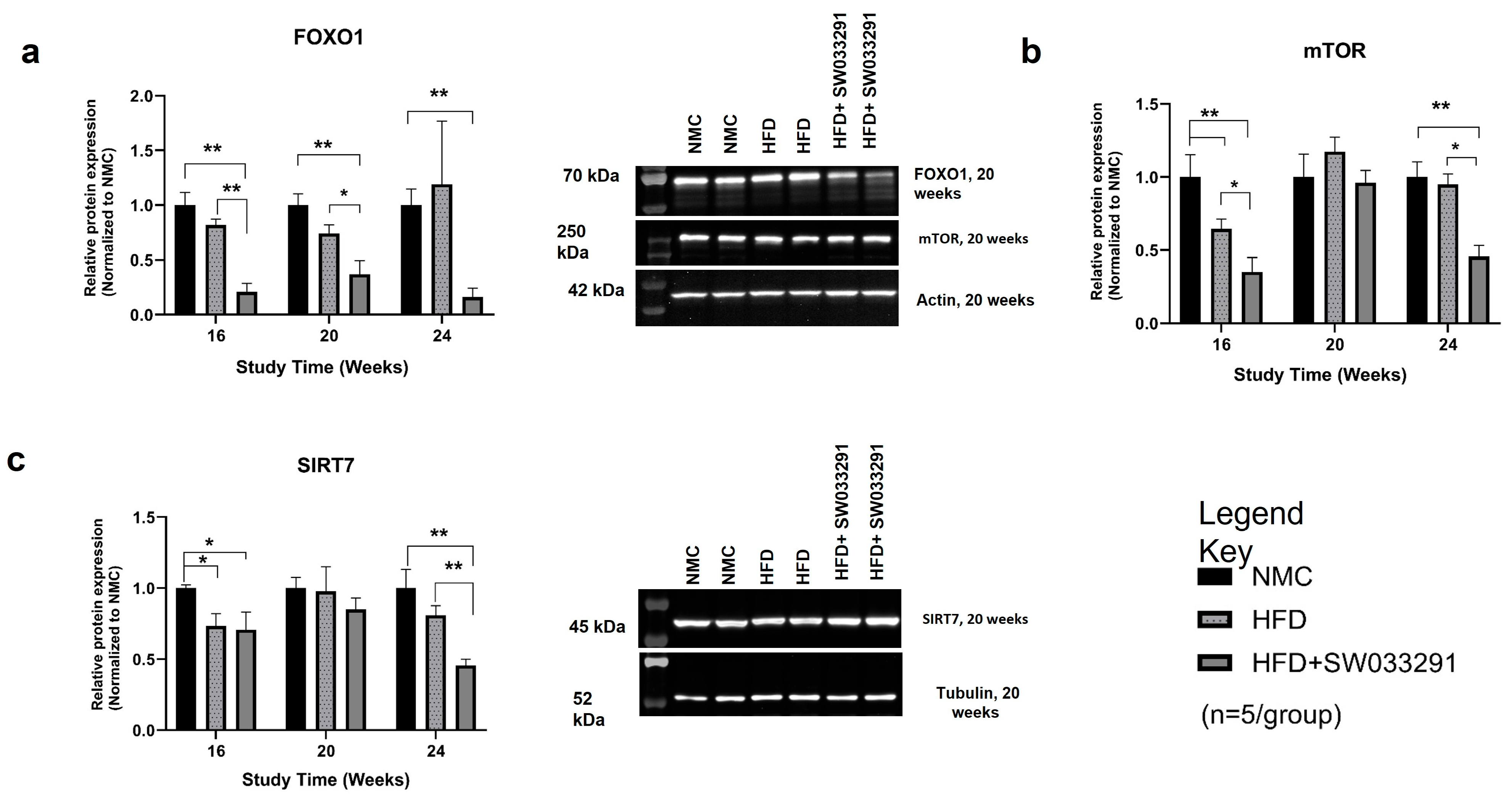
3.5. Protein Expression
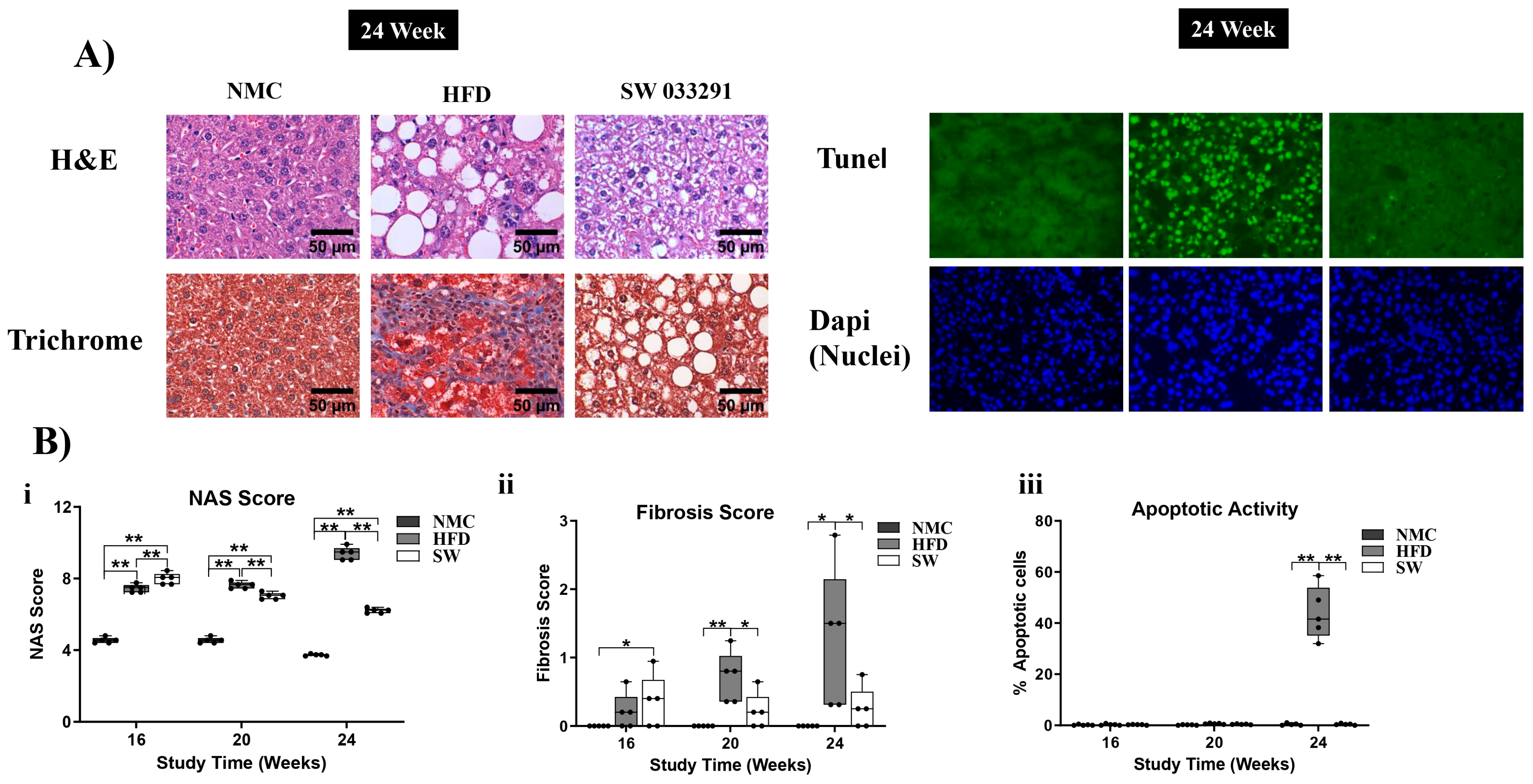
3.6. Liver Morphology
3.6.1. The NAS Score
3.6.2. Fibrosis Score
3.6.3. Apoptotic Activity
4. DISCUSSION
5. CONCLUSIONS
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Devarbhavi, H.; Asrani, S.K.; Arab, J.P.; Nartey, Y.A.; Pose, E.; Kamath, P.S. Global burden of liver disease: 2023 update. J. Hepatol. 2023, 79, 516–537. [Google Scholar] [CrossRef] [PubMed]
- Sepanlou, S.G.; Safiri, S.; Bisignano, C.; Ikuta, K.S.; Merat, S.; Saberifiroozi, M.; Poustchi, H.; Tsoi, D.; Colombara, D.V.; Abdoli, A.; et al. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 245–266. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Wong, G.; Anstee, Q.M.; Henry, L. The Global Burden of Liver Disease. Clin. Gastroenterol. Hepatol. 2023, 21, 1978–1991. [Google Scholar] [CrossRef]
- Henry, L.; Paik, J.; Younossi, Z.M. Review article: The epidemiologic burden of non-alcoholic fatty liver disease across the world. Aliment. Pharmacol. Ther. 2022, 56, 942–956. [Google Scholar] [CrossRef]
- Koo, S.H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Fazel, Y.; Koenig, A.B.; Sayiner, M.; Goodman, Z.D.; Younossi, Z.M. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism 2016, 65, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Bugianesi, E.; McCullough, A.J.; Marchesini, G. Insulin resistance: A metabolic pathway to chronic liver disease. Hepatology 2005, 42, 987–1000. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular Pathogenesis of Nonalcoholic Steatohepatitis- (NASH-) Related Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 8543763. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, F.; Wang, J.; Wong, R.J. Nonalcoholic Steatohepatitis Becomes the Leading Indication for Liver Transplant Registrants Among US Adults Born Between 1945 and 1965. J. Clin. Exp. Hepatol. 2020, 10, 30–36. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Henry, L. Epidemiology of non-alcoholic fatty liver disease and hepatocellular carcinoma. JHEP Rep. 2021, 3, 100305. [Google Scholar] [CrossRef]
- Swinburn, B.A.; Kraak, V.I.; Allender, S.; Atkins, V.J.; Baker, P.I.; Bogard, J.R.; Brinsden, H.; Calvillo, A.; De Schutter, O.; Devarajan, R.; et al. The Global Syndemic of Obesity, Undernutrition, and Climate Change: The Lancet Commission report. Lancet 2019, 393, 791–846. [Google Scholar] [CrossRef] [PubMed]
- Ward, Z.J.; Bleich, S.N.; Cradock, A.L.; Barrett, J.L.; Giles, C.M.; Flax, C.; Long, M.W.; Gortmaker, S.L. Projected U.S. State-Level Prevalence of Adult Obesity and Severe Obesity. N. Engl. J. Med. 2019, 381, 2440–2450. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, X.J.; Li, H. Progress and challenges in the prevention and control of nonalcoholic fatty liver disease. Med. Res. Rev. 2019, 39, 328–348. [Google Scholar] [CrossRef]
- Udoh, U.A.; Sanabria, J.D.; Rajan, P.K.; Banerjee, M.; Schade, M.; Sanabria, J.A.; Smith, G.; Udoh, G.; Sodhi, K.; Pierre, S.; et al. Non-Alcoholic Fatty Liver Disease Progression to Non-Alcoholic Steatohepatitis-Related Primary Liver Cancer. In Liver Cancer; Sergi, C.M., Ed.; Exon Publications: Brisbane, Australia, 2021. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-N.; Xue, F.; Zhang, N.; Zhang, W.; Hou, J.-J.; Lv, Y.; Xiang, J.-X.; Zhang, X.-F. Global burden of liver cirrhosis and other chronic liver diseases caused by specific etiologies from 1990 to 2019. BMC Public Health 2024, 24, 363. [Google Scholar] [CrossRef]
- Keam, S.J. Resmetirom: First Approval. Drugs 2024, 84, 729–735. [Google Scholar] [CrossRef]
- An effective MASH drug is good, but biotech can make it better. Nat. Biotechnol. 2024, 42, 685–686. [CrossRef]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef]
- Hoggatt, J.; Singh, P.; Sampath, J.; Pelus, L.M. Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood 2009, 113, 5444–5455. [Google Scholar] [CrossRef]
- Hoggatt, J.; Mohammad, K.S.; Singh, P.; Hoggatt, A.F.; Chitteti, B.R.; Speth, J.M.; Hu, P.; Poteat, B.A.; Stilger, K.N.; Ferraro, F.; et al. Differential stem- and progenitor-cell trafficking by prostaglandin E2. Nature 2013, 495, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Desai, A.; Yang, S.Y.; Bae, K.B.; Antczak, M.I.; Fink, S.P.; Tiwari, S.; Willis, J.E.; Williams, N.S.; Dawson, D.M.; et al. TISSUE REGENERATION. Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science 2015, 348, aaa2340. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clin. Immunol. 2006, 119, 229–240. [Google Scholar] [CrossRef]
- Mallipeddi, P.L.; Zhang, Y.; Li, H.; Markowitz, S.D.; Posner, B. Structural Insights into Novel 15-Prostaglandin Dehydrogenase Inhibitors. Molecules 2021, 26, 5903. [Google Scholar] [CrossRef] [PubMed]
- Antczak, M.I.; Zhang, Y.; Wang, C.; Doran, J.; Naidoo, J.; Voruganti, S.; Williams, N.S.; Markowitz, S.D.; Ready, J.M. Inhibitors of 15-Prostaglandin Dehydrogenase to Potentiate Tissue Repair. J. Med. Chem. 2017, 60, 3979–4001. [Google Scholar] [CrossRef]
- Xie, M.; He, R.; Wang, H.; Zhao, S.; Zheng, J.; Wu, J.; Wen, H. Effects of small molecule inhibitor SW033291 on hepatic ischemia-reperfusion injury in mice. Biochem. Biophys. Res. Commun. 2022, 615, 70–74. [Google Scholar] [CrossRef]
- Wang, W.; Liang, M.; Wang, L.; Bei, W.; Guo, J. 15-Hydroxyprostaglandin dehydrogenase inhibitor SW033291 ameliorates hepatic abnormal lipid metabolism, ER stress, and inflammation through PGE(2)/EP4 in T2DM mice. Bioorgan. Chem. 2023, 137, 106646. [Google Scholar] [CrossRef]
- Rubino, M.; Travers, J.G.; Headrick, A.L.; Enyart, B.T.; Lemieux, M.E.; Cavasin, M.A.; Schwisow, J.A.; Hardy, E.J.; Kaltenbacher, K.J.; Felisbino, M.B.; et al. Inhibition of Eicosanoid Degradation Mitigates Fibrosis of the Heart. Circ. Res. 2023, 132, 10–29. [Google Scholar] [CrossRef]
- Bärnthaler, T.; Theiler, A.; Zabini, D.; Trautmann, S.; Stacher-Priehse, E.; Lanz, I.; Klepetko, W.; Sinn, K.; Flick, H.; Scheidl, S.; et al. Inhibiting eicosanoid degradation exerts antifibrotic effects in a pulmonary fibrosis mouse model and human tissue. J. Allergy Clin. Immunol. 2020, 145, 818–833.e811. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Yokotobi, A.; Yamamoto, N.; Takada, M.; Kawase, A.; Nakanishi, T.; Iwaki, M. Inhibition of 15-prostaglandin dehydrogenase attenuates acetaminophen-induced liver injury via suppression of apoptosis in liver endothelial cells. Prostaglandins Leukot. Essent. Fat. Acids 2024, 202, 102640. [Google Scholar] [CrossRef]
- Li, J.; Liu, H.; Jia, Y.; Tuniyazi, X.; Liao, X.; Zhao, J.; Du, Y.; Fang, Z.; Lü, G. SW033291 promotes liver regeneration after acetaminophen-induced liver injury in mice. Biochem. Biophys. Res. Commun. 2025, 749, 151365. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Zhang, G.F.; Kasumov, T.; Roe, C.R.; Brunengraber, H. Interrelations between C4 ketogenesis, C5 ketogenesis, and anaplerosis in the perfused rat liver. J. Biol. Chem. 2009, 284, 27799–27807. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Jiang, E.; Wang, C.; Li, Y.-Y.; Wang, X.-C.; An, J.-D.; Wang, Y.-J.; Wang, X.-J. Mistletoe alkaloid fractions alleviates carbon tetrachloride-induced liver fibrosis through inhibition of hepatic stellate cell activation via TGF-β/Smad interference. J. Ethnopharmacol. 2014, 158, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Zheng, B.; Chen, T.; Chang, X.; Yin, B.; Huang, Z.; Shuai, P.; Han, L. Semen Brassicae ameliorates hepatic fibrosis by regulating transforming growth factor-β1/Smad, nuclear factor-κB, and AKT signaling pathways in rats. Drug Des. Dev. Ther. 2018, 12, 1205–1213. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Kalhan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, A.J.; Hanson, R.W.; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism 2011, 60, 404–413. [Google Scholar] [CrossRef]
- Roberto, P.; Miriam, L.; Adele, A.; Erika, P.; Annalisa, C.; Marica, M.; Paola, D. Old-fashioned and newly discovered biomarkers: The future of NAFLD-related HCC screening and monitoring. Hepatoma Res. 2022, 8, 37. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Liang, M.; Wang, L.; Wang, W. The 15-hydroxyprostaglandin dehydrogenase inhibitor SW033291 ameliorates abnormal hepatic glucose metabolism through PGE2–EP4 receptor–AKT signaling in a type 2 diabetes mellitus mouse model. Cell. Signal. 2023, 108, 110707. [Google Scholar] [CrossRef] [PubMed]
- Vairetti, M.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Ferrigno, A.; Berardo, C. Changes in Glutathione Content in Liver Diseases: An Update. Antioxidants 2021, 10, 364. [Google Scholar] [CrossRef]
- Dou, X.; Li, S.; Hu, L.; Ding, L.; Ma, Y.; Ma, W.; Chai, H.; Song, Z. Glutathione disulfide sensitizes hepatocytes to TNFα-mediated cytotoxicity via IKK-β S-glutathionylation: A potential mechanism underlying non-alcoholic fatty liver disease. Exp. Mol. Med. 2018, 50, 1–16. [Google Scholar] [CrossRef]
- Santacroce, G.; Gentile, A.; Soriano, S.; Novelli, A.; Lenti, M.V.; Di Sabatino, A. Glutathione: Pharmacological aspects and implications for clinical use in non-alcoholic fatty liver disease. Front. Med. 2023, 10, 1124275. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Dysregulation of glutathione synthesis in liver disease. Liver Res. 2020, 4, 64–73. [Google Scholar] [CrossRef]
- Soga, T.; Baran, R.; Suematsu, M.; Ueno, Y.; Ikeda, S.; Sakurakawa, T.; Kakazu, Y.; Ishikawa, T.; Robert, M.; Nishioka, T.; et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006, 281, 16768–16776. [Google Scholar] [CrossRef]
- Andres Ibarra, R.; Abbas, R.; Kombu, R.S.; Zhang, G.F.; Jacobs, G.; Lee, Z.; Brunengraber, H.; Sanabria, J.R. Disturbances in the glutathione/ophthalmate redox buffer system in the woodchuck model of hepatitis virus-induced hepatocellular carcinoma. HPB Surg. 2011, 2011, 789323. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Solis Herruzo, J.A.; Garcia Ruiz, I.; Perez Carreras, M.; Munoz Yague, M.T. Non-alcoholic fatty liver disease. From insulin resistance to mitochondrial dysfunction. Rev. Esp. Enferm. Dig. 2006, 98, 844–874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Agarwal, K.C.; Beylot, M.; Soloviev, M.V.; David, F.; Reider, M.W.; Anderson, V.E.; Tserng, K.Y.; Brunengraber, H. Nonhomogeneous labeling of liver extra-mitochondrial acetyl-CoA. Implications for the probing of lipogenic acetyl-CoA via drug acetylation and for the production of acetate by the liver. J. Biol. Chem. 1994, 269, 11025–11029. [Google Scholar] [CrossRef]
- Kasumov, T.; Adams, J.E.; Bian, F.; David, F.; Thomas, K.R.; Jobbins, K.A.; Minkler, P.E.; Hoppel, C.L.; Brunengraber, H. Probing peroxisomal beta-oxidation and the labelling of acetyl-CoA proxies with [1-(13C)]octanoate and [3-(13C)]octanoate in the perfused rat liver. Biochem. J. 2005, 389, 397–401. [Google Scholar] [CrossRef]
- Des Rosiers, C.; David, F.; Garneau, M.; Brunengraber, H. Nonhomogeneous labeling of liver mitochondrial acetyl-CoA. J. Biol. Chem. 1991, 266, 1574–1578. [Google Scholar] [CrossRef] [PubMed]
- van de Wier, B.; Balk, J.M.; Haenen, G.R.; Giamouridis, D.; Bakker, J.A.; Bast, B.C.; den Hartog, G.J.; Koek, G.H.; Bast, A. Elevated citrate levels in non-alcoholic fatty liver disease: The potential of citrate to promote radical production. FEBS Lett. 2013, 587, 2461–2466. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.P.; Hazlehurst, J.M.; Tomlinson, J.W. Glucocorticoids and non-alcoholic fatty liver disease. J. Steroid Biochem. Mol. Biol. 2015, 154, 94–103. [Google Scholar] [CrossRef]
- Yoshizawa, T.; Karim, M.F.; Sato, Y.; Senokuchi, T.; Miyata, K.; Fukuda, T.; Go, C.; Tasaki, M.; Uchimura, K.; Kadomatsu, T.; et al. SIRT7 Controls Hepatic Lipid Metabolism by Regulating the Ubiquitin-Proteasome Pathway. Cell Metab. 2014, 19, 712–721. [Google Scholar] [CrossRef]
- Gosis, B.S.; Wada, S.; Thorsheim, C.; Li, K.; Jung, S.; Rhoades, J.H.; Yang, Y.; Brandimarto, J.; Li, L.; Uehara, K.; et al. Inhibition of nonalcoholic fatty liver disease in mice by selective inhibition of mTORC1. Science 2022, 376, eabf8271. [Google Scholar] [CrossRef]
- Arora, M.; Pavlíková, Z.; Kučera, T.; Kozlík, P.; Šopin, T.; Vacík, T.; Ľupták, M.; Duda, M.; Slanař, O.; Kutinová Canová, N. Pharmacological effects of mTORC1/C2 inhibitor in a preclinical model of NASH progression. Biomed. Pharmacother. 2023, 167, 115447. [Google Scholar] [CrossRef]
- Sha, X.; Zou, X.; Liu, S.; Guan, C.; Shi, W.; Gao, J.; Zhong, X.; Jiang, X. Forkhead box O1 in metabolic dysfunction-associated fatty liver disease: Molecular mechanisms and drug research. Front. Nutr. 2024, 11. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.C.; Turner, N. FOX01 Is the Headline Akt Regulating Hepatic Glucose Metabolism. Endocrinology 2017, 158, 2436–2438. [Google Scholar] [CrossRef]
- Kousteni, S. FoxO1, the transcriptional chief of staff of energy metabolism. Bone 2012, 50, 437–443. [Google Scholar] [CrossRef]
- Albert, V.; Hall, M.N. mTOR signaling in cellular and organismal energetics. Curr. Opin. Cell Biol. 2015, 33, 55–66. [Google Scholar] [CrossRef]
- Okuno, T.; Kakehashi, A.; Ishii, N.; Fujioka, M.; Gi, M.; Wanibuchi, H. mTOR Activation in Liver Tumors Is Associated with Metabolic Syndrome and Non-Alcoholic Steatohepatitis in Both Mouse Models and Humans. Cancers 2018, 10, 465. [Google Scholar] [CrossRef]
- Zhou, W.; Ye, S. Rapamycin improves insulin resistance and hepatic steatosis in type 2 diabetes rats through activation of autophagy. Cell Biol. Int. 2018, 42, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Sirtuins and nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 10084–10092. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. SIRT7 and hepatic lipid metabolism. Front. Cell Dev. Biol. 2015, 3. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Smith-Cortinez, N.; van Eunen, K.; Heegsma, J.; Serna-Salas, S.A.; Sydor, S.; Bechmann, L.P.; Moshage, H.; Bakker, B.M.; Faber, K.N. Simultaneous Induction of Glycolysis and Oxidative Phosphorylation during Activation of Hepatic Stellate Cells Reveals Novel Mitochondrial Targets to Treat Liver Fibrosis. Cells 2020, 9, 2456. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Wang, X.; Jiao, Q.; Shao, Y.; Luo, R.; Zhang, J.; Zheng, X.; Zhou, S.; Chen, Y. Selective inhibition of glycolysis in hepatic stellate cells and suppression of liver fibrogenesis with vitamin A-derivative decorated camptothecin micelles. Acta Biomater. 2023, 168, 497–514. [Google Scholar] [CrossRef] [PubMed]
- Marra, F. Hepatic stellate cells and the regulation of liver inflammation. J. Hepatol. 1999, 31, 1106–1119. [Google Scholar] [CrossRef]
- Najar, M.; Fayyad-Kazan, H.; Faour, W.H.; El Taghdouini, A.; Raicevic, G.; Najimi, M.; Toungouz, M.; van Grunsven, L.A.; Sokal, E.; Lagneaux, L. Human hepatic stellate cells and inflammation: A regulated cytokine network balance. Cytokine 2017, 90, 130–134. [Google Scholar] [CrossRef]
- Gupta, G.; Khadem, F.; Uzonna, J.E. Role of hepatic stellate cell (HSC)-derived cytokines in hepatic inflammation and immunity. Cytokine 2019, 124, 154542. [Google Scholar] [CrossRef]
- Cheng, H.; Huang, H.; Guo, Z.; Chang, Y.; Li, Z. Role of prostaglandin E2 in tissue repair and regeneration. Theranostics 2021, 11, 8836–8854. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Cichoż-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Urtasun, R.; Conde de la Rosa, L.; Nieto, N. Oxidative and nitrosative stress and fibrogenic response. Clin. Liver Dis. 2008, 12, 769–790, viii. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Kahn, S.E. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef]
- Nogueira, J.P.; Cusi, K. Role of Insulin Resistance in the Development of Nonalcoholic Fatty Liver Disease in People with Type 2 Diabetes: From Bench to Patient Care. Diabetes Spectr. 2024, 37, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Negi, C.K.; Babica, P.; Bajard, L.; Bienertova-Vasku, J.; Tarantino, G. Insights into the molecular targets and emerging pharmacotherapeutic interventions for nonalcoholic fatty liver disease. Metabolism 2022, 126, 154925. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.A.; Gallego, P.; Grande, L. Role of inflammatory response in liver diseases: Therapeutic strategies. World J. Hepatol. 2018, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pratim Das, P.; Medhi, S. Role of inflammasomes and cytokines in immune dysfunction of liver cirrhosis. Cytokine 2023, 170, 156347. [Google Scholar] [CrossRef]
- Zang, S.; Ma, X.; Wu, Y.; Liu, W.; Cheng, H.; Li, J.; Liu, J.; Huang, A. PGE2 synthesis and signaling in malignant transformation and progression of human hepatocellular carcinoma. Human Pathol. 2017, 63, 120–127. [Google Scholar] [CrossRef]
- Hasan, S.; Satake, M.; Dawson, D.W.; Funahashi, H.; Angst, E.; Go, V.L.; Reber, H.A.; Hines, O.J.; Eibl, G. Expression analysis of the prostaglandin E2 production pathway in human pancreatic cancers. Pancreas 2008, 37, 121–127. [Google Scholar] [CrossRef]
- Schmidt, A.; Sinnett-Smith, J.; Young, S.; Chang, H.H.; Hines, O.J.; Dawson, D.W.; Rozengurt, E.; Eibl, G. Direct growth-inhibitory effects of prostaglandin E2 in pancreatic cancer cells in vitro through an EP4/PKA-mediated mechanism. Surgery 2017, 161, 1570–1578. [Google Scholar] [CrossRef]
- Wang, D.; Fu, L.; Sun, H.; Guo, L.; DuBois, R.N. Prostaglandin E2 Promotes Colorectal Cancer Stem Cell Expansion and Metastasis in Mice. Gastroenterology 2015, 149, 1884–1895.e1884. [Google Scholar] [CrossRef]
- Karpisheh, V.; Nikkhoo, A.; Hojjat-Farsangi, M.; Namdar, A.; Azizi, G.; Ghalamfarsa, G.; Sabz, G.; Yousefi, M.; Yousefi, B.; Jadidi-Niaragh, F. Prostaglandin E2 as a potent therapeutic target for treatment of colon cancer. Prostaglandins Other Lipid Mediat. 2019, 144, 106338. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | ||||||||||||||
| Experimental Group | ||||||||||||||
| METABOLITE NAME | Class | NMC | HFD | NMC vs. HFD | SW033291 | HFD vs. SW033291 | ||||||||
| 24 Week | 24 Week | p-value * | 24 Week | p-value * | ||||||||||
| GLUTATHIONE (OXIDIZED) | Amino acid | 19.58 ± 12.0 | 58.31 ± 28.2 | NS | 25.95 ± 7.9 | <0.05 | ||||||||
| HIPPURIC ACID | Amino acid | 22,131.74 ± 19.9 | 22,456.87 ± 2210.2 | NS | 21,955.22 ± 2117.0 | NS | ||||||||
| ISOLEUCINE | Amino acid | 212.67 ± 45.4 | 186.67 ± 24.3 | NS | 165.58 ± 50.2 | NS | ||||||||
| L-HISTIDINE | Amino acid | 8.28 ± 3.3 | 7.55 ± 3.2 | NS | 8.21 ± 4.1 | NS | ||||||||
| L-TRYPTOPHAN | Amino acid | 526.58 ± 58.2 | 521.39 ± 52.2 | NS | 247.50 ± 215.5 | <0.05 | ||||||||
| LYSINE | Amino acid | 8.44 ± 2.2 | 10.98 ± 0.9 | NS | 10.83 ± 3.8 | NS | ||||||||
| N-ACETYL-L-LEUCINE | Amino acid | 130.31 ± 44.5 | 105.02 ± 29.9 | NS | 97.87 ± 28.6 | NS | ||||||||
| N-ACETYL-L-PHENYLALANINE | Amino acid | 61.05 ± 11.8 | 38.19 ± 6.4 | <0.05 | 42.01 ± 6.0 | NS | ||||||||
| PHENYLALANINE | Amino acid | 341.33 ± 50.1 | 364.37 ± 46.3 | NS | 271.41 ± 25.8 | <0.05 | ||||||||
| TYROSINE | Amino acid | 223.15 ± 20.1 | 184.14 ± 63.3 | NS | 102.57 ± 25.4 | <0.05 | ||||||||
| (b) | ||||||||||||||
| Experimental Group | ||||||||||||||
| METABOLITE NAME | Class | NMC | HFD | NMC vs. HFD | SW033291 | HFD vs. SW033291 | ||||||||
| 24 Week | 24 Week | p-value * | 24 Week | p-value * | ||||||||||
| BENZOIC ACID | Carbohydrate | 52.51 ± 18.5 | 96.71 ± 6.4 | NS | 79.76 ± 31.4 | NS | ||||||||
| CITRAMALIC ACID | Carbohydrate | 1700.75 ± 578.1 | 1864.37 ± 856.9 | NS | 1347.37 ± 566.5 | NS | ||||||||
| CITRIC ACID | Carbohydrate | 2708.11 ± 201.4 | 3746.76 ± 460.4 | <0.05 | 2775.55 ± 669.8 | <0.05 | ||||||||
| GLUCOSE | Carbohydrate | 282.89 ± 282.0 | 345.56 ± 150.7 | NS | 368.92 ± 308.1 | NS | ||||||||
| GLUTARIC ACID | Carbohydrate | 23.68 ± 7.3 | 47.88 ± 19.2 | NS | 27.78 ± 12.5 | <0.05 | ||||||||
| HOMOVANILLIC ACID | carbohydrate | 13.69 ± 1.0 | 10.05 ± 3.1 | <0.05 | 7.64 ± 1.5 | <0.05 | ||||||||
| INDOLE-3-PYRUVIC ACID | carbohydrate | 5357 ± 3622 | 4.77 ± 2.6 | NS | 15.25 ± 9.7 | NS | ||||||||
| ISOCITRIC ACID | carbohydrate | 3562.14 ± 250.8 | 4110.98 ± 995.2 | NS | 3773.45 ± 559.7 | NS | ||||||||
| MALEIC ACID | carbohydrate | 42.93 ± 15.0 | 73.77 ± 24.5 | <0.05 | 48.92 ± 19.2 | NS | ||||||||
| MALIC ACID | carbohydrate | 46.12 ± 11.0 | 57.99 ± 16.3 | NS | 92.63 ± 36.6 | NS | ||||||||
| OXALOACETIC ACID | carbohydrate | 412.77 ± 48.4 | 388.33 ± 39.3 | NS | 391.72 ± 21.9 | NS | ||||||||
| SUCCINIC ACID | carbohydrate | 20.83 ± 153.9 | 551.80 ± 356.4 | NS | 230.69 ± 179.6 | <0.05 | ||||||||
| TRANS-ACONITIC ACID | carbohydrate | 234.20 ± 210.3 | 313.10 ± 221.0 | NS | 231.61 ± 150.4 | NS | ||||||||
| (c) | ||||||||||||||
| Experimental Group | ||||||||||||||
| Class | NMC | HFD | NMC vs. HFD | SW033291 | HFD vs. SW033291 | |||||||||
| METABOLITE NAME | 24 Week | 24 Week | p-value * | 24 Week | p-value * | |||||||||
| ADIPIC ACID | Lipid | 9.02 ± 2.1 | 14.02 ± 5.7 | NS | 9.59 ± 7.5 | NS | ||||||||
| CAPRYLIC ACID | Lipid | 56.22 ± 20.2 | 187.55 ± 89.1 | NS | 135.93 ± 106.6 | NS | ||||||||
| CIS-11-EICOSENOIC ACID | Lipid | 6.65 ± 1.9 | 7.08 ± 2.2 | NS | 8.33 ± 3.5 | NS | ||||||||
| CORTICOSTERONE | Lipid | 51.34 ± 5.7 | 85.03 ± 11.3 | <0.05 | 55.76 ± 21.3 | <0.05 | ||||||||
| DOCOSAHEXAENOIC ACID | Lipid | 140.46 ± 91.9 | 125.75 ± 111.5 | NS | 111.24 ± 130.0 | NS | ||||||||
| EPIBRASSINOLIDE | Lipid | 430.64 ± 36.3 | 392.40 ± 39.4 | <0.05 | 377.29 ± 50.7 | NS | ||||||||
| LIGNOCERIC ACID | Lipid | 2.02 ± 0.7 | 1.95 ± 0.8 | NS | 2.40 ± 0.2 | NS | ||||||||
| LINOLEIC ACID | Lipid | 102.82 ± 66.7 | 114.24 ± 34.9 | NS | 129.82 ± 114.1 | NS | ||||||||
| MYRISTIC ACID | Lipid | 4.04 ± 1.5 | 3.55 ± 1.0 | NS | 4.19 ± 2.0 | NS | ||||||||
| NERVONIC ACID | Lipid | 1.96 ± 0.5 | 1.8 ± 0.3 | NS | 1.77 ± 0.2 | NS | ||||||||
| OLEIC ACID | Lipid | 177.67 ± 100.0 | 236.90 ± 51.5 | NS | 296.92 ± 178.9 | NS | ||||||||
| PALMITIC ACID | Lipid | 56.99 ± 32.5 | 63.56 ± 8.9 | NS | 97.62 ± 40.2 | NS | ||||||||
| SEBACIC ACID | Lipid | 20.57 ± 16.1 | 27.06 ± 7.9 | NS | 35.80 ± 26.9 | NS | ||||||||
| STEARIC ACID | Lipid | 57.71 ± 20.6 | 75.40 ± 15.2 | NS | 93.62 ± 23.5 | NS | ||||||||
| (d) | ||||||||||||||
| Metabolite Name | NMC | HFD | SW033291 | p-value | Benjamini–Hochberg corrected p-value | |||||||||
| 16:0 LYSO PC (M+Cl)- | 999,071 ± 67,793 | 871,748 ± 65,482 | 787,031 ± 163,053 | 0.0137 | NS | |||||||||
| 16:0–18:0 PC (M+Cl)- | 47,443 ± 5741 | 140,776 ± 25,533 | 132,632 ± 29,631 | 0.0049 | 0.0441 | |||||||||
| 18:0 LYSO-PE (M-H)- | 141,905 ± 76,160 | 306,758 ± 169,087 | 251,645 ± 148,057 | NS | NS | |||||||||
| 20:0 LYSO PC (M+Cl)- | 9055 ± 4134 | 12,829 ± 9749 | 8540 ± NA | NS | NS | |||||||||
| 24:0 SM (D18:1/24:0) (M+Cl)- | 72,815 ± 16,580 | 42,977 ± 9509 | 75,725 ± 30,083 | 0.0416 | NS | |||||||||
| 2-HYDROXY-3-METHYLBUTYRIC ACID (M-H)- | 136,900 ± 14,513 | 238,877 ± 40,419 | 253,001 ± 24,659 | 0.0014 | 0.0336 | |||||||||
| 2-HYDROXYBUTYRIC ACID (M-H)- | NaN ± NA | NaN ± NA | 48,141 ± NA | NP | NP | |||||||||
| 3-(4-HYDROXYPHENYL)LACTIC ACID (M-H)- | 233,017 ± 53,067 | 306,087 ± 80,924 | 159,901 ± 56,999 | 0.0066 | 0.0464 | |||||||||
| 3′-CMP (M-H)- | NaN ± NA | 3695 ± 3003 | 6276 ± NA | NS | NS | |||||||||
| 3-HYDROXY-3-METHYLGLUTARIC ACID (M-H)- | 31,353 ± 25,717 | 102,719 ± 56,648 | 153,170 ± 114,368 | 0.0069 | 0.0464 | |||||||||
| 3-METHYL-2-OXOPENTANOIC ACID (M-H)- | 1,062,442 ± 174,392 | 809,583 ± 202,873 | 859,039 ± 160,885 | NS | NS | |||||||||
| 3-UREIDOPROPIONIC ACID (M-H)- | 13,633 ± 4566 | 27,704 ± 25,280 | 23,812 ± 6769 | NS | NS | |||||||||
| 4-COUMARIC ACID (M-H)- | 31,824 ± 14,756 | 15,533 ± 8403 | 14,592 ± 10,303 | NS | NS | |||||||||
| 4-HYDROXYBENZOIC ACID (M-H)- | 9132 ± 1185 | 5225 ± NA | NaN ± NA | NS | NS | |||||||||
| 4-METHYL-2-OXOPENTANOIC ACID (M-H)- | 651,191 ± 187,117 | 468,621 ± 142,524 | 561,698 ± 137,764 | NS | NS | |||||||||
| ADIPIC ACID (M-H)- | 9023 ± 2135 | 14,024 ± 5700 | 9589 ± 7496 | NS | NS | |||||||||
| ARACHIDIC ACID (M-H)- | 17,808 ± 7593 | 16,941 ± 10,347 | 10,695 ± 5785 | NS | NS | |||||||||
| ARACHIDONIC ACID (M-H)- | 56,224 ± 20,232 | 187,550 ± 89,136 | 135,930 ± 106,642 | 0.0047 | 0.0441 | |||||||||
| BEHENIC ACID (M-H)- | 17,921 ± 2014 | 14,981 ± 3108 | 13,695 ± 3147 | NS | NS | |||||||||
| BENZOIC ACID (M-H)- | 52,510 ± 18,477 | 96,710 ± 6365 | 79,761 ± 31,390 | 0.0213 | NS | |||||||||
| CAPRYLIC ACID (M-H)- | 8235 ± NA | 14,695 ± 5273 | 91,301 ± 71,223 | NS | NS | |||||||||
| CIS-11-EICOSENOIC ACID (M-H)- | 6653 ± 1896 | 7079 ± 2194 | 8326 ± 3508 | NS | NS | |||||||||
| CITRAMALIC ACID (M-H)- | 1,700,753 ± 578074 | 1,864,369 ± 856,907 | 1,347,370 ± 566,523 | NS | NS | |||||||||
| CITRIC ACID (M-H)- | 2,708,109 ± 201,367 | 3,746,763 ± 460,431 | 2,775,550 ± 669,761 | 0.0022 | 0.036 | |||||||||
| CORTICOSTERONE (M+Cl)- | 51,373 ± 5709 | 85,029 ± 11,330 | 55,762 ± 21,315 | 0.0119 | NS | |||||||||
| DEHYDROASCORBIC ACID (M-H)- | 135,278 ± 73,273 | 348,721 ± 319,377 | 203,330 ± 91,806 | NS | NS | |||||||||
| DEOXYCHOLIC ACID (2M-H)- | 2967 ± 2017 | 7326 ± 6067 | 4896 ± 1959 | NS | NS | |||||||||
| DEOXYCHOLIC ACID (M+Cl)- | 34,177 ± 12,878 | 55,698 ± 25,533 | 40,464 ± 18,047 | NS | NS | |||||||||
| DEOXYCHOLIC ACID (M-H)- | 113,724 ± 46,601 | 199,481 ± 105,148 | 133,817 ± 69,673 | NS | NS | |||||||||
| DEOXYURIDINE (M+Cl)- | 154,902 ± 23,677 | 103,335 ± 48,760 | 131,551 ± 13,136 | NS | NS | |||||||||
| DEOXYURIDINE (M-H)- | 32,078 ± NA | 29,972 ± 7838 | 67,584 ± NA | NS | NS | |||||||||
| D-GLUCOSAMINE 6-SULFATE (M-H)-[-H2O] | 18,374 ± 8325 | 17,577 ± 1312 | 18,702 ± 24,299 | NS | NS | |||||||||
| DOCOSAHEXAENOIC ACID (M-H)- | 140,460 ± 91,921 | 125,748 ± 111,514 | 111,240 ± 130,038 | NS | NS | |||||||||
| DUMP (M-H)- | 47,184 ± 11,690 | 37,025 ± 15,323 | 36,035 ± 11,244 | NS | NS | |||||||||
| EPIBRASSINOLIDE [ISTD] (M+Cl)- | 430,639 ± 36,260 | 392,399 ± 39,397 | 377,287 ± 50,648 | NS | NS | |||||||||
| ERUCIC ACID (M-H)- | 6393 ± 3349 | 7132 ± 3663 | 4177 ± 2520 | NS | NS | |||||||||
| FLAVIN ADENINE DINUCLEOTIDE (M-H)- | 5672 ± 2621 | 12,029 ± NA | 6807 ± NA | NS | NS | |||||||||
| GLUCOSE (M+Cl)- | 282,885 ± 282,005 | 345,560 ± 150,718 | 368,916 ± 308,115 | NS | NS | |||||||||
| GLUTAMINE (M-H)- | 22,951 ± 9624 | 20,569 ± 9510 | 25,922 ± 17,703 | NS | NS | |||||||||
| GLUTARIC ACID (M-H)- | 23,683 ± 7280 | 47,883 ± 19,225 | 27,778 ± 12,511 | 0.0414 | NS | |||||||||
| GLUTATHIONE (OXIDIZED) (M-H)- | 19,578 ± 12,000 | 58,391 ± 28,237 | 25,952 ± 7853 | 0.0053 | 0.0441 | |||||||||
| HEPTADECANOIC ACID (M-H)- | 3545 ± 722 | 4146 ± 1220 | 3343 ± 972 | NS | NS | |||||||||
| HIPPURIC ACID (M-H)- | 22,131,742 ± 1,988,863 | 22,456,867 ± 2,210,184 | 21,955,215 ± 2,117,015 | NS | NS | |||||||||
| HOMOVANILLIC ACID (2M-H)- | 13,689 ± 1028 | 10,045 ± 3139 | 7643 ± 1505 | 0.0028 | 0.0336 | |||||||||
| HYODEOXYCHOLIC ACID (M-H)- | 74,010 ± 56,633 | 864,680 ± 1,358,876 | 748,160 ± 955,799 | NS | NS | |||||||||
| INDOLE-3-PYRUVIC ACID (M-H)- | 5357 ± 3622 | 4765 ± 2582 | 15,253 ± 9718 | NS | NS | |||||||||
| INOSINE (M+Cl)- | 5817 ± 3002 | 13,905 ± 17,966 | 4782 ± 2270 | NS | NS | |||||||||
| INOSINE (M-H)- | 26,120 ± 14,532 | 65,914 ± 52,076 | 29,321 ± 17,353 | NS | NS | |||||||||
| INOSINE 5′-DIPHOSPHATE (M-H)- | 74,867 ± 10,956 | 83,878 ± 3826 | 75,515 ± 4497 | 0.0293 | NS | |||||||||
| ISOCITRIC ACID (M-H)- | 3,562,142 ± 250,803 | 4,110,980 ± 995,231 | 3,773,454 ± 559,709 | NS | NS | |||||||||
| ISOLEUCINE (M-H)- | 212,672 ± 45,422 | 186,668 ± 24,327 | 165,577 ± 50,201 | NS | NS | |||||||||
| KYNURENIC ACID (M-H)- | 15,956 ± 5823 | 9186 ± 4342 | 7267 ± 1166 | NS | NS | |||||||||
| L-HISTIDINE (M-H)- | 8280 ± 3336 | 7551 ± 3216 | 8214 ± 4073 | NS | NS | |||||||||
| LIGNOCERIC ACID (M-H)- | 2016 ± 669 | 1951 ± 750 | 2400 ± 228 | NS | NS | |||||||||
| LINOLEIC ACID (M-H)- | 102,817 ± 66,691 | 114,235 ± 34,869 | 129,823 ± 114,118 | NS | NS | |||||||||
| L-TRYPTOPHAN-15N2 [ISTD] (M-H)- | 526,582 ± 58,182 | 521,388 ± 52,165 | 247,497 ± 215,531 | 0.0431 | NS | |||||||||
| LYSINE (M-H)- | 8474 ± 2194 | 10,975 ± 922 | 10,832 ± 3802 | NS | NS | |||||||||
| MALEIC ACID (M-H)- | 42,927 ± 15,026 | 73,767 ± 24,525 | 48,916 ± 19,167 | 0.0492 | NS | |||||||||
| MALIC ACID (M-H)- | 46,118 ± 11,016 | 57,993 ± 16,328 | 92,673 ± 36,594 | 0.0213 | NS | |||||||||
| METHYL BETA-D-GALACTOSIDE (M+Cl)- | 139,948 ± 15,875 | 15,084 ± 9067 | 18,351 ± 5849 | 0.0027 | 0.0336 | |||||||||
| METHYLMALONIC ACID (M-H)- | 18,467 ± 6095 | 21,842 ± 1848 | 16,695 ± 6137 | NS | NS | |||||||||
| MYRISTIC ACID (M-H)- | 4038 ± 1518 | 3553 ± 995 | 4185 ± 2014 | NS | NS | |||||||||
| MYRISTOLEIC ACID (M-H)- | 8435 ± 4072 | 10,032 ± 2060 | 5925 ± 5062 | NS | NS | |||||||||
| N-ACETYLGLYCINE (M-H)- | 19,086 ± 16,253 | 6539 ± NA | NaN ± NA | NS | NS | |||||||||
| N-ACETYL-L-ALANINE (M-H)- | 2428 ± NA | 20,516 ± 10,736 | 61,836 ± NA | NS | NS | |||||||||
| N-ACETYL-L-LEUCINE (M-H)- | 130,301 ± 44,499 | 105,017 ± 29,922 | 97,866 ± 28,641 | NS | NS | |||||||||
| N-ACETYL-L-PHENYLALANINE (M-H)- | 61,046 ± 11,829 | 38,194 ± 6372 | 42,010 ± 6002 | 0.0029 | 0.0336 | |||||||||
| NERVONIC ACID (M-H)- | 1959 ± 482 | 1798 ± 334 | 1766 ± 165 | NS | NS | |||||||||
| OLEIC ACID (M-H)- | 177,669 ± 100,039 | 236,903 ± 51,510 | 296,922 ± 178,847 | NS | NS | |||||||||
| OXALOACETIC ACID (M-H)-[-H2O] | 412,772 ± 48,365 | 388,334 ± 39,291 | 391,721 ± 21,886 | NS | NS | |||||||||
| PALMITIC ACID (M-H)- | 56,987 ± 32,515 | 63,557 ± 8874 | 97,618 ± 40,172 | NS | NS | |||||||||
| PALMITOLEIC ACID (M-H)- | 20,565 ± 16,069 | 27,060 ± 7841 | 35,802 ± 26,925 | NS | NS | |||||||||
| PANTOTHENIC ACID (M+Cl)- | 33,766 ± 4352 | 19,796 ± 3323 | 18,831 ± 1855 | 0.0013 | 0.0336 | |||||||||
| PANTOTHENIC ACID (M-H)- | 319,857 ± 24,878 | 175,161 ± 26,571 | 158,159 ± 19,156 | 0.0011 | 0.0336 | |||||||||
| PHENYLALANINE (M-H)- | 341,328 ± 50,115 | 364,373 ± 46,304 | 271,410 ± 25,772 | 0.0071 | 0.0464 | |||||||||
| PHTHALIC ACID (M-H)- | 22,434 ± NA | 11,568 ± 2642 | 12,377 ± 1856 | NS | NS | |||||||||
| PIMELIC ACID (M-H)- | 7642 ± 1255 | 8244 ± 360 | 11,072 ± NA | NS | NS | |||||||||
| SEBACIC ACID (M-H)- | 28,480 ± 8595 | NaN ± NA | 7867 ± NA | NS | NS | |||||||||
| STEARIC ACID (M-H)- | 57,709 ± 20,613 | 75,404 ± 15,206 | 93,624 ± 23,480 | NS | NS | |||||||||
| SUCCINIC ACID (M-H)- | 209,834 ± 153,853 | 551,798 ± 356,372 | 230,686 ± 179,562 | NS | NS | |||||||||
| TAURINE (M-H)- | 19,104 ± 6139 | 20,171 ± 6428 | 17,516 ± 3764 | NS | NS | |||||||||
| THYMIDINE (M+Cl)- | 97,088 ± 19,468 | 90,952 ± 15,171 | 84,750 ± 6181 | NS | NS | |||||||||
| THYMIDINE (M-H)- | 49,977 ± 10,993 | 45,691 ± 9062 | 42,271 ± 3163 | NS | NS | |||||||||
| THYMINE-D4(METHYL-D3,6-D1) [ISTD] (M-H)- | 263,379 ± 10,027 | 245,320 ± 22,888 | 246,512 ± 34,562 | NS | NS | |||||||||
| TRANS-ACONITIC ACID (M-H)- | 234,201 ± 210,269 | 313,096 ± 221,031 | 231,614 ± 150,408 | NS | NS | |||||||||
| TYROSINE (M-H)- | 223,150 ± 20,045 | 184,140 ± 63,255 | 102,568 ± 25,374 | 0.0018 | 0.0336 | |||||||||
| URIC ACID (M-H)- | 226,627 ± 245,663 | 597,501 ± 781,901 | 401,973 ± 423,309 | NS | NS | |||||||||
| URIDINE (M+Cl)- | 130,771 ± 73,528 | 303,107 ± 229,645 | 172,537 ± 93,266 | NS | NS | |||||||||
| URIDINE (M-H)- | 41,396 ± 14,784 | 95,222 ± 33,938 | 63,181 ± 18,945 | 0.0153 | NS | |||||||||
| XANTHINE (M-H)- | 32,567 ± NA | 63,862 ± 58,166 | 53,646 ± 79,311 | NS | NS | |||||||||
| XANTHOSINE (M-H)- | 15,451 ± NA | 23,618 ± 13,508 | NaN ± NA | NS | NS | |||||||||
| ZEATIN [ISTD] (M+Cl)- | 95,865 ± 13,149 | 87,654 ± 6336 | 79,413 ± 10,396 | NS | NS | |||||||||
| ZEATIN [ISTD] (M-H)- | 71,790 ± 11,292 | 62,437 ± 5294 | 56,018 ± 8993 | 0.045 | NS | |||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udoh, U.-A.S.; Schade, M.S.; Sanabria, J.A.; Rajan, P.K.; Aguilar, R.; Andryka, M.; Gorka, A.; Pierre, S.V.; Sanabria, J. INHIBITION OF THE PROSTAGLANDIN-DEGRADING ENZYME 15-PGDH AMELIORATES MASH-ASSOCIATED APOPTOSIS AND FIBROSIS IN MICE. Cells 2025, 14, 987. https://doi.org/10.3390/cells14130987
Udoh U-AS, Schade MS, Sanabria JA, Rajan PK, Aguilar R, Andryka M, Gorka A, Pierre SV, Sanabria J. INHIBITION OF THE PROSTAGLANDIN-DEGRADING ENZYME 15-PGDH AMELIORATES MASH-ASSOCIATED APOPTOSIS AND FIBROSIS IN MICE. Cells. 2025; 14(13):987. https://doi.org/10.3390/cells14130987
Chicago/Turabian StyleUdoh, Utibe-Abasi S., Mathew Steven Schade, Jacqueline A. Sanabria, Pradeep Kumar Rajan, Rodrigo Aguilar, Micheal Andryka, Alexei Gorka, Sandrine V. Pierre, and Juan Sanabria. 2025. "INHIBITION OF THE PROSTAGLANDIN-DEGRADING ENZYME 15-PGDH AMELIORATES MASH-ASSOCIATED APOPTOSIS AND FIBROSIS IN MICE" Cells 14, no. 13: 987. https://doi.org/10.3390/cells14130987
APA StyleUdoh, U.-A. S., Schade, M. S., Sanabria, J. A., Rajan, P. K., Aguilar, R., Andryka, M., Gorka, A., Pierre, S. V., & Sanabria, J. (2025). INHIBITION OF THE PROSTAGLANDIN-DEGRADING ENZYME 15-PGDH AMELIORATES MASH-ASSOCIATED APOPTOSIS AND FIBROSIS IN MICE. Cells, 14(13), 987. https://doi.org/10.3390/cells14130987