
Effects of Obeticholic Acid Treatment on Primary Human Hepatocytes in a Novel Tri-Culture Model System

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. TruVivo® and OCA Treatment
2.2. Morphological Assessment and PHH Attachment
2.3. Nile Red Staining
2.4. Gene Expression
2.5. Triglycerides Assay
2.6. Albumin, Urea, and ELISAs
2.7. Assay for Basal CYP3A4 Activity
2.8. Immunofluorescence
2.9. 5-(and-6)-Carboxy-2′,7′-Dichlorofluorescein Diacetate (CDFDA) Staining
2.10. Statistical Analysis
3. Results
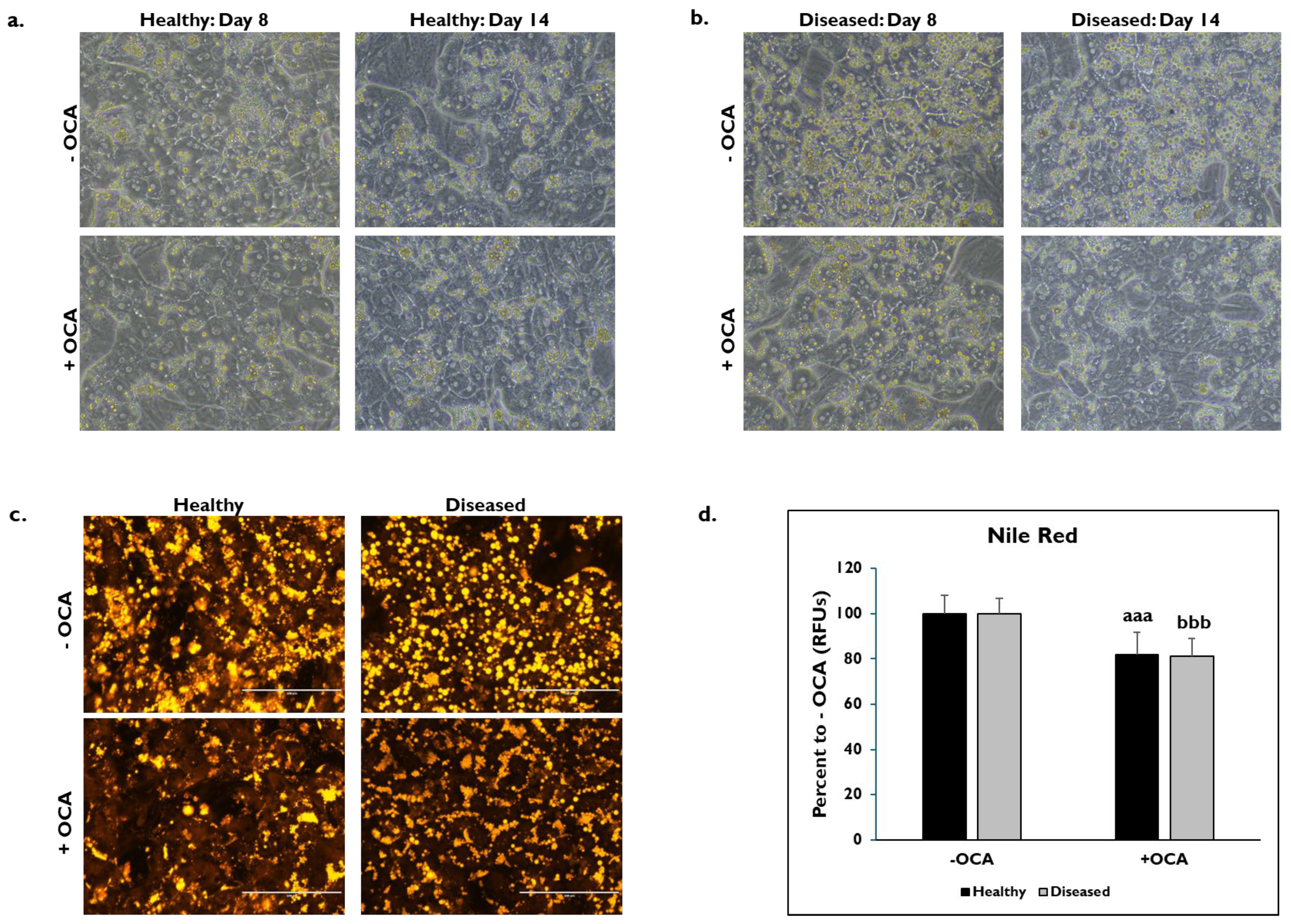
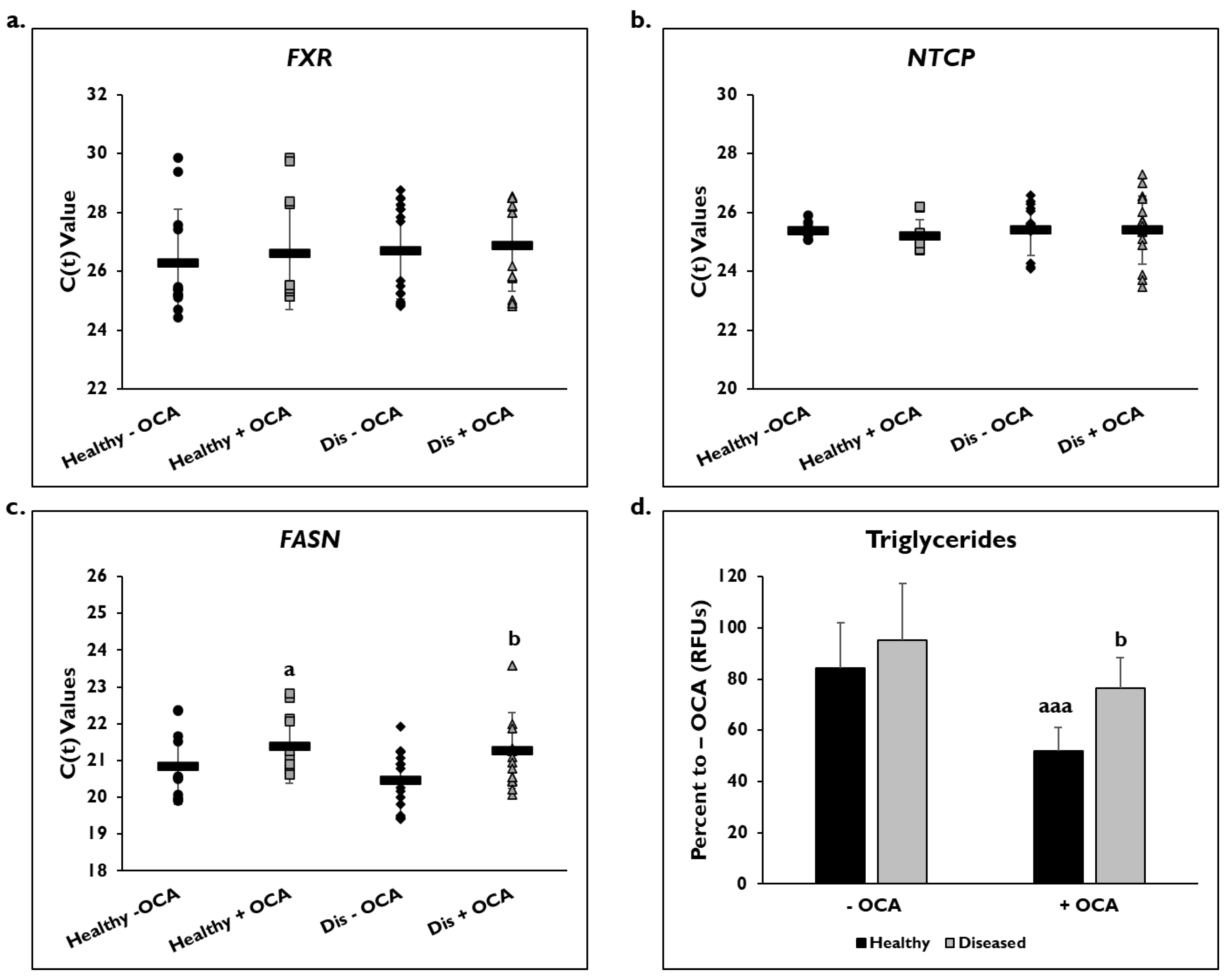
3.1. OCA Treatment and PHH Lipogenesis
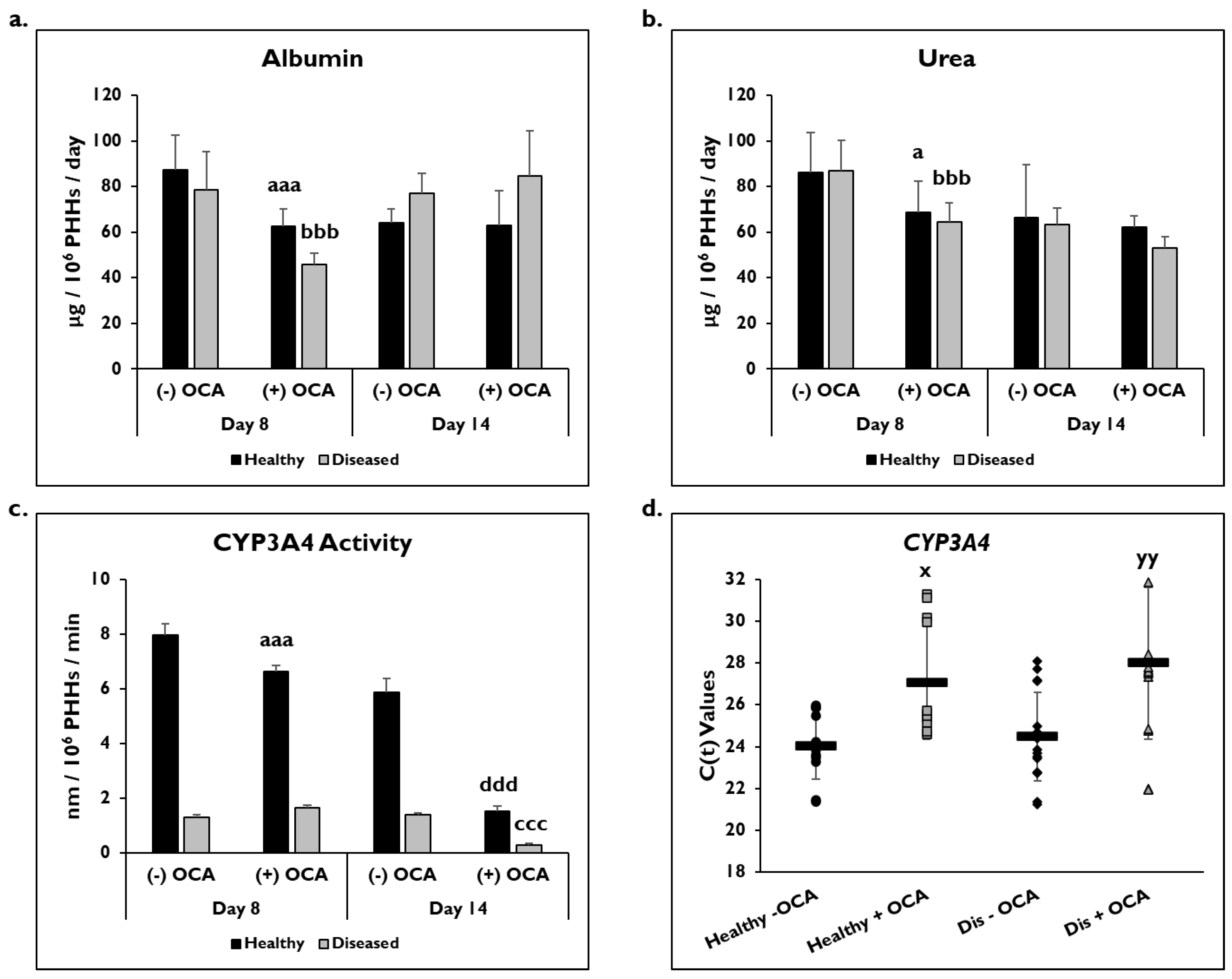
3.2. OCA Treatment and PHH Functionality
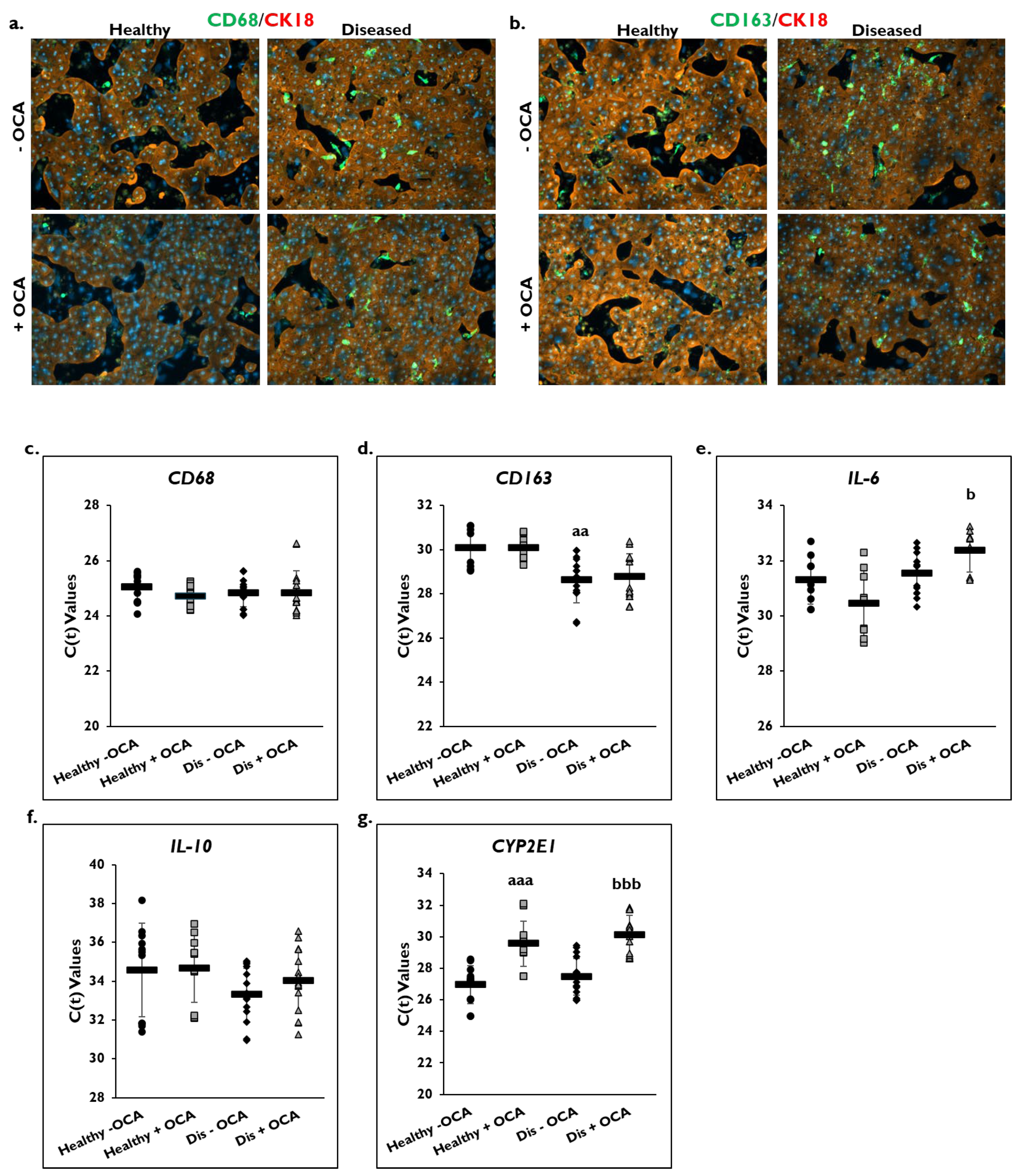
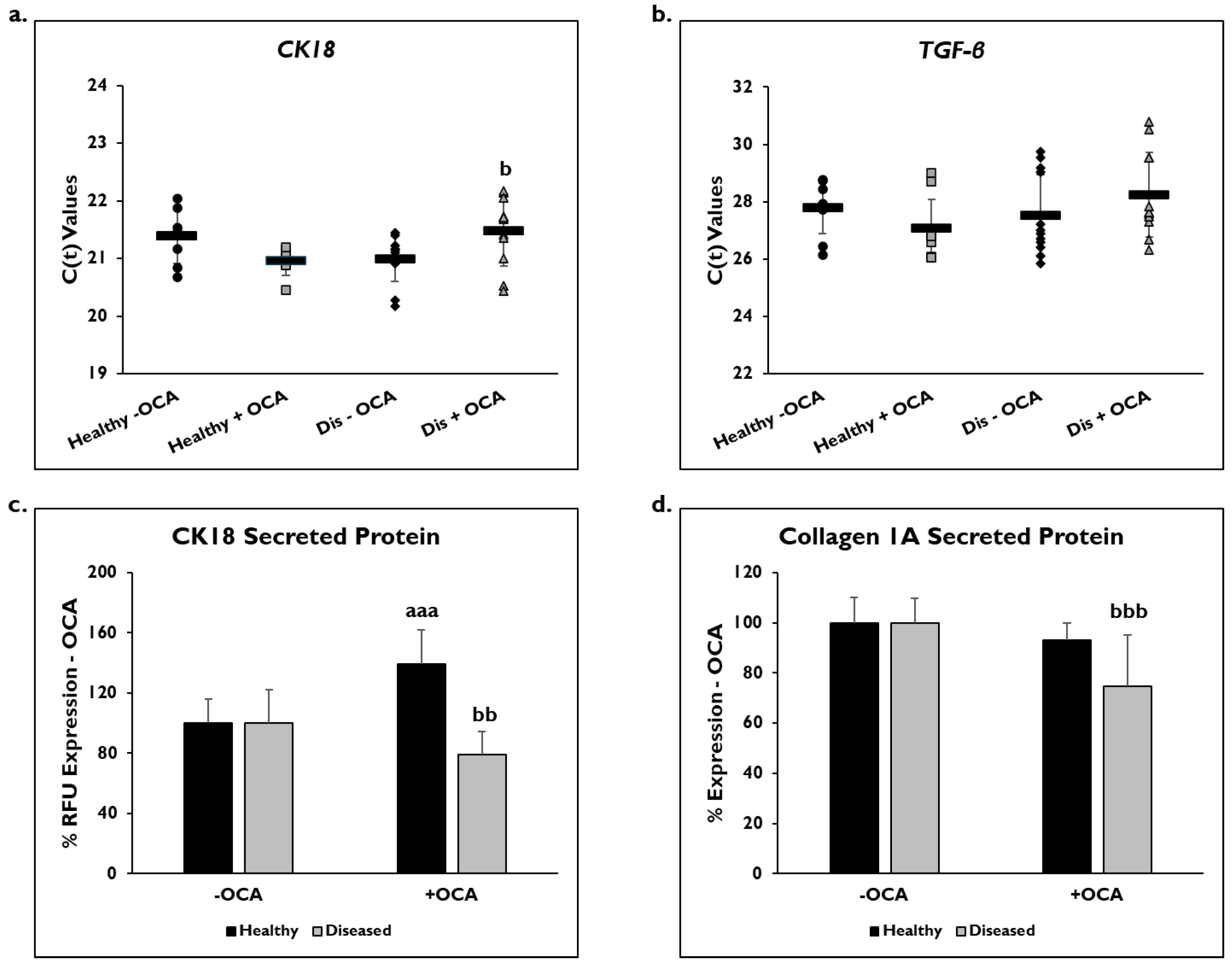
3.3. OCA Treatment and Inflammation
3.4. OCA Treatment and Fibrogenic Markers
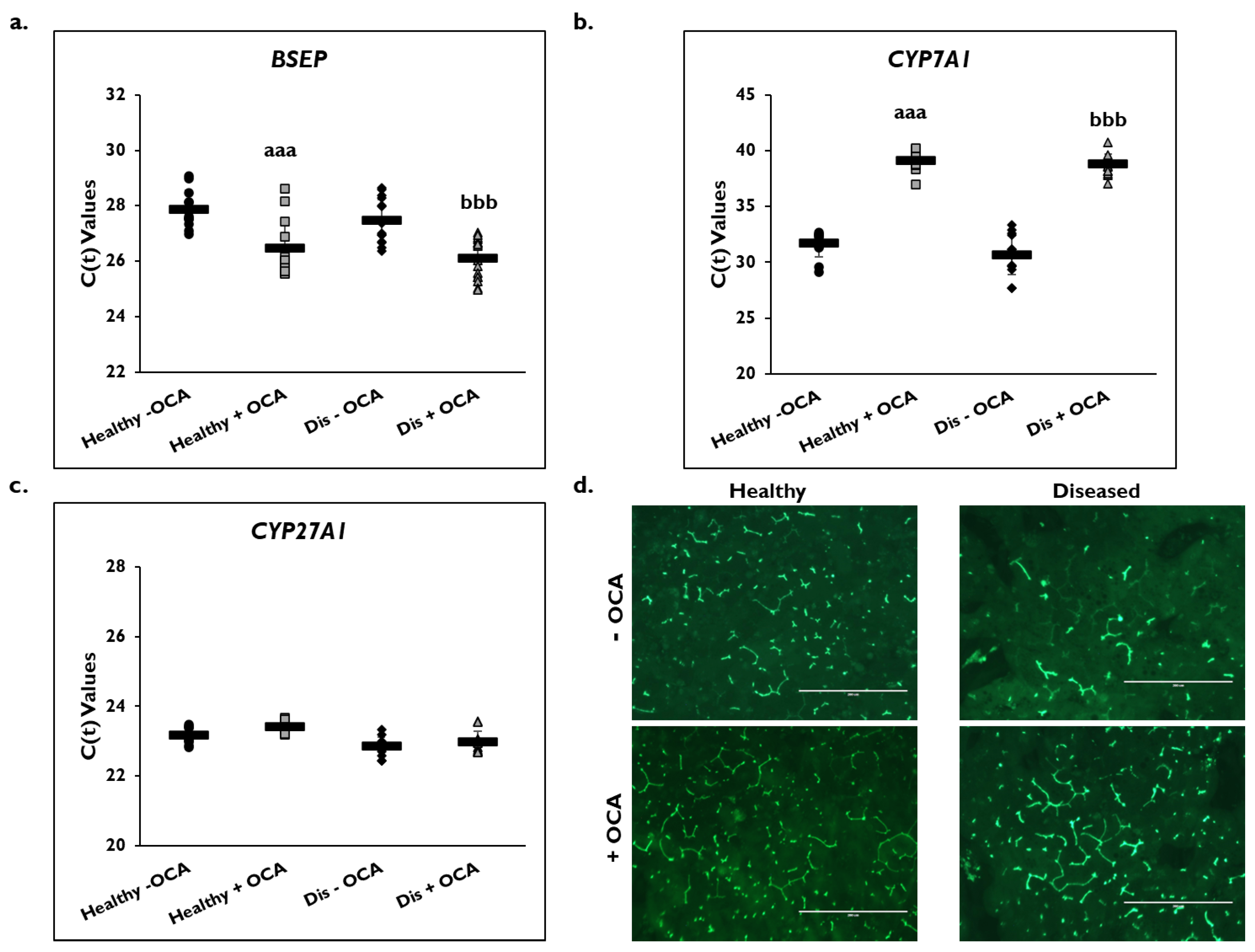
3.5. Effects of OCA on BA Synthesis Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BA | Bile acids |
| BSEP | Bile salt export pump |
| CDFDA | 5-(and-6)-Carboxy-2′,7′-Dichlorofluorescein Diacetate |
| COL1A1 | Collagen 1A |
| CYP | Cytochrome P450 |
| CYP7A1 | Cholesterol 7-α-hydroxylase |
| CK18 | Cytokeratin-18 |
| DPBS | Dulbecco’s PBS |
| FASN | Fatty Acid Synthase |
| FXR | Farnesoid X Receptor |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| IL-6 | Interleukin-6 |
| IL-10 | Interleukin-10 |
| KC | Kupffer Cell |
| LSEC | Liver Sinusoidal Endothelial Cell |
| MAFLD | Metabolic Dysfunction-Associated Fatty Liver Disease |
| MASH | Metabolic Dysfunction-Associated Steatohepatitis |
| NAFLD | Nonalcoholic Fatty Liver Disease |
| NASH | Nonalcoholic steatohepatitis |
| NTCP | sodium taurocholate co-transporting polypeptide |
| OCA | Obeticholic Acid |
| PBC | Primary biliary cholangitis |
| PHH | Primary Human Hepatocyte |
| RFU | Relative Fluorescent Unit |
| SC | Stellate Cell |
| TGF-β | Transforming Growth Factor-beta |
References
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.-A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2023, 7, 851–861. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease- Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Weaver, J.R.; Odanga, J.J.; Wolf, K.K.; Piekos, S.; Biven, M.; Taub, M.; LaRocca, J.; Thomas, C.; Byer-Alcorace, A.; Chen, J.; et al. The morphology, functionality, and longevity of a novel all human hepatic cell-based tri-culture system. Toxicol. In Vitro 2023, 86, 105504. [Google Scholar] [CrossRef]
- Odanga, J.J.; Gianulis, E.; Whaley, L.; LeCluyse, E.L.; Presnell, S.; Weaver, J.R. An All-Human Hepatic Culture System for Drug Development Applications. J. Vis. Exp. 2023, 200, e65992. [Google Scholar] [CrossRef]
- Odanga, J.J.; Anderson, S.M.; Breathwaite, E.K.; Presnell, S.C.; LeCluyse, E.L.; Chen, J.; Weaver, J.R. Characterization of diseased primary human hepatocytes in an all-human cell-based triculture system. Sci. Rep. 2024, 14, 6772. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Q.; He, J.; Li, Y. Novel Therapeutic Targets in Liver Fibrosis. Front. Mol. Biosci. 2021, 8, 766855. [Google Scholar] [CrossRef]
- Trauner, M.; Fuchs, C.D. Novel therapeutic targets for cholestatic and fatty liver disease. Gut 2022, 71, 194–209. [Google Scholar] [CrossRef]
- Lee, F.Y.; Lee, H.; Hubbert, M.L.; Edwards, P.A.; Zhang, Y. FXR, a multipurpose nuclear receptor. Trends Biochem. Sci. 2006, 31, 572–580. [Google Scholar] [CrossRef]
- Salhab, A.; Amer, J.; Lu, Y.; Safadi, R. Sodium +/Taurocholate Cotransporting Polypeptide as Target Therapy for Liver Fibrosis. Gut 2022, 71, 1373–1385. [Google Scholar] [CrossRef]
- Halilbasic, E.; Claudel, T.; Trauner, M. Bile Acid Transporters and Regulatory Nuclear Receptors in the Liver and Beyond. J. Hepatol. 2013, 58, 155–168. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, H.; Xiao, D.; Wei, H.; Chen, Y. Farnesoid X receptor (FXR): Structures and ligands. Comput. Struct. Biotechnol. J. 2021, 19, 2148–2159. [Google Scholar] [CrossRef]
- Myant, N.B.; Mitropoulos, K.A. Cholesterol 7 alpha-hydroxylase. J. Lipid Res. 1977, 18, 135–153. [Google Scholar] [CrossRef]
- Kubitz, R.; Dröge, C.; Stindt, J.; Weissenberger, K.; Häussinger, D. The bile salt export pump (BSEP) in health and disease. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 536–553. [Google Scholar] [CrossRef]
- Floerl, S.; Kuehne, A.; Geyer, J.; Brockmoeller, J.; Tzvetkov, M.V.; Hagos, Y. Functional and Pharmacological Comparison of Human and Mouse Na+/Taurocholate Cotransporting Polypeptide (NTCP). SLAS Discov. 2021, 26, 1055–1064. [Google Scholar] [CrossRef]
- Jiao, Y.; Lu, Y.; Li, X. Farnesoid X receptor: A master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol. Sin. 2015, 36, 44–50. [Google Scholar] [CrossRef]
- Porez, G.; Prawitt, J.; Gross, B.; Staels, B. Bile acid receptors as targets for the treatment of dylipidemia and cardiovascular disease. J. Lipid Res. 2012, 53, 1723–1737. [Google Scholar] [CrossRef]
- Pellicciari, R.; Costantino, G.; Camaioni, E.; Sadeghpour, B.M.; Entrena, A.; Willson, T.M.; Fiorucci, S.; Clerici, C.; Gioiello, A. Bile Acid Derivatives as Ligands of the Farnesoid X Receptor. Synthesis, Evaluation, and Structure-Activity Relationship of a Series of Body and Side Chain Modified Analogues of Chenodeoxycholic Acid. J. Med. Chem. 2004, 47, 4559–4569. [Google Scholar] [CrossRef]
- Floreani, A.; Gabbia, D.; De Martin, S. Obeticholic Acid for Primary Cholangitis. Biomedicines 2022, 10, 2464. [Google Scholar] [CrossRef]
- Gulamhusein, A.F.; Hirschfield, G.M. Primary biliary cholangitis: Pathogenesis and therapeutic opportunities. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 93–110. [Google Scholar] [CrossRef]
- Ronca, V.; Davies, S.P.; Oo, Y.H.; Lleo, A. The immunological landscape of primary biliary cholangitis: Mechanisms and therapeutic prospects. Hepatology 2025, 10-1097. [Google Scholar] [CrossRef]
- Verbeke, L.; Mannaerts, I.; Schierwagen, R.; Govaere, O.; Klein, S.; Vander Elst, I.; Windmolders, P.; Farre, R.; Wenes, M.; Mazzone, M.; et al. FXR Agonist Obeticholic Acid Reduces Hepatic Inflammation and Fibrosis in a Rat Model of Toxic Cirrhosis. Sci. Rep. 2016, 6, 33453. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 959–965. [Google Scholar] [CrossRef]
- Alkhouri, N.; LaCerte, C.; Edwards, J.; Poordad, F.; Lawitz, E.; Lee, L.; Karan, S.; Sawhney, S.; Erickson, M.; MacConell, L.; et al. Safety, pharmacokinetics and pharmacodynamics of obeticholic acid in subjects with fibrosis or cirrhosis from NASH. Liver Int. 2024, 44, 966–978. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582. [Google Scholar] [CrossRef]
- Ghabril, M.; Vuppalanchi, R.; Chalasani, N. Drug-Induced Liver Injury in Patients With Chronic Liver Disease. Liver Int. 2025, 45, e70019. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration Drug Safety and Availability. “Serious Liver Injury Being Observed in Patients Without Cirrhosis Taking Ocaliva (Obeticholic Acid) to Treat Primary Biliary Cholangitis.” 12-12-2024 FDA Drug Safety Communication. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/serious-liver-injury-being-observed-patients-without-cirrhosis-taking-ocaliva-obeticholic-acid-treat (accessed on 12 December 2024).
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Mason, A.; Luketic, V.; Lindor, K.; Gordon, S.C.; Mayo, M.; Kowdley, K.V.; Vincent, C.; Bodhenheimer, H.C., Jr.; Parés, A.; et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 2015, 148, 751–761.e8. [Google Scholar] [CrossRef]
- Feaver, R.E.; Cole, B.K.; Lawson, M.J.; Hoang, S.A.; Marukian, S.; Blackman, B.R.; Figler, R.A.; Sanyal, A.J.; Wamhoff, B.R. and Dash, A. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. JCI Insight 2016, 1, e90954. [Google Scholar] [CrossRef] [PubMed]
- Anfuso, B.; Tiribelli, C.; Adorini, L.; Rosso, N. Obeticholic acid and INT-767 modulate collagen deposition in a NASH in vitro model. Sci. Rep. 2020, 10, 1699. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef]
- Shimano, H.; Horton, J.D.; Shimomura, I.; Hammer, R.E.; Brown, M.S.; Goldstein, J.L. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J. Clin. Investig. 1997, 99, 846–854. [Google Scholar] [CrossRef]
- Dash, A.; Figler, R.A.; Blackman, B.R.; Marukian, S.; Collado, M.S.; Lawson, M.J.; Hoang, S.A.; Mackey, A.J.; Manka, D.; Cole, B.K.; et al. Pharmacotoxicology of clinically-relevant concentrations of obeticholic acid in an organotypic human hepatocyte system. Toxicol. In Vitro 2017, 39, 93–103. [Google Scholar] [CrossRef]
- Smith, S.M.; Pegram, A.H. Obeticholic Acid: A Farnesoid X Receptor Agonist for Primary Biliary Cholangitis. J. Pharm. Technol. 2017, 2, 66–71. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; Cipriani, S.; D’Amore, C.; Zampella, A.; Monti, M.C.; Distrutti, E.; Fiorucci, S. The nuclear receptor FXR regulates hepatic transport and metabolism of glutamine and glutamate. Biochim. Biophys. Acta 2011, 1812, 1522–1531. [Google Scholar] [CrossRef]
- Massafra, V.; Milona, A.; Vos, H.R.; Ramos, R.J.; Gerrits, J.; Willemsen, E.C.; Pittol, J.M.R.; Ijssennagger, N.; Houweling, M.; Prinsen, H.C.; et al. Farnesoid X Receptor Activation Promotes Hepatic Amino Acid Catabolism and Ammonium Clearance in Mice. Gastroenterology 2017, 152, 1462–1476. [Google Scholar] [CrossRef]
- Nørgaard, R.A.; Bhatt, D.K.; Järvinen, E.; Stage, T.B.; Gabel-Jensen, C.; Galetin, A.; Säll, C. Evaluating Drug-Drug Interaction Risk Associated with Peptide Analogs Using advanced In Vitro Systems. Drug Metab. Dispos. 2024, 52, 1170–1180. [Google Scholar] [CrossRef]
- Zhang, Y.; LaCerte, C.; Kansra, S.; Jackson, J.P.; Brouwer, K.R.; Edwards, J.E. Comparative potency of obeticholic acid and natural bile acids on FXR in hepatic and intestinal in vitro cell models. Pharmacol. Res. Perspect. 2017, 5, e00368. [Google Scholar] [CrossRef]
- Edwards, J.E.; Eliot, L.; Parkinson, A.; Karan, S.; MacConell, L. Assessment of Pharmacokinetic Interactions Between Obeticholic Acid and Caffeine, Midazolam, Warfarin, Dextromethorphan, Omeprazole, Rosuvastatin, and Digoxin in Phase 1 Studies in Healthy Subjects. Adv. Ther. 2017, 34, 2120–2138. [Google Scholar] [CrossRef]
- Gnerre, C.; Blättler, S.; Kaufmann, M.R.; Looser, R.; Meyer, U.A. Regulation of CYP3A4 by the bile acid receptor FXR: Evidence for functional binding sites in the CYP3A4 gene. Pharmacogenetics 2004, 14, 635–645. [Google Scholar] [CrossRef]
- Zhang, S.; Pan, X.; Jeong, H. GW4064, an agonist of farnesoid X receptor, represses CYP3A4 expression in human hepatocytes by inducing small heterodimer partner expression. Drug Metab. Dispos. 2015, 43, 743–748. [Google Scholar] [CrossRef]
- Alisi, A.; Carpino, G.; Oliveira, F.L.; Panera, N.; Nobili, V.; Gaudio, E. The Role of Tissue Macrophage-Mediated Inflammation on NAFLD Pathogenesis and Its Clinical Implications. Mediators Inflamm. 2017, 8162421. [Google Scholar] [CrossRef]
- Huang, S.; Wu, Y.; Zhao, Z.; Wu, B.; Sun, K.; Wang, H.; Qin, L.; Bai, F.; Leng, Y.; Tang, W. A new mechanism of obeticholic acid on NASH treatment by inhibiting NLRP3 inflammasome activation in macrophage. Metabolism 2021, 120, 154797. [Google Scholar] [CrossRef]
- Odanga, J.J.; Anderson, S.M.; Presnell, S.C.; LeCluyse, E.L.; Chen, J.; Weaver, J.R. Impact of Post-Thaw Enrichment of Primary Human Hepatocytes on Steatosis, Inflammation, and Fibrosis in the TruVivo® System. Pharmaceuticals 2024, 17, 1624. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Ratziu, V.; Loomba, R.; Anstee, Q.M.; Kowdley, K.V.; Rinella, M.E.; Sheikh, M.Y.; Trotter, J.F.; Knapple, W.; Lawitz, E.J.; et al. Results from a new efficacy and safety analysis of the REGENERATE trial of obeticholic acid for treatment of pre-cirrhotic fibrosis due to non-alcoholic steatohepatitis. J. Hepatol. 2023, 79, 1110–1120. [Google Scholar] [CrossRef]
- Zhou, J.; Huang, N.; Guo, Y.; Cui, S.; Ge, C.; He, Q.; Pan, X.; Wang, G.; Wang, H.P.; Hao, H. Combined obeticholic acid and apoptosis inhibitor treatment alleviates liver fibrosis. Acta Pharm. Sin. B 2019, 9, 526–536. [Google Scholar] [CrossRef]
- Azizsoltani, A.; Hatami, B.; Zali, M.R.; Mahdavi, V.; Baghaei, K.; Alizadeh, E. Obeticholic acid-loaded exosomes attenuate liver fibrosis through dual targeting of the FXR signaling pathway and ECM remodeling. Biomed. Pharmacother. 2023, 168, 115777. [Google Scholar] [CrossRef]
- Tan, X.; Xiang, Y.; Shi, J.; Chen, L.; Yu, D. Targeting NTCP for liver disease treatment: A promising strategy. J. Pharm. Anal. 2024, 14, 100979. [Google Scholar] [CrossRef]
- Nell, P.; Kattler, K.; Feuerborn, D.; Hellwig, B.; Rieck, A.; Salhab, A.; Lepikhov, K.; Gasparoni, G.; Thomitzek, A.; Belgasmi, K.; et al. Identification of an FXR-modulated liver-intestine hybrid state in iPSC-derived hepatocyte-like cells. J. Hepatol. 2022, 77, 1386–1398. [Google Scholar] [CrossRef]
- Kosters, A.; Karpen, S.J. Bile acid transporters in health and disease. Xenobiotica 2008, 38, 1043–1071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jackson, J.P.; St Claire, R.L., 3rd; Freeman, K.; Brouwer, K.R.; Edwards, J.E. Obeticholic acid, a selective farnesoid X receptor agonist, regulates bile acid homeostasis in sandwich-cultured human hepatocytes. Pharmacol. Res. Perspect. 2017, 5, e00329. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor (Lot #) | Age | Sex | BMI | NAS Score | Steatosis Score | Lobular Inflammation Score | Hepatocyte Ballooning Score | Fibrosis Stage | Health Category |
|---|---|---|---|---|---|---|---|---|---|
| 16117 | 41 | F | 30 | 0 | 0 | 0 | 0 | 0 | Healthy |
| 1814680 | 52 | F | 23 | 0 | 0 | 0 | 0 | 0 | Healthy |
| 1919613 | 60 | M | 26 | 0 | 0 | 0 | 0 | 1–2 | Healthy |
| 2117885 | 23 | F | 23.7 | 1 | 1 | 0 | 0 | 0 | Healthy |
| 2021736 | 50 | F | 25.6 | 6 | 3 | 1 | 2 | 0 | Diseased |
| 2111401 | 28 | M | 21.8 | 5 | 3 | 0 | 2 | 1 | Diseased |
| 2116167 | 51 | M | 29.8 | 4 | 1 | 1 | 2 | 1 | Diseased |
| 2222437 | 30 | M | 26.8 | 4 | 1 | 1 | 2 | 1 | Diseased |
| Primer Name | Forward Sequence (5′-3′) | Reverse Sequence (5′-3′) |
|---|---|---|
| GAPDH | GGTCACCAGGGCTGCTTTTA | GGATCTCGCTCCTGGAAGATG |
| FASN | AAGGACCTGTCTAGGTTTGATGC | TGGCTTCATAGGTGACTTCCA |
| IL-6 | ACAACCTGAACCTTCCAAAGA | TCAGCAGGCTGGCATTT |
| IL-10 | GGCTACGGCGCTGTCATCGATT | GCATTCTTCACCTGCTCCACGG |
| CK18 | GGCATCCAGAACGAGAAGGA | AGTGCTCCCGGATTTTGCT |
| TGF-β | TTCCCTCGAGGCCCTCCTA | GCCGCAGCTTGGACAGGATC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Odanga, J.J.; Anderson, S.M.; LeCluyse, E.L.; Presnell, S.C.; Chen, J.; Weaver, J.R. Effects of Obeticholic Acid Treatment on Primary Human Hepatocytes in a Novel Tri-Culture Model System. Cells 2025, 14, 968. https://doi.org/10.3390/cells14130968
Odanga JJ, Anderson SM, LeCluyse EL, Presnell SC, Chen J, Weaver JR. Effects of Obeticholic Acid Treatment on Primary Human Hepatocytes in a Novel Tri-Culture Model System. Cells. 2025; 14(13):968. https://doi.org/10.3390/cells14130968
Chicago/Turabian StyleOdanga, Justin J., Sharon M. Anderson, Edward L. LeCluyse, Sharon C. Presnell, Jingsong Chen, and Jessica R. Weaver. 2025. "Effects of Obeticholic Acid Treatment on Primary Human Hepatocytes in a Novel Tri-Culture Model System" Cells 14, no. 13: 968. https://doi.org/10.3390/cells14130968
APA StyleOdanga, J. J., Anderson, S. M., LeCluyse, E. L., Presnell, S. C., Chen, J., & Weaver, J. R. (2025). Effects of Obeticholic Acid Treatment on Primary Human Hepatocytes in a Novel Tri-Culture Model System. Cells, 14(13), 968. https://doi.org/10.3390/cells14130968