
Metabolic Reprogramming in Toll-like Receptor-Mediated Platelet Activation
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Platelet Isolation
2.2. Murine Platelet Isolation
2.3. Light Transmission Platelet Aggregation
2.4. Bioenergetic Profiles
2.5. Hexokinase Activity Assay
2.6. Statistical Analyses
3. Results and Discussion
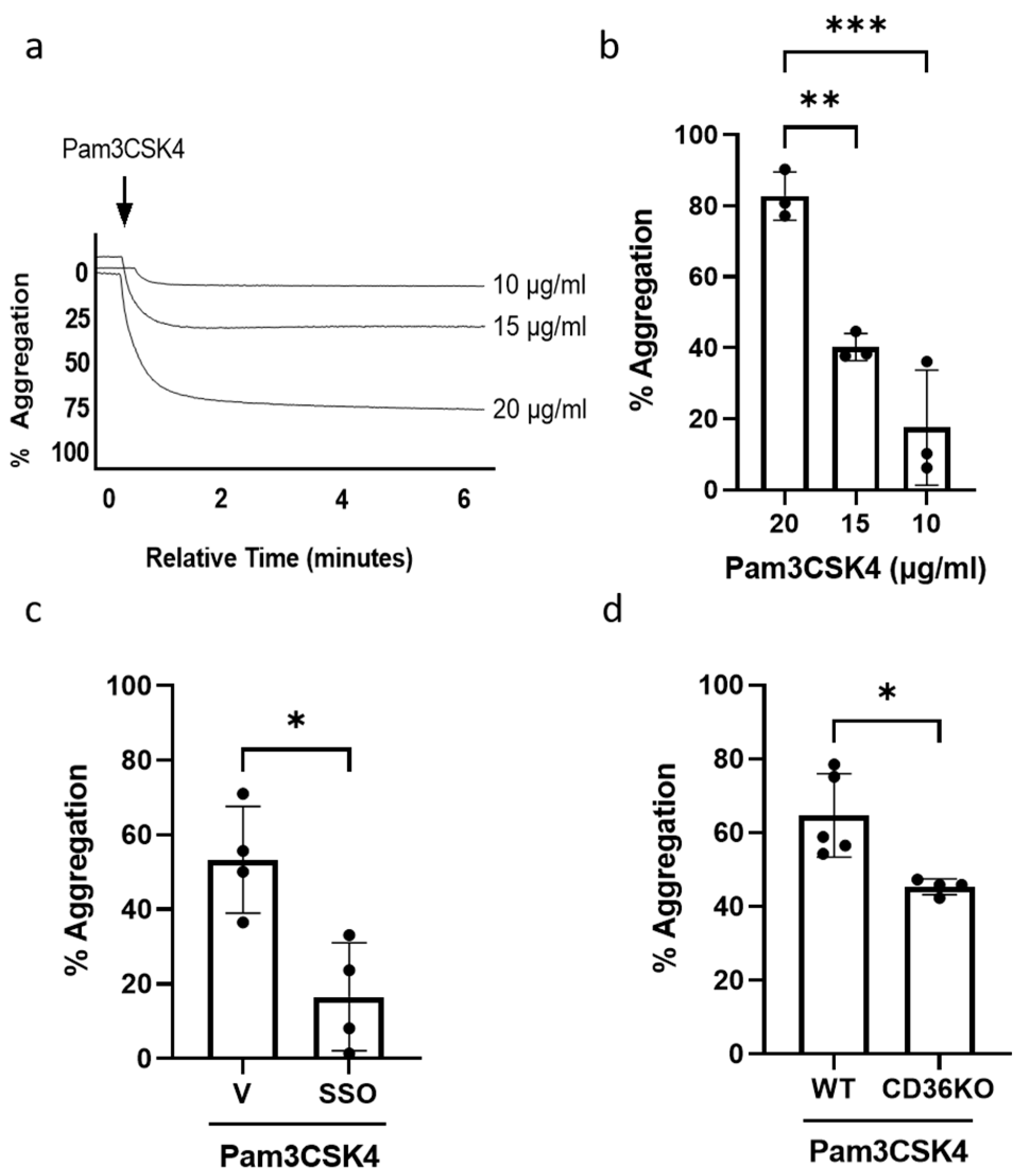
3.1. TLR1/TLR2 Engagement Induces Platelet Aggregation via CD36-Mediated Mechanism
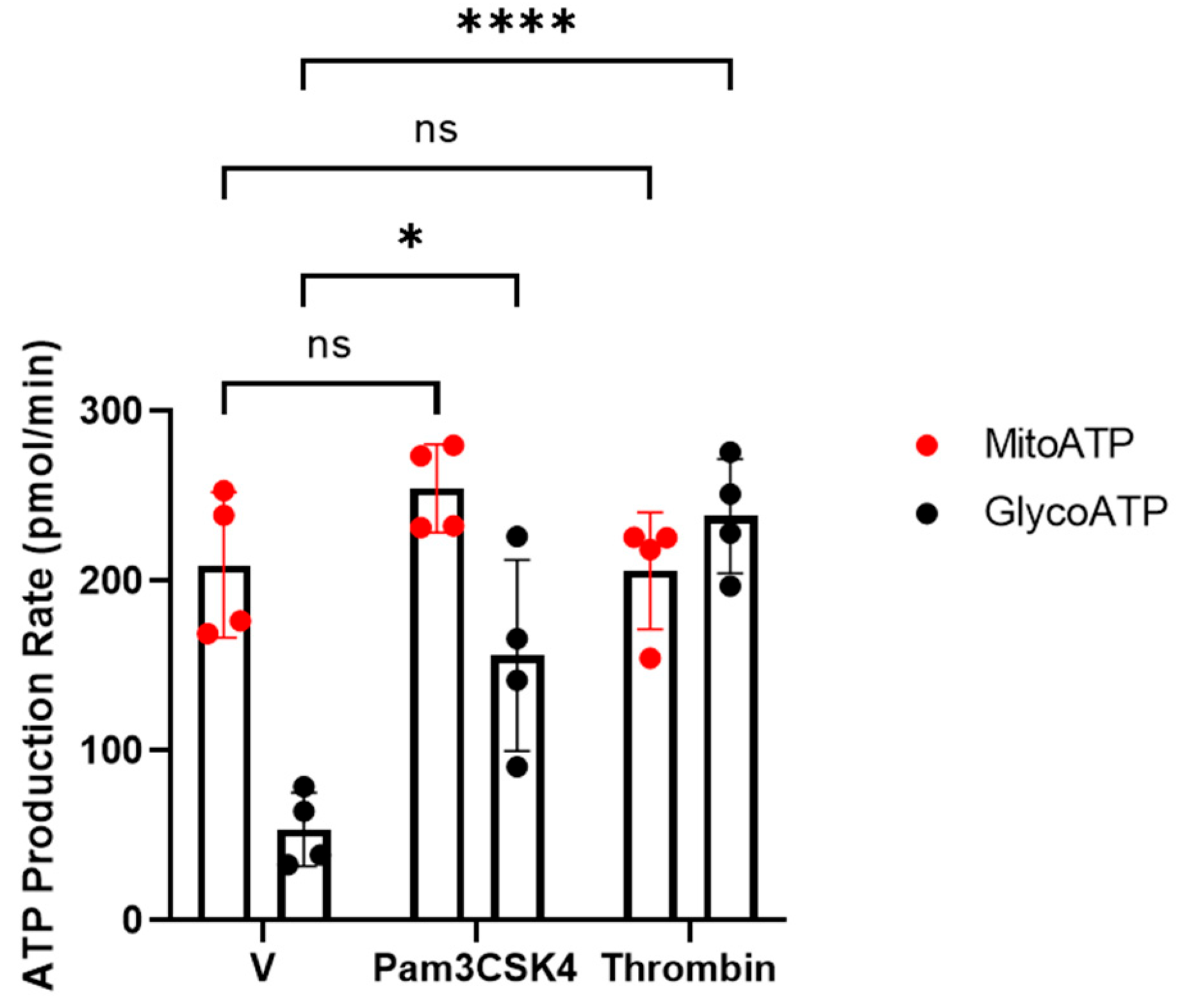
3.2. TLR1/TLR2-Mediated Platelet Activation Is Glycolytically Driven
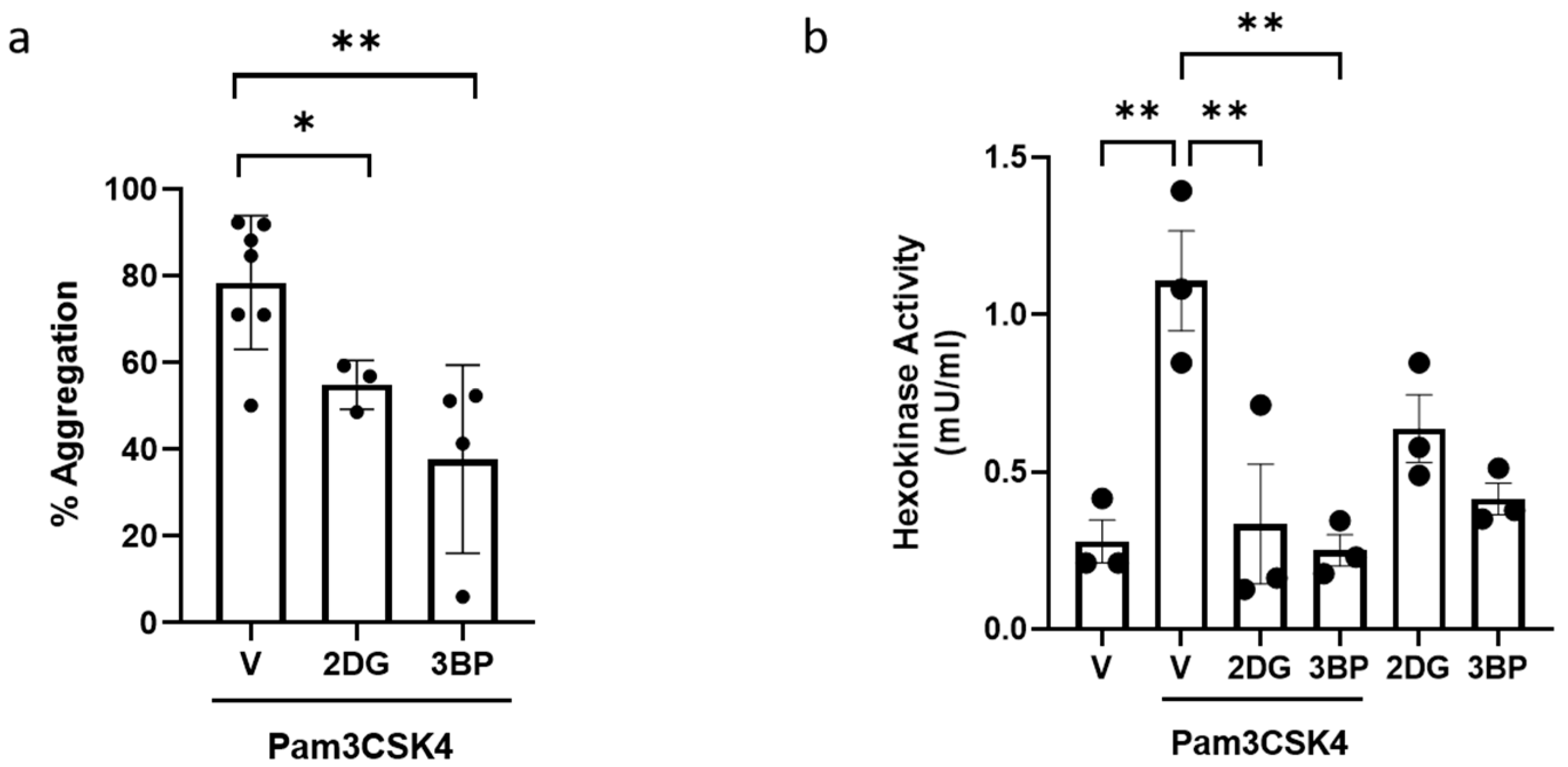
3.3. Inhibition of HK Prevents TLR1/TLR2-Mediated Platelet Aggregation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scherlinger, M.; Richez, C.; Tsokos, G.C.; Boilard, E.; Blanco, P. The role of platelets in immune-mediated inflammatory diseases. Nat. Rev. Immunol. 2023, 23, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Assinger, A.; Schrottmaier, W.C.; Salzmann, M.; Rayes, J. Platelets in Sepsis: An Update on Experimental Models and Clinical Data. Front. Immunol. 2019, 10, 1687. [Google Scholar] [CrossRef]
- von Hundelshausen, P.; Weber, C. Platelets as immune cells: Bridging inflammation and cardiovascular disease. Circ. Res. 2007, 100, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.R.; Foster, T.J.; Cox, D. The interaction of bacterial pathogens with platelets. Nat. Rev. Microbiol. 2006, 4, 445–457. [Google Scholar] [CrossRef]
- Hally, K.; Fauteux-Daniel, S.; Hamzeh-Cognasse, H.; Larsen, P.; Cognasse, F. Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis. Int. J. Mol. Sci. 2020, 21, 6150. [Google Scholar] [CrossRef] [PubMed]
- Rivadeneyra, L.; Carestia, A.; Etulain, J.; Pozner, R.G.; Fondevila, C.; Negrotto, S.; Schattner, M. Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB. Thromb. Res. 2014, 133, 235–243. [Google Scholar] [CrossRef]
- Marín Oyarzún, C.P.; Glembotsky, A.C.; Goette, N.P.; Lev, P.R.; De Luca, G.; Baroni Pietto, M.C.; Moiraghi, B.; Castro Ríos, M.A.; Vicente, A.; Marta, R.F.; et al. Platelet Toll-Like Receptors Mediate Thromboinflammatory Responses in Patients with Essential Thrombocythemia. Front. Immunol. 2020, 11, 705. [Google Scholar] [CrossRef]
- Hally, K.E.; La Flamme, A.C.; Harding, S.A.; Larsen, P.D. The effects of aspirin and ticagrelor on Toll-like receptor (TLR)-mediated platelet activation: Results of a randomized, cross-over trial. Platelets 2019, 30, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Keane, C.; Tilley, D.; Cunningham, A.; Smolenski, A.; Kadioglu, A.; Cox, D.; Jenkinson, H.F.; Kerrigan, S.W. Invasive Streptococcus pneumoniae trigger platelet activation via Toll-like receptor 2. J. Thromb. Haemost. 2010, 8, 2757–2765. [Google Scholar] [CrossRef]
- Fälker, K.; Klarström-Engström, K.; Bengtsson, T.; Lindahl, T.L.; Grenegård, M. The toll-like receptor 2/1 (TLR2/1) complex initiates human platelet activation via the src/Syk/LAT/PLCγ2 signalling cascade. Cell Signal 2014, 26, 279–286. [Google Scholar] [CrossRef]
- Klarström Engström, K.; Brommesson, C.; Kälvegren, H.; Bengtsson, T. Toll like receptor 2/1 mediated platelet adhesion and activation on bacterial mimetic surfaces is dependent on src/Syk-signaling and purinergic receptor P2X1 and P2Y12 activation. Biointerphases 2014, 9, 041003. [Google Scholar] [CrossRef]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Cui, W.; Silverstein, R.L. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J. Exp. Med. 2022, 219, e20211314. [Google Scholar] [CrossRef] [PubMed]
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.; McNeil, C.; Wheatcroft, S.; Yuldasheva, N.; Febbriao, M.; et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015, 125, 2693–2703. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Raslan, Z.; Aburima, A.; Magwenzi, S.; Wraith, K.S.; Spurgeon, B.E.J.; Hindle, M.S.; Law, R.; Febbraio, M.; Naseem, K.M. Atherogenic lipid stress induces platelet hyperactivity through CD36-mediated hyposensitivity to prostacyclin: The role of phosphodiesterase 3A. Haematologica 2020, 105, 808–819. [Google Scholar] [CrossRef]
- Biswas, S.; Zimman, A.; Gao, D.; Byzova, T.V.; Podrez, E.A. TLR2 Plays a Key Role in Platelet Hyperreactivity and Accelerated Thrombosis Associated With Hyperlipidemia. Circ. Res. 2017, 121, 951–962. [Google Scholar] [CrossRef]
- Hoebe, K.; Georgel, P.; Rutschmann, S.; Du, X.; Mudd, S.; Crozat, K.; Sovath, S.; Shamel, L.; Hartung, T.; Zähringer, U.; et al. CD36 is a sensor of diacylglycerides. Nature 2005, 433, 523–527. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef]
- Valerio-Rojas, J.C.; Jaffer, I.J.; Kor, D.J.; Gajic, O.; Cartin-Ceba, R. Outcomes of severe sepsis and septic shock patients on chronic antiplatelet treatment: A historical cohort study. Crit. Care Res. Pract. 2013, 2013, 782573. [Google Scholar] [CrossRef]
- Holmsen, H.; Kaplan, K.L.; Dangelmaier, C.A. Differential energy requirements for platelet responses. A simultaneous study of aggregation, three secretory processes, arachidonate liberation, phosphatidylinositol breakdown and phosphatidate production. Biochem. J. 1982, 208, 9–18. [Google Scholar] [CrossRef]
- Doery, J.C.; Hirsh, J.; Cooper, I. Energy metabolism in human platelets: Interrelationship between glycolysis and oxidative metabolism. Blood 1970, 36, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, A.J.; Gorter, G.; Mommersteeg, M.E.; Akkerman, J.W. The energetics of early platelet responses. Energy consumption during shape change and aggregation with special reference to protein phosphorylation and the polyphosphoinositide cycle. Biochem. J. 1985, 228, 451–462. [Google Scholar] [CrossRef]
- Verhoeven, A.J.; Mommersteeg, M.E.; Akkerman, J.W. Comparative studies on the energetics of platelet responses induced by different agonists. Biochem. J. 1986, 236, 879–887. [Google Scholar] [CrossRef]
- Aibibula, M.; Naseem, K.M.; Sturmey, R.G. Glucose metabolism and metabolic flexibility in blood platelets. J. Thromb. Haemost. 2018, 16, 2300–2314. [Google Scholar] [CrossRef] [PubMed]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.A.; Chacko, B.K.; Ravi, S.; Johnson, M.S.; Mitchell, T.; Darley-Usmar, V.M. Bioenergetics and the oxidative burst: Protocols for the isolation and evaluation of human leukocytes and platelets. J. Vis. Exp. 2014, 85, 51301. [Google Scholar] [CrossRef]
- Mai, K.; Chui, J.J.; Di Girolamo, N.; McCluskey, P.J.; Wakefield, D. Role of toll-like receptors in human iris pigment epithelial cells and their response to pathogen-associated molecular patterns. J. Inflamm. 2014, 11, 20. [Google Scholar] [CrossRef]
- Cioanca, A.V.; McCluskey, P.J.; Eamegdool, S.S.; Madigan, M.C. Human choroidal melanocytes express functional Toll-like receptors (TLRs). Exp. Eye Res. 2018, 173, 73–84. [Google Scholar] [CrossRef]
- Claushuis, T.A.M.; Van Der Veen, A.I.P.; Horn, J.; Schultz, M.J.; Houtkooper, R.H.; Van ‘t Veer, C.; Van Der Poll, T. Platelet Toll-like receptor expression and activation induced by lipopolysaccharide and sepsis. Platelets 2019, 30, 296–304. [Google Scholar] [CrossRef]
- Kuda, O.; Pietka, T.A.; Demianova, Z.; Kudova, E.; Cvacka, J.; Kopecky, J.; Abumrad, N.A. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164: SSO also inhibits oxidized low density lipoprotein uptake by macrophages. J. Biol. Chem. 2013, 288, 15547–15555. [Google Scholar] [CrossRef]
- Wilson, J.E. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J. Exp. Biol. 2003, 206, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Fidler, T.P.; Middleton, E.A.; Rowley, J.W.; Boudreau, L.H.; Campbell, R.A.; Souvenir, R.; Funari, T.; Tessandier, N.; Boilard, E.; Weyrich, A.S.; et al. Glucose Transporter 3 Potentiates Degranulation and Is Required for Platelet Activation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1628–1639. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tian, H.; Luo, H.; Fu, J.; Jiao, Y.; Li, Y. Prognostic Significance and Related Mechanisms of Hexokinase 1 in Ovarian Cancer. Onco Targets Ther. 2020, 13, 11583–11594. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Zhang, Y.; Wang, J.; Deng, Y.; Lin, D. Inhibition of glycolytic enzyme hexokinase II (HK2) suppresses lung tumor growth. Cancer Cell Int. 2016, 16, 9. [Google Scholar] [CrossRef]
- Pedersen, P.L. 3-Bromopyruvate (3BP) a fast acting, promising, powerful, specific, and effective “small molecule” anti-cancer agent taken from labside to bedside: Introduction to a special issue. J. Bioenerg. Biomembr. 2012, 44, 1–6. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheah, L.T.; Khalil, J.S.; McKay, M.; Ali, M.; Duval, C.; Unsworth, A.J.; Naseem, K.M. Metabolic Reprogramming in Toll-like Receptor-Mediated Platelet Activation. Cells 2025, 14, 906. https://doi.org/10.3390/cells14120906
Cheah LT, Khalil JS, McKay M, Ali M, Duval C, Unsworth AJ, Naseem KM. Metabolic Reprogramming in Toll-like Receptor-Mediated Platelet Activation. Cells. 2025; 14(12):906. https://doi.org/10.3390/cells14120906
Chicago/Turabian StyleCheah, Lih T., Jawad S. Khalil, Mary McKay, Mohammad Ali, Cedric Duval, Amanda J. Unsworth, and Khalid M. Naseem. 2025. "Metabolic Reprogramming in Toll-like Receptor-Mediated Platelet Activation" Cells 14, no. 12: 906. https://doi.org/10.3390/cells14120906
APA StyleCheah, L. T., Khalil, J. S., McKay, M., Ali, M., Duval, C., Unsworth, A. J., & Naseem, K. M. (2025). Metabolic Reprogramming in Toll-like Receptor-Mediated Platelet Activation. Cells, 14(12), 906. https://doi.org/10.3390/cells14120906