
Molecular Mechanisms of Cellular Senescence in Age-Related Endometrial Dysfunction

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
Search Strategy and Selection Criteria
3. Factors Involved in Endometrial Cellular Senescence
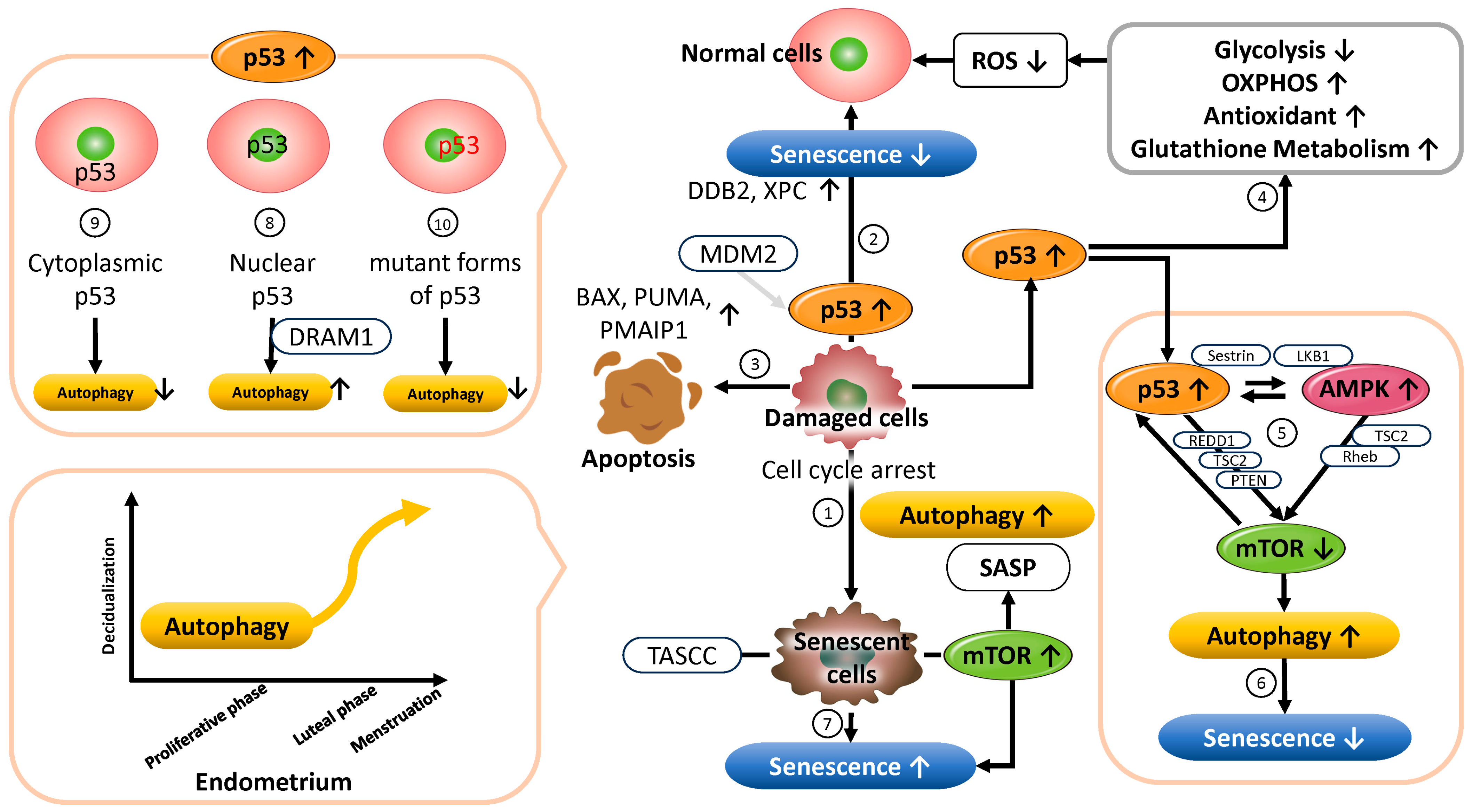
3.1. p53
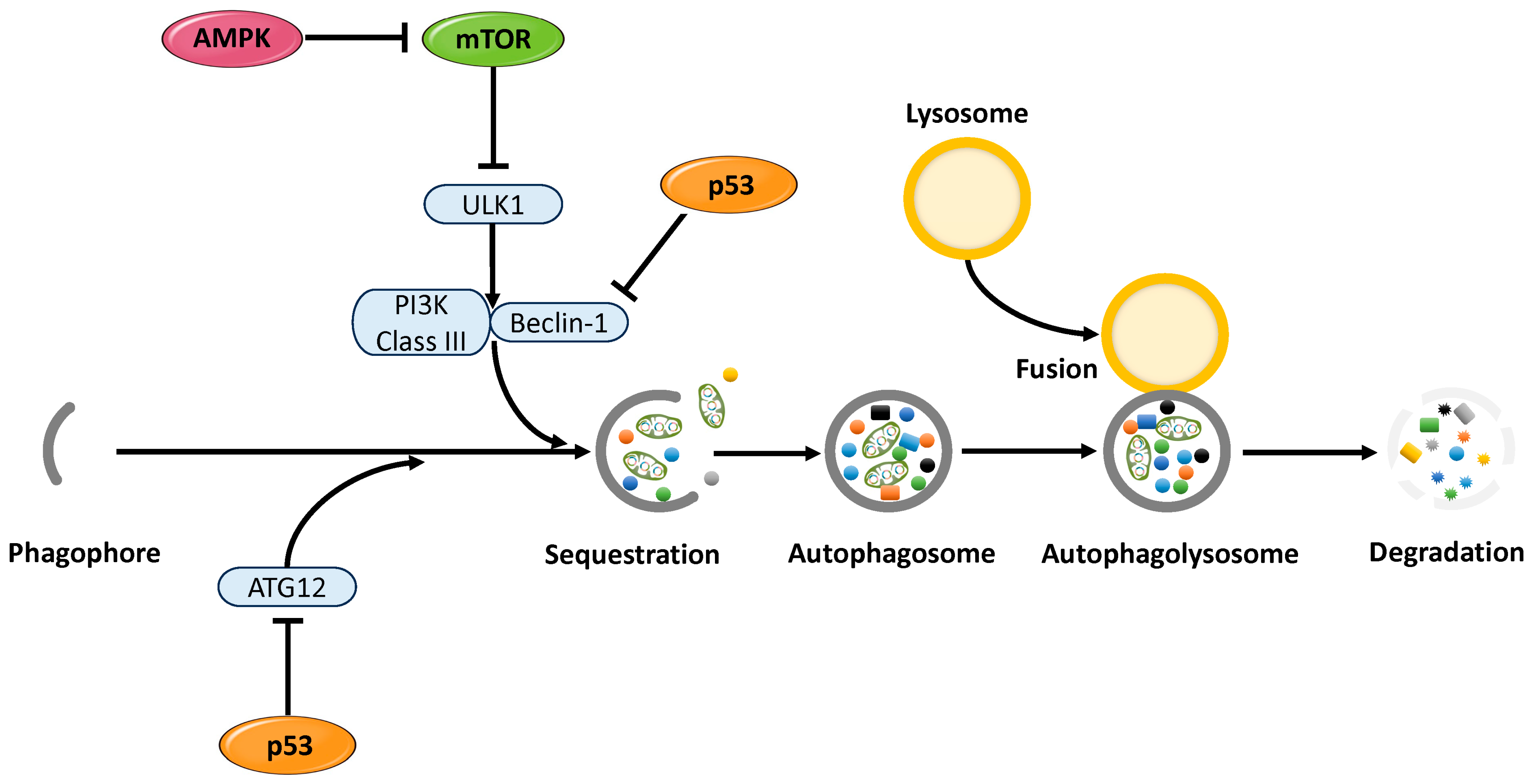
3.1.1. Functional Role of p53 in Cellular Senescence and Autophagy Regulation
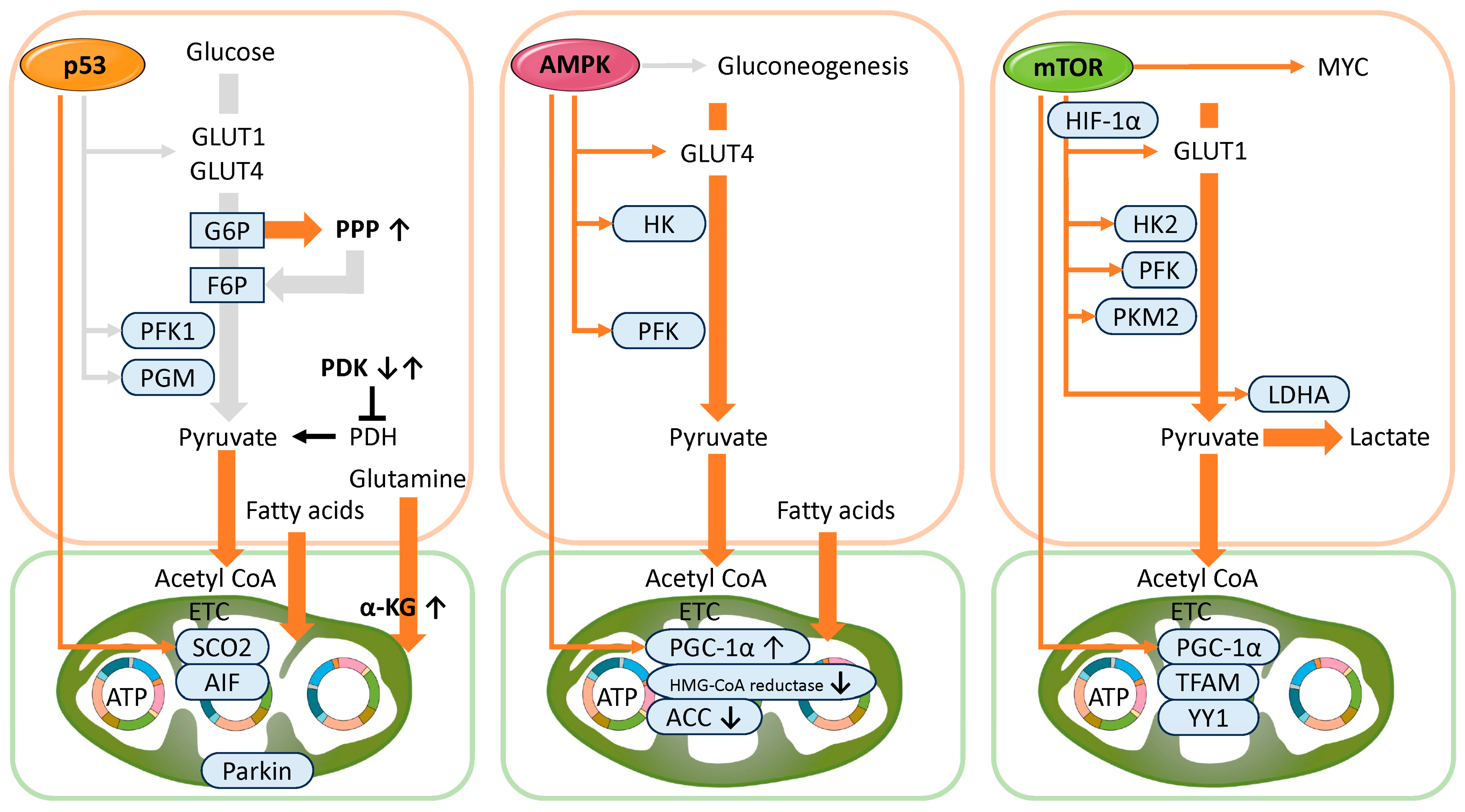
3.1.2. Functional Role of p53 in Metabolism
3.1.3. Functional Role of p53 in Maintaining Reproductive Function
3.2. AMPK
3.2.1. Functional Roles of AMPK in Cellular Senescence and Autophagy Regulation
3.2.2. Functional Role of AMPK in Metabolism
3.2.3. Functional Role of AMPK in the Maintenance of Reproductive Function
3.3. mTOR
3.3.1. Functional Role of mTOR in Cellular Senescence and Autophagy Regulation
3.3.2. Functional Role of mTOR in Metabolism
3.3.3. Functional Role of mTOR in the Maintenance of Reproductive Function
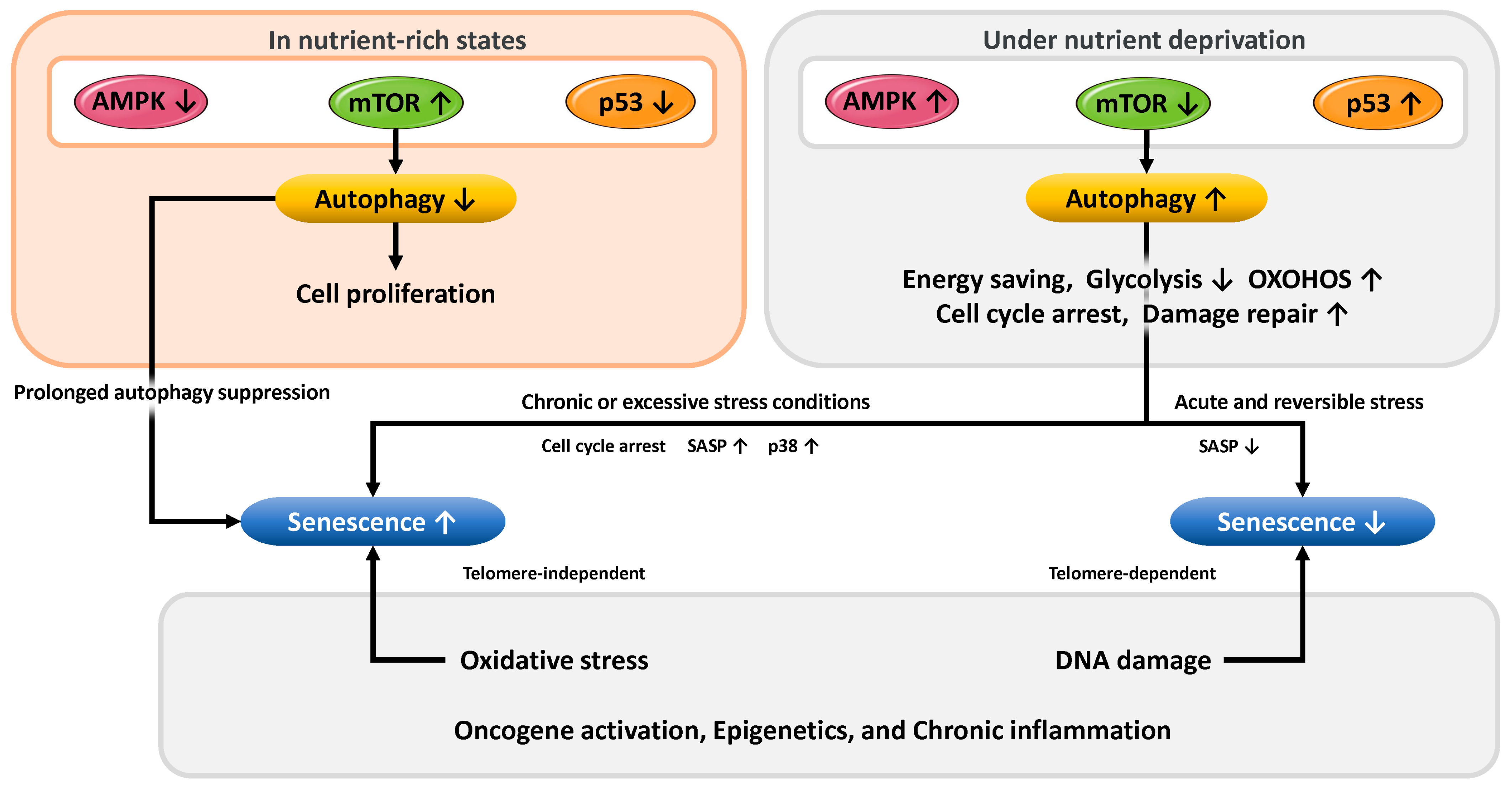
3.4. Mechanisms of Cellular Stress Response and Senescence Regulation via Autophagy
3.5. The Relationship Between Endometrial Cellular Senescence and Reproductive Capacity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sheldon, I.M.; Bromfield, J.J. Innate immunity in the human endometrium and ovary. Am. J. Reprod. Immunol. 2011, 66, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, A.K.; Buck, V.U.; Classen-Linke, I.; Leube, R.E. How Mechanical Forces Change the Human Endometrium during the Menstrual Cycle in Preparation for Embryo Implantation. Cells 2021, 10, 2008. [Google Scholar] [CrossRef]
- Agostinis, C.; Mangogna, A.; Bossi, F.; Ricci, G.; Kishore, U.; Bulla, R. Uterine Immunity and Microbiota: A Shifting Paradigm. Front. Immunol. 2019, 10, 2387. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Yoshimura, Y. Molecular and cellular mechanisms for differentiation and regeneration of the uterine endometrium. Endocr. J. 2008, 55, 795–810. [Google Scholar] [CrossRef]
- Varela-Eirín, M.; Demaria, M. Cellular senescence. Curr. Biol. 2022, 32, R448–R452. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of senescence and aging. Biochem. Med. 2019, 29, 030501. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. From Ancient Pathways to Aging Cells-Connecting Metabolism and Cellular Senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef]
- Regulski, M.J. Cellular Senescence: What, Why, and How. Wounds 2017, 29, 168–174. [Google Scholar]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Armstrong, J.L.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype in age-related chronic diseases. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Hirota, Y.; Cha, J.; Yoshie, M.; Daikoku, T.; Dey, S.K. Heightened uterine mammalian target of rapamycin complex 1 (mTORC1) signaling provokes preterm birth in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 18073–18078. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Yu, Q. Role of mTOR Signaling in Female Reproduction. Front. Endocrinol. 2019, 10, 692. [Google Scholar] [CrossRef]
- Borodkina, A.; Shatrova, A.; Abushik, P.; Nikolsky, N.; Burova, E. Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging 2014, 6, 481–495. [Google Scholar] [CrossRef]
- Kawamura, K.; Matsumura, Y.; Kawamura, T.; Araki, H.; Hamada, N.; Kuramoto, K.; Yagi, H.; Onoyama, I.; Asanoma, K.; Kato, K. Endometrial senescence is mediated by interleukin 17 receptor B signaling. Cell Commun. Signal. 2024, 22, 363. [Google Scholar] [CrossRef]
- Taylor, R.N.; Berga, S.L.; Zou, E.; Washington, J.; Song, S.; Marzullo, B.J.; Bagchi, I.C.; Bagchi, M.K.; Yu, J. Interleukin-1beta induces and accelerates human endometrial stromal cell senescence and impairs decidualization via the c-Jun N-terminal kinase pathway. Cell Death Discov. 2024, 10, 288. [Google Scholar] [CrossRef]
- Griukova, A.; Deryabin, P.; Shatrova, A.; Burova, E.; Severino, V.; Farina, A.; Nikolsky, N.; Borodkina, A. Molecular basis of senescence transmitting in the population of human endometrial stromal cells. Aging 2019, 11, 9912–9931. [Google Scholar] [CrossRef]
- Vassilieva, I.; Kosheverova, V.; Vitte, M.; Kamentseva, R.; Shatrova, A.; Tsupkina, N.; Skvortsova, E.; Borodkina, A.; Tolkunova, E.; Nikolsky, N.; et al. Paracrine senescence of human endometrial mesenchymal stem cells: A role for the insulin-like growth factor binding protein 3. Aging 2020, 12, 1987–2004. [Google Scholar] [CrossRef]
- Pathare, A.D.S.; Loid, M.; Saare, M.; Gidlöf, S.B.; Zamani Esteki, M.; Acharya, G.; Peters, M.; Salumets, A. Endometrial receptivity in women of advanced age: An underrated factor in infertility. Hum. Reprod. Update 2023, 29, 773–793. [Google Scholar] [CrossRef]
- Saito, Y.; Yamamoto, S.; Chikenji, T.S. Role of cellular senescence in inflammation and regeneration. Inflamm. Regen. 2024, 44, 28. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.I.; Choi, S.; Roh, W.S.; Lee, J.H.; Kim, T.G. Cellular Senescence and Inflammaging in the Skin Microenvironment. Int. J. Mol. Sci. 2021, 22, 3849. [Google Scholar] [CrossRef]
- Lu, Y.; Jarrahi, A.; Moore, N.; Bartoli, M.; Brann, D.W.; Baban, B.; Dhandapani, K.M. Inflammaging, cellular senescence, and cognitive aging after traumatic brain injury. Neurobiol. Dis. 2023, 180, 106090. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Goyal, A.; Pathak, S.; Ganguly, P. Cellular Senescence and Inflammaging in the Bone: Pathways, Genetics, Anti-Aging Strategies and Interventions. Int. J. Mol. Sci. 2024, 25, 7411. [Google Scholar] [CrossRef]
- Li, C.; Yuan, Y.; Jia, Y.; Zhou, Q.; Wang, Q.; Jiang, X. Cellular senescence: From homeostasis to pathological implications and therapeutic strategies. Front. Immunol. 2025, 16, 1534263. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wei, X.; Wu, J.; Xu, Y.; Hu, J.; Qin, C.; Chen, C.; Lin, Y. CBLL1 promotes endometrial stromal cell senescence via inhibiting PTEN in recurrent spontaneous abortion. FASEB J. 2024, 38, e23833. [Google Scholar] [CrossRef]
- Tang, X.; Zhu, Y.; Cao, Z.; Wang, X.; Cai, X.; Tang, Y.; Zhou, J.; Wu, M.; Zhen, X.; Ding, L.; et al. CDC42 deficiency leads to endometrial stromal cell senescence in recurrent implantation failure. Hum. Reprod. 2024, 39, 2768–2784. [Google Scholar] [CrossRef]
- Umbayev, B.; Safarova Yantsen, Y.; Yermekova, A.; Nessipbekova, A.; Syzdykova, A.; Askarova, S. Role of a small GTPase Cdc42 in aging and age-related diseases. Biogerontology 2023, 24, 27–46. [Google Scholar] [CrossRef]
- Ito, T.K.; Yokoyama, M.; Yoshida, Y.; Nojima, A.; Kassai, H.; Oishi, K.; Okada, S.; Kinoshita, D.; Kobayashi, Y.; Fruttiger, M.; et al. A crucial role for CDC42 in senescence-associated inflammation and atherosclerosis. PLoS ONE 2014, 9, e102186. [Google Scholar] [CrossRef]
- Johmura, Y.; Shimada, M.; Misaki, T.; Naiki-Ito, A.; Miyoshi, H.; Motoyama, N.; Ohtani, N.; Hara, E.; Nakamura, M.; Morita, A.; et al. Necessary and sufficient role for a mitosis skip in senescence induction. Mol. Cell 2014, 55, 73–84. [Google Scholar] [CrossRef]
- Soussi, T. The p53 tumor suppressor gene: From molecular biology to clinical investigation. Ann. N. Y. Acad. Sci. 2000, 910, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Qu, R.; Liu, D.; Xiong, X.; Liang, T.; Zhao, Y. The Cross Talk Between p53 and mTOR Pathways in Response to Physiological and Genotoxic Stresses. Front. Cell Dev. Biol. 2021, 9, 775507. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef]
- Qian, Y.; Chen, X. Senescence regulation by the p53 protein family. Methods Mol. Biol. 2013, 965, 37–61. [Google Scholar] [CrossRef]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Christy, B.; Demaria, M.; Campisi, J.; Huang, J.; Jones, D.; Dodds, S.G.; Williams, C.; Hubbard, G.; Livi, C.B.; Gao, X.; et al. p53 and rapamycin are additive. Oncotarget 2015, 6, 15802–15813. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures, and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef]
- Giono, L.E.; Manfredi, J.J. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J. Cell. Physiol. 2006, 209, 13–20. [Google Scholar] [CrossRef]
- Adimoolam, S.; Ford, J.M. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc. Natl. Acad. Sci. USA 2002, 99, 12985–12990. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Puzio-Kuter, A.M. The Role of p53 in Metabolic Regulation. Genes Cancer 2011, 2, 385–391. [Google Scholar] [CrossRef]
- Simabuco, F.M.; Morale, M.G.; Pavan, I.C.B.; Morelli, A.P.; Silva, F.R.; Tamura, R.E. p53 and metabolism: From mechanism to therapeutics. Oncotarget 2018, 9, 23780–23823. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Demidenko, Z.N.; Korotchkina, L.G.; Gudkov, A.V.; Blagosklonny, M.V. Paradoxical suppression of cellular senescence by p53. Proc. Natl. Acad. Sci. USA 2010, 107, 9660–9664. [Google Scholar] [CrossRef]
- Devis-Jauregui, L.; Eritja, N.; Davis, M.L.; Matias-Guiu, X.; Llobet-Navàs, D. Autophagy in the physiological endometrium and cancer. Autophagy 2021, 17, 1077–1095. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.; Kim, H.; Lee, H.; Seong, K.M.; Youn, H.; Youn, B. Autophagic Organelles in DNA Damage Response. Front. Cell Dev. Biol. 2021, 9, 668735. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181–185. [Google Scholar] [CrossRef]
- Deng, W.; Cha, J.; Yuan, J.; Haraguchi, H.; Bartos, A.; Leishman, E.; Viollet, B.; Bradshaw, H.B.; Hirota, Y.; Dey, S.K. p53 coordinates decidual sestrin 2/AMPK/mTORC1 signaling to govern parturition timing. J. Clin. Investig. 2016, 126, 2941–2954. [Google Scholar] [CrossRef]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.S.; Mowla, S.N.; Arora, P.; Jat, P.S. Tumour suppressors and cellular senescence. IUBMB Life 2014, 66, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Kim, J.W.; Jeoung, J.A.; Kim, M.S.; Kang, C. Autophagy Is Pro-Senescence When Seen in Close-Up, but Anti-Senescence in Long-Shot. Mol. Cells 2017, 40, 607–612. [Google Scholar] [CrossRef]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef]
- Narita, M.; Young, A.R.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy ments secretory phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, J.; Kim, M.S.; Kwon, Y.; Shin, S.; Yi, H.; Kim, H.; Chang, M.J.; Chang, C.B.; Kang, S.B.; et al. Coordinate regulation of the senescent state by selective autophagy. Dev. Cell 2021, 56, 1512–1525.e7. [Google Scholar] [CrossRef]
- Shim, D.; Duan, L.; Maki, C.G. P53-regulated autophagy and its impact on drug resistance and cell fate. Cancer Drug Resist. 2021, 4, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef]
- Cordani, M.; Butera, G.; Pacchiana, R.; Donadelli, M. Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 19–28. [Google Scholar] [CrossRef]
- Hajimohammadebrahim-Ketabforoush, M.; Zali, A.; Shahmohammadi, M.; Hamidieh, A.A. Metformin and its potential influence on cell fate decision between apoptosis and senescence in cancer, with a special emphasis on glioblastoma. Front. Oncol. 2024, 14, 1455492. [Google Scholar] [CrossRef]
- Stabenow, L.K.; Zibrova, D.; Ender, C.; Helbing, D.L.; Spengler, K.; Marx, C.; Wang, Z.Q.; Heller, R. Oxidative Glucose Metabolism Promotes Senescence in Vascular Endothelial Cells. Cells 2022, 11, 2213. [Google Scholar] [CrossRef] [PubMed]
- Sabbatinelli, J.; Prattichizzo, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Giuliani, A. Where Metabolism Meets Senescence: Focus on Endothelial Cells. Front. Physiol. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Liu, B.; Meng, Q.; Gao, X.; Sun, H.; Xu, Z.; Wang, Y.; Zhou, H. Lipid and glucose metabolism in senescence. Front. Nutr. 2023, 10, 1157352. [Google Scholar] [CrossRef] [PubMed]
- Abukwaik, R.; Vera-Siguenza, E.; Tennant, D.; Spill, F. p53 Orchestrates Cancer Metabolism: Unveiling Strategies to Reverse the Warburg Effect. Bull. Math. Biol. 2024, 86, 124. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wu, M.; Zhu, G.; Xu, Y. Emerging Roles of the Tumor Suppressor p53 in Metabolism. Front. Cell Dev. Biol. 2022, 9, 762742. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, J.; Feng, Z. The regulation of cellular metabolism by tumor suppressor p53. Cell Biosci. 2013, 3, 9. [Google Scholar] [CrossRef]
- Dou, X.; Fu, Q.; Long, Q.; Liu, S.; Zou, Y.; Fu, D.; Xu, Q.; Jiang, Z.; Ren, X.; Zhang, G.; et al. PDK4-dependent hypercatabolism and lactate production of senescent cells promotes cancer malignancy. Nat. Metab. 2023, 5, 1887–1910. [Google Scholar] [CrossRef] [PubMed]
- Olenchock, B.A.; Vander Heiden, M.G. Pyruvate as a pivot point for oncogene-induced senescence. Cell 2013, 153, 1429–1430. [Google Scholar] [CrossRef]
- Gomes, A.S.; Ramos, H.; Soares, J.; Saraiva, L. p53 and glucose metabolism: An orchestra to be directed in cancer therapy. Pharmacol. Res. 2018, 131, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Assaily, W.; Rubinger, D.A.; Wheaton, K.; Lin, Y.; Ma, W.; Xuan, W.; Brown-Endres, L.; Tsuchihara, K.; Mak, T.W.; Benchimol, S. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol. Cell 2011, 44, 491–501. [Google Scholar] [CrossRef]
- Ide, T.; Chu, K.; Aaronson, S.A.; Lee, S.W. GAMT joins the p53 network: Branching into metabolism. Cell Cycle 2010, 9, 1706–1710. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef]
- Vousden, K.H. Alternative fuel--another role for p53 in the regulation of metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 7117–7118. [Google Scholar] [CrossRef]
- Stambolsky, P.; Weisz, L.; Shats, I.; Klein, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Regulation of AIF expression by p53. Cell Death Differ. 2006, 13, 2140–2149. [Google Scholar] [CrossRef]
- Kuo, M.L.; Sy, A.J.; Xue, L.; Chi, M.; Lee, M.T.; Yen, T.; Chiang, M.I.; Chang, L.; Chu, P.; Yen, Y. RRM2B suppresses activation of the oxidative stress pathway and is up-regulated by p53 during senescence. Sci. Rep. 2012, 2, 822. [Google Scholar] [CrossRef]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef]
- Huang, W.; Xie, W.; Zhong, H.; Cai, S.; Huang, Q.; Liu, Y.; Zeng, Z.; Liu, Y. Cytosolic p53 Inhibits Parkin-Mediated Mitophagy and Promotes Acute Liver Injury Induced by Heat Stroke. Front. Immunol. 2022, 13, 859231. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 2020, 33, 2–22. [Google Scholar] [CrossRef]
- Shen, H.H.; Zhang, T.; Yang, H.L.; Lai, Z.Z.; Zhou, W.J.; Mei, J.; Shi, J.W.; Zhu, R.; Xu, F.Y.; Li, D.J.; et al. Ovarian hormones-autophagy-immunity axis in menstruation and endometriosis. Theranostics 2021, 11, 3512–3526. [Google Scholar] [CrossRef]
- Mestre Citrinovitz, A.C.; Strowitzki, T.; Germeyer, A. Decreased Autophagy Impairs Decidualization of Human Endometrial Stromal Cells: A Role for ATG Proteins in Endometrial Physiology. Int. J. Mol. Sci. 2019, 20, 3066. [Google Scholar] [CrossRef]
- Park, H.; Cho, M.; Do, Y.; Park, J.K.; Bae, S.J.; Joo, J.; Ha, K.T. Autophagy as a Therapeutic Target of Natural Products Enhancing Embryo Implantation. Pharmaceuticals 2021, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, X.; Lv, Y. HMGB1 Mediated Inflammation and Autophagy Contribute to Endometriosis. Front. Endocrinol. 2021, 12, 616696. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Wada-Hiraike, O. The Two-Faced Role of Autophagy in Endometrial Cancer. Front. Cell Dev. Biol. 2022, 10, 839416. [Google Scholar] [CrossRef]
- Yang, S.; Wang, H.; Li, D.; Li, M. Role of Endometrial Autophagy in Physiological and Pathophysiological Processes. J. Cancer 2019, 10, 3459–3471. [Google Scholar] [CrossRef]
- Zhou, S.; Zhao, L.; Yi, T.; Wei, Y.; Zhao, X. Menopause-induced uterine epithelium atrophy results from arachidonic acid/prostaglandin E2 axis inhibition-mediated autophagic cell death. Sci. Rep. 2016, 6, 31408. [Google Scholar] [CrossRef]
- Berger, C.; Qian, Y.; Chen, X. The p53-estrogen receptor loop in cancer. Curr. Mol. Med. 2013, 13, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Maia, H., Jr.; Maltez, A.; Studart, E.; Athayde, C.; Coutinho, E.M. Ki-67, Bcl-2 and p53 expression in endometrial polyps and in the normal endometrium during the menstrual cycle. BJOG Int. J. Obstet. Gynaecol. 2004, 111, 1242–1247. [Google Scholar] [CrossRef]
- Hirota, Y.; Daikoku, T.; Tranguch, S.; Xie, H.; Bradshaw, H.B.; Dey, S.K. Uterine-specific p53 deficiency confers premature uterine senescence and promotes preterm birth in mice. J. Clin. Investig. 2010, 120, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.; Fang, Q.J.; Zhao, X.B. A research on the protein expression of p53, p16, and MDM2 in endometriosis. Medicine 2019, 98, e14776. [Google Scholar] [CrossRef]
- Hu, W.; Feng, Z.; Teresky, A.K.; Levine, A.J. p53 regulates maternal reproduction through LIF. Nature 2007, 450, 721–724. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, C.; Kang, H.J.; Sun, Y.; Wang, H.; Naqvi, A.; Frank, A.K.; Rosenwaks, Z.; Murphy, M.E.; Levine, A.J. Regulation of female reproduction by p53 and its family members. FASEB J. 2011, 25, 2245–2255. [Google Scholar] [CrossRef]
- Delenko, J.; Xue, X.; Chatterjee, P.K.; Hyman, N.; Shih, A.J.; Adelson, R.P.; Safaric Tepes, P.; Gregersen, P.K.; Metz, C.N. Quercetin enhances decidualization through AKT-ERK-p53 signaling and supports a role for senescence in endometriosis. Reprod. Biol. Endocrinol. 2024, 22, 100. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Ditch, S.; Paull, T.T. The ATM protein kinase and cellular redox signaling: Beyond the DNA damage response. Trends Biochem. Sci. 2012, 37, 15–22. [Google Scholar] [CrossRef]
- Alao, J.P.; Legon, L.; Dabrowska, A.; Tricolici, A.M.; Kumar, J.; Rallis, C. Interplays of AMPK and TOR in Autophagy Regulation in Yeast. Cells 2023, 12, 519. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H. Contrasting views on the role of AMPK in autophagy. Bioessays 2024, 46, e2300211. [Google Scholar] [CrossRef]
- Ranek, M.J.; Kokkonen-Simon, K.M.; Chen, A.; Dunkerly-Eyring, B.L.; Vera, M.P.; Oeing, C.U.; Patel, C.H.; Nakamura, T.; Zhu, G.; Bedja, D.; et al. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature 2019, 566, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Lee, D.H.; Kim, D.H. Redefining the role of AMPK in autophagy and the energy stress response. Nat. Commun. 2023, 14, 2994. [Google Scholar] [CrossRef]
- Nwadike, C.; Williamson, L.E.; Gallagher, L.E.; Guan, J.L.; Chan, E.Y.W. AMPK Inhibits ULK1-Dependent Autophagosome Formation and Lysosomal Acidification via Distinct Mechanisms. Mol. Cell. Biol. 2018, 38, e00023-18. [Google Scholar] [CrossRef]
- Gong, H.; Chen, H.; Xiao, P.; Huang, N.; Han, X.; Zhang, J.; Yang, Y.; Li, T.; Zhao, T.; Tai, H.; et al. miR-146a impedes the anti-aging effect of AMPK via NAMPT suppression and NAD(+)/SIRT inactivation. Signal Transduct. Target. Ther. 2022, 7, 66. [Google Scholar] [CrossRef]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef]
- Han, Y.; Liu, Y.; Zhang, Y.; Wang, W.; Lv, T.; Huang, J.; Peng, X. The Role and Application of the AMPK-Sirtuins Network in Cellular Senescence. Front. Biosci.-Landmark 2023, 28, 250. [Google Scholar] [CrossRef]
- Han, X.; Zhang, T.; Zhang, X.; Zhang, R.; Lao, K.; Mi, Y.; Gou, X. AMPK alleviates oxidative stress-induced premature senescence via inhibition of NF-kappaB/STAT3 axis-mediated positive feedback loop. Mech. Ageing Dev. 2020, 191, 111347. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.Y.; Li, Y.Y.; Huang, C.; Li, J.; Yao, H.W. AMP-activated protein kinase reduces inflammatory responses and cellular senescence in pulmonary emphysema. Oncotarget 2017, 8, 22513–22523. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, F. Coordination of the AMPK, Akt, mTOR, and p53 Pathways under Glucose Starvation. Int. J. Mol. Sci. 2022, 23, 14945. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.L.; Veprintsev, D.B.; Bycroft, M.; Fersht, A.R. Comparative binding of p53 to its promoter and DNA recognition elements. J. Mol. Biol. 2005, 348, 589–596. [Google Scholar] [CrossRef]
- Liebl, M.C.; Hofmann, T.G. Cell Fate Regulation upon DNA Damage: p53 Serine 46 Kinases Pave the Cell Death Road. Bioessays 2019, 41, e1900127. [Google Scholar] [CrossRef]
- Sun, Y.; Connors, K.E.; Yang, D.Q. AICAR induces phosphorylation of AMPK in an ATM-dependent, LKB1-independent manner. Mol. Cell. Biochem. 2007, 306, 239–245. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. The metabolic roots of senescence: Mechanisms and opportunities for intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef]
- Pieles, O.; Hartmann, M.; Morsczeck, C. AMP-activated protein kinase and the down-stream activated process of autophagy regulate the osteogenic differentiation of human dental follicle cells. Arch. Oral Biol. 2021, 122, 104951. [Google Scholar] [CrossRef]
- Morsczeck, C.; Reck, A.; Reichert, T.E. Changes in AMPK activity induces cellular senescence in human dental follicle cells. Exp. Gerontol. 2023, 172, 112071. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, C.; Zeng, H.; Xia, Y.; Weng, C.; Deng, Y.; Wang, L.; Wang, H. Glycolysis maintains AMPK activation in sorafenib-induced Warburg effect. Mol. Metab. 2023, 77, 101796. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Seo, W.Y.; Song, K.H.; Chanda, D.; Kim, Y.D.; Kim, D.K.; Lee, M.W.; Ryu, D.; Kim, Y.H.; Noh, J.R.; et al. AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB.CRTC2 complex by orphan nuclear receptor small heterodimer partner. J. Biol. Chem. 2010, 285, 32182–32191. [Google Scholar] [CrossRef]
- Kim, Y.D.; Park, K.G.; Lee, Y.S.; Park, Y.Y.; Kim, D.K.; Nedumaran, B.; Jang, W.G.; Cho, W.J.; Ha, J.; Lee, I.K.; et al. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes 2008, 57, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Zhang, P.; Fu, H.J.; Lv, L.X.; Liu, C.F.; Han, C.; Zhao, X.F.; Wang, J.X. WSSV exploits AMPK to activate mTORC2 signaling for proliferation by enhancing aerobic glycolysis. Commun. Biol. 2023, 6, 361. [Google Scholar] [CrossRef]
- Clarke, P.R.; Hardie, D.G. Regulation of HMG-CoA reductase: Identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J. 1990, 9, 2439–2446. [Google Scholar] [CrossRef]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Viollet, B.; Bhat, M.K. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1alpha-mediated mitochondrial biogenesis. Cell Death Discov. 2015, 1, 15063. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, M.; Zhang, J.; Wang, S. Unveiling uterine aging: Much more to learn. Ageing Res. Rev. 2023, 86, 101879. [Google Scholar] [CrossRef]
- Borodkina, A.V.; Shatrova, A.N.; Deryabin, P.I.; Griukova, A.A.; Abushik, P.A.; Antonov, S.M.; Nikolsky, N.N.; Burova, E.B. Calcium alterations signal either to senescence or to autophagy induction in stem cells upon oxidative stress. Aging 2016, 8, 3400–3418. [Google Scholar] [CrossRef]
- Kawano, Y.; Sato, H.; Goto, K.; Nishida, M.; Nasu, K. The inhibitory effect of AMP-activated protein kinase (AMPK) on chemokine and prostaglandin production in human endometrial stromal cells. Reprod. Biol. Endocrinol. 2021, 19, 188. [Google Scholar] [CrossRef]
- McCallum, M.L.; Pru, C.A.; Smith, A.R.; Kelp, N.C.; Foretz, M.; Viollet, B.; Du, M.; Pru, J.K. A functional role for AMPK in female fertility and endometrial regeneration. Reproduction 2018, 156, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, R.M.; Pru, C.A.; Behura, S.K.; Cronrath, A.R.; McCallum, M.L.; Kelp, N.C.; Winuthayanon, W.; Spencer, T.E.; Pru, J.K. AMPK is required for uterine receptivity and normal responses to steroid hormones. Reproduction 2020, 159, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, R.; Zhang, L.; Geng, Y.; Chen, Q.; Chen, X.; Liu, X.; Mu, X.; Ding, Y.; Wang, Y.; et al. AMPK/mTOR downregulated autophagy enhances aberrant endometrial decidualization in folate-deficient pregnant mice. J. Cell. Physiol. 2021, 236, 7376–7389. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef]
- Hasty, P.; Sharp, Z.D.; Curiel, T.J.; Campisi, J. mTORC1 and p53: Clash of the gods? Cell Cycle 2013, 12, 20–25. [Google Scholar] [CrossRef]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823.e12. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xie, W.; Xie, W.; Wei, W.; Guo, J. Beyond controlling cell size: Functional analyses of S6K in tumorigenesis. Cell Death Dis. 2022, 13, 646. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.; Sitzmann, J.M.; Dastidar, S.G.; Rodriguez, A.A.; Vu, S.L.; McDonald, C.E.; Academia, E.C.; O’Leary, M.N.; Ashe, T.D.; La Spada, A.R.; et al. Muscle-specific 4E-BP1 signaling activation improves metabolic parameters during aging and obesity. J. Clin. Investig. 2015, 125, 2952–2964. [Google Scholar] [CrossRef]
- Bodine, S.C. The role of mTORC1 in the regulation of skeletal muscle mass. Fac. Rev. 2022, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.; Nelson, G.; Rabanal-Ruiz, Y.; Kucheryavenko, O.; Dunhill-Turner, N.A.; Chesterman, C.C.; Zahari, Q.; Zhang, T.; Conduit, S.E.; Mitchell, C.A.; et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J. Cell Biol. 2017, 216, 1949–1957. [Google Scholar] [CrossRef]
- Lee, C.H.; Inoki, K.; Karbowniczek, M.; Petroulakis, E.; Sonenberg, N.; Henske, E.P.; Guan, K.L. Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. EMBO J. 2007, 26, 4812–4823. [Google Scholar] [CrossRef]
- Lai, K.P.; Leong, W.F.; Chau, J.F.; Jia, D.; Zeng, L.; Liu, H.; He, L.; Hao, A.; Zhang, H.; Meek, D.; et al. S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO J. 2010, 29, 2994–3006. [Google Scholar] [CrossRef]
- Jung, S.H.; Hwang, H.J.; Kang, D.; Park, H.A.; Lee, H.C.; Jeong, D.; Lee, K.; Park, H.J.; Ko, Y.G.; Lee, J.S. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene 2019, 38, 1639–1650. [Google Scholar] [CrossRef]
- Chibaya, L.; Karim, B.; Zhang, H.; Jones, S.N. Mdm2 phosphorylation by Akt regulates the p53 response to oxidative stress to promote cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2003193118. [Google Scholar] [CrossRef]
- Pavlova, J.A.; Guseva, E.A.; Dontsova, O.A.; Sergiev, P.V. Natural Activators of Autophagy. Biochemistry 2024, 89, 1–26. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Dalla Pozza, E.; Nadal-Serrano, M.; Oliver, J.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 2016, 10, 1008–1029. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Fan, L.; Han, Y.; Guo, J.; Hao, Z.; Cao, L.; Kang, J.; Wang, X.; He, J.; Li, J. The mTORC1/eIF4E/HIF-1alpha Pathway Mediates Glycolysis to Support Brain Hypoxia Resistance in the Gansu Zokor, Eospalax cansus. Front. Physiol. 2021, 12, 626240. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Wu, Y.; Yu, S.; Li, X.; Wang, A.; Wang, S.; Chen, W.; Lu, Y. Critical role of mTOR in regulating aerobic glycolysis in carcinogenesis (Review). Int. J. Oncol. 2021, 58, 9–19. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef]
- Magaway, C.; Kim, E.; Jacinto, E. Targeting mTOR and Metabolism in Cancer: Lessons and Innovations. Cells 2019, 8, 1584. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef]
- de la Cruz López, K.G.; Toledo Guzmán, M.E.; Sánchez, E.O.; García Carrancá, A. mTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1373. [Google Scholar] [CrossRef]
- Chrienova, Z.; Nepovimova, E.; Kuca, K. The role of mTOR in age-related diseases. J. Enzym. Inhib. Med. Chem. 2021, 36, 1679–1693. [Google Scholar] [CrossRef]
- Zhang, H.; Su, X.; Burley, S.K.; Zheng, X.F.S. mTOR regulates aerobic glycolysis through NEAT1 and nuclear paraspeckle-mediated mechanism in hepatocellular carcinoma. Theranostics 2022, 12, 3518–3533. [Google Scholar] [CrossRef]
- Levy, T.; Voeltzke, K.; Hruby, L.; Alasad, K.; Bas, Z.; Snaebjörnsson, M.; Marciano, R.; Scharov, K.; Planque, M.; Vriens, K.; et al. mTORC1 regulates cell survival under glucose starvation through 4EBP1/2-mediated translational reprogramming of fatty acid metabolism. Nat. Commun. 2024, 15, 4083. [Google Scholar] [CrossRef]
- Li, Q.; Lin, Y.; Liang, G.; Xiao, N.; Zhang, H.; Yang, X.; Yang, J.; Liu, A. Autophagy and Senescence: The Molecular Mechanisms and Implications in Liver Diseases. Int. J. Mol. Sci. 2023, 24, 16880. [Google Scholar] [CrossRef]
- Zhai, P.; Sung, E.A.; Shiheido-Watanabe, Y.; Takayama, K.; Tian, Y.; Sadoshima, J. Suppression of autophagy induces senescence in the heart. J. Mol. Cell. Cardiol. 2024, 195, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Liu, B.; Gan, L. Autophagy prevents microglial senescence. Nat. Cell Biol. 2023, 25, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, N.K.; Lim, H.J.; Ji, S.T.; Kim, Y.J.; Jang, W.B.; Kim, D.Y.; Kang, S.; Yun, J.; Ha, J.S.; et al. Pharmacological inhibition of mTOR attenuates replicative cell senescence and improves cellular function via regulating the STAT3-PIM1 axis in human cardiac progenitor cells. Exp. Mol. Med. 2020, 52, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Gelino, S.; Hansen, M. Autophagy—An Emerging Anti-Aging Mechanism. J. Clin. Exp. Pathol. 2012, S4, 006. [Google Scholar] [CrossRef]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef]
- Wilkinson, J.E.; Burmeister, L.; Brooks, S.V.; Chan, C.C.; Friedline, S.; Harrison, D.E.; Hejtmancik, J.F.; Nadon, N.; Strong, R.; Wood, L.K.; et al. Rapamycin slows aging in mice. Aging Cell 2012, 11, 675–682. [Google Scholar] [CrossRef]
- Xu, S.; Cai, Y.; Wei, Y. mTOR Signaling from Cellular Senescence to Organismal Aging. Aging Dis. 2013, 5, 263–273. [Google Scholar] [CrossRef]
- Walters, H.E.; Deneka-Hannemann, S.; Cox, L.S. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging 2016, 8, 231–244. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct. Target. Ther. 2023, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.O.; Song, H.I.; Han, J.S.; Yoon, M.S. Differential regulation of mTORC1 and mTORC2 is critical for 8-Br-cAMP-induced decidualization. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Levine, B. Autophagy in cellular growth control. FEBS Lett. 2010, 584, 1417–1426. [Google Scholar] [CrossRef]
- Luo, Y.; Zou, P.; Zou, J.; Wang, J.; Zhou, D.; Liu, L. Autophagy regulates ROS-induced cellular senescence via p21 in a p38 MAPKalpha dependent manner. Exp. Gerontol. 2011, 46, 860–867. [Google Scholar] [CrossRef]
- Kang, C.; Elledge, S.J. How autophagy both activates and inhibits cellular senescence. Autophagy 2016, 12, 898–899. [Google Scholar] [CrossRef]
- Pantelis, P.; Theocharous, G.; Lagopati, N.; Veroutis, D.; Thanos, D.F.; Lampoglou, G.P.; Pippa, N.; Gatou, M.A.; Tremi, I.; Papaspyropoulos, A.; et al. The Dual Role of Oxidative-Stress-Induced Autophagy in Cellular Senescence: Comprehension and Therapeutic Approaches. Antioxidants 2023, 12, 169. [Google Scholar] [CrossRef]
- Xu, Y.; Li, N.; Xiang, R.; Sun, P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends Biochem. Sci. 2014, 39, 268–276. [Google Scholar] [CrossRef]
- Slobodnyuk, K.; Radic, N.; Ivanova, S.; Llado, A.; Trempolec, N.; Zorzano, A.; Nebreda, A.R. Autophagy-induced senescence is regulated by p38alpha signaling. Cell Death Dis. 2019, 10, 376. [Google Scholar] [CrossRef]
- Kwong, J.; Hong, L.; Liao, R.; Deng, Q.; Han, J.; Sun, P. p38α and p38γ mediate oncogenic ras-induced senescence through differential mechanisms. J. Biol. Chem. 2009, 284, 11237–11246. [Google Scholar] [CrossRef]
- Borodkina, A.V.; Shatrova, A.N.; Nikolsky, N.N.; Burova, E.B. Role of P38 Map-Kinase in the Stress-Induced Senescence Progression of Human Endometrium-Derived Mesenchymal Stem Cells. Tsitologiia 2016, 58, 429–435. [Google Scholar]
- Jin, J.; Richardson, L.; Sheller-Miller, S.; Zhong, N.; Menon, R. Oxidative stress induces p38MAPK-dependent senescence in the feto-maternal interface cells. Placenta 2018, 67, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Mann, D.J.; Hara, E. Cellular senescence: Its role in tumor suppression and aging. Cancer Sci. 2009, 100, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Toniyan, K.A.; Malkov, A.A.; Biryukov, N.S.; Gorbacheva, E.Y.; Boyarintsev, V.V.; Ogneva, I.V. The Cellular Respiration of Endometrial Biopsies from Patients with Various Forms of Endometriosis. Int. J. Mol. Sci. 2024, 25, 3680. [Google Scholar] [CrossRef]
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 2022, 132, e158447. [Google Scholar] [CrossRef]
- Huang, Y.; Zhu, Q.; Sun, Y. Glucose metabolism and endometrium decidualization. Front. Endocrinol. 2025, 16, 1546335. [Google Scholar] [CrossRef]
- Velarde, M.C.; Menon, R. Positive and negative effects of cellular senescence during female reproductive aging and pregnancy. J. Endocrinol. 2016, 230, R59–R76. [Google Scholar] [CrossRef]
- Tomari, H.; Kawamura, T.; Asanoma, K.; Egashira, K.; Kawamura, K.; Honjo, K.; Nagata, Y.; Kato, K. Contribution of senescence in human endometrial stromal cells during proliferative phase to embryo receptivity. Biol. Reprod. 2020, 103, 104–113. [Google Scholar] [CrossRef]
- Parvanov, D.; Ganeva, R.; Arsov, K.; Decheva, I.; Handzhiyska, M.; Ruseva, M.; Vidolova, N.; Scarpellini, F.; Metodiev, D.; Stamenov, G. Association between endometrial senescent cells and immune cells in women with repeated implantation failure. J. Assist. Reprod. Genet. 2023, 40, 1631–1638. [Google Scholar] [CrossRef]
- Takasugi, M.; Yoshida, Y.; Hara, E.; Ohtani, N. The role of cellular senescence and SASP in tumour microenvironment. FEBS J. 2023, 290, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Qin, D.; Hou, X.; Tian, L.; Yu, Y.; Zhang, R.; Lyu, H.; Guo, D.; Chen, X.Z.; Zhou, C.; et al. Cellular senescence: A double-edged sword in cancer therapy. Front. Oncol. 2023, 13, 1189015. [Google Scholar] [CrossRef] [PubMed]
- Kusama, K.; Yamauchi, N.; Yoshida, K.; Azumi, M.; Yoshie, M.; Tamura, K. Senolytic treatment modulates decidualization in human endometrial stromal cells. Biochem. Biophys. Res. Commun. 2021, 571, 174–180. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, H.; Umetani, M.; Nishio, M.; Shigetomi, H.; Imanaka, S.; Hashimoto, H. Molecular Mechanisms of Cellular Senescence in Age-Related Endometrial Dysfunction. Cells 2025, 14, 858. https://doi.org/10.3390/cells14120858
Kobayashi H, Umetani M, Nishio M, Shigetomi H, Imanaka S, Hashimoto H. Molecular Mechanisms of Cellular Senescence in Age-Related Endometrial Dysfunction. Cells. 2025; 14(12):858. https://doi.org/10.3390/cells14120858
Chicago/Turabian StyleKobayashi, Hiroshi, Mai Umetani, Miki Nishio, Hiroshi Shigetomi, Shogo Imanaka, and Hiratsugu Hashimoto. 2025. "Molecular Mechanisms of Cellular Senescence in Age-Related Endometrial Dysfunction" Cells 14, no. 12: 858. https://doi.org/10.3390/cells14120858
APA StyleKobayashi, H., Umetani, M., Nishio, M., Shigetomi, H., Imanaka, S., & Hashimoto, H. (2025). Molecular Mechanisms of Cellular Senescence in Age-Related Endometrial Dysfunction. Cells, 14(12), 858. https://doi.org/10.3390/cells14120858