
B10 Promotes Polarization and Pro-Resolving Functions of Bone Marrow Derived Macrophages (BMDM) Through PD-1 Activation

 , , ,
, , ,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Splenocyte Isolation and Culture
2.3. Bone Marrow-Derived Macrophage Isolation and Culture
2.4. B-10 Culture and Isolation
2.5. Macrophage-B-10 Co-Culture
2.6. Flow Cytometry
2.7. Western Blotting
2.8. Real-Time RT-PCR
2.9. Phagocytosis Assay
2.10. LC-MS/MS-Based Lipidomics
2.11. Statistical Analysis
3. Results
3.1. B-10 Cells Induce Macrophage Polarization Towards M2 Through Direct Cell–Cell Interaction
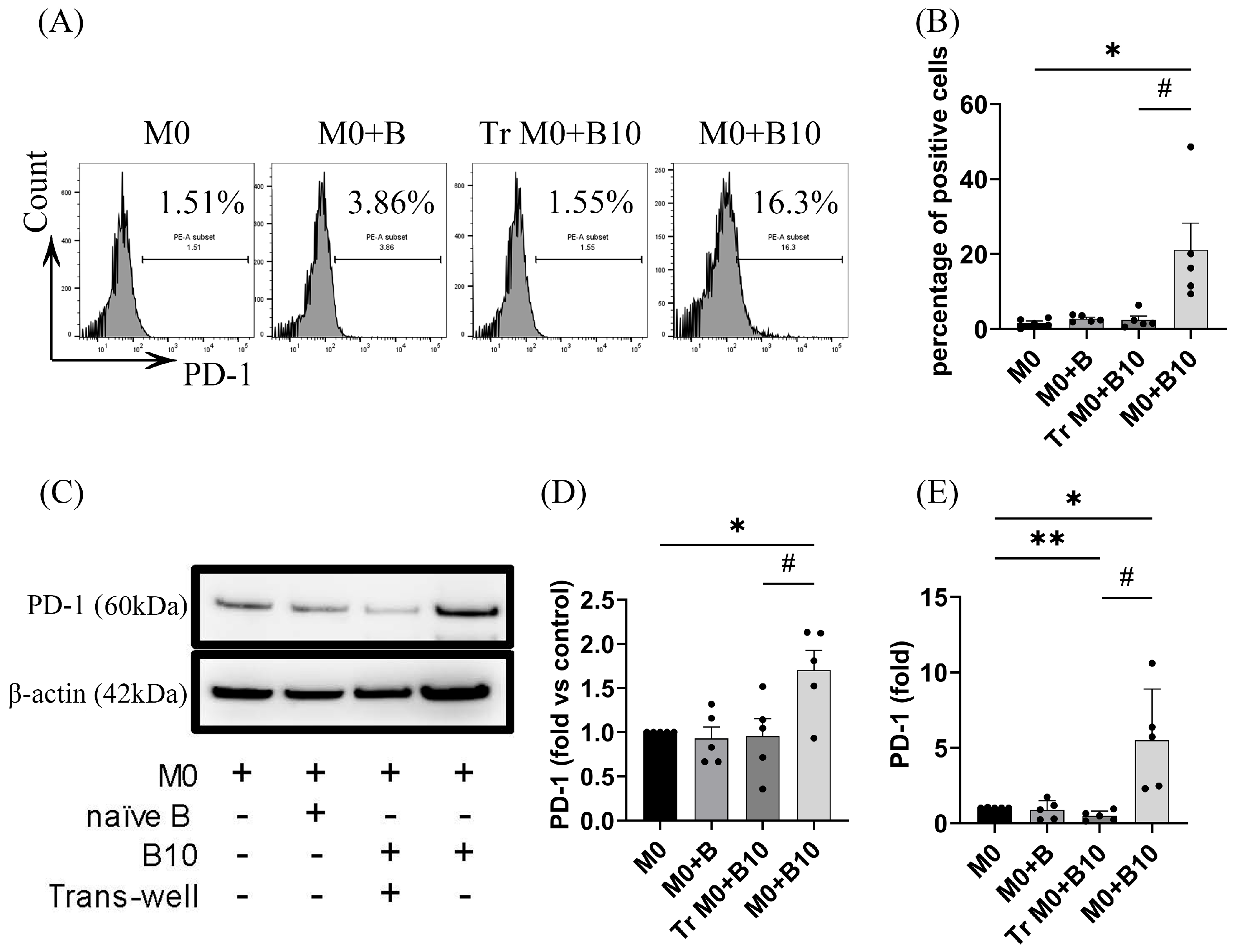
3.2. B-10 Cells Upregulate PD-1 on Macrophages via Direct Cell–Cell Interaction
3.3. B-10 Cells Enhance M2-Related Cytokines in Macrophages
3.4. B-10 Cells Enhance Macrophage Phagocytosis Through Direct Cell–Cell Contact
3.5. B-10 Cells Induce M2 Macrophage Polarization from M0 Through PD-L1/PD-1 Ligation
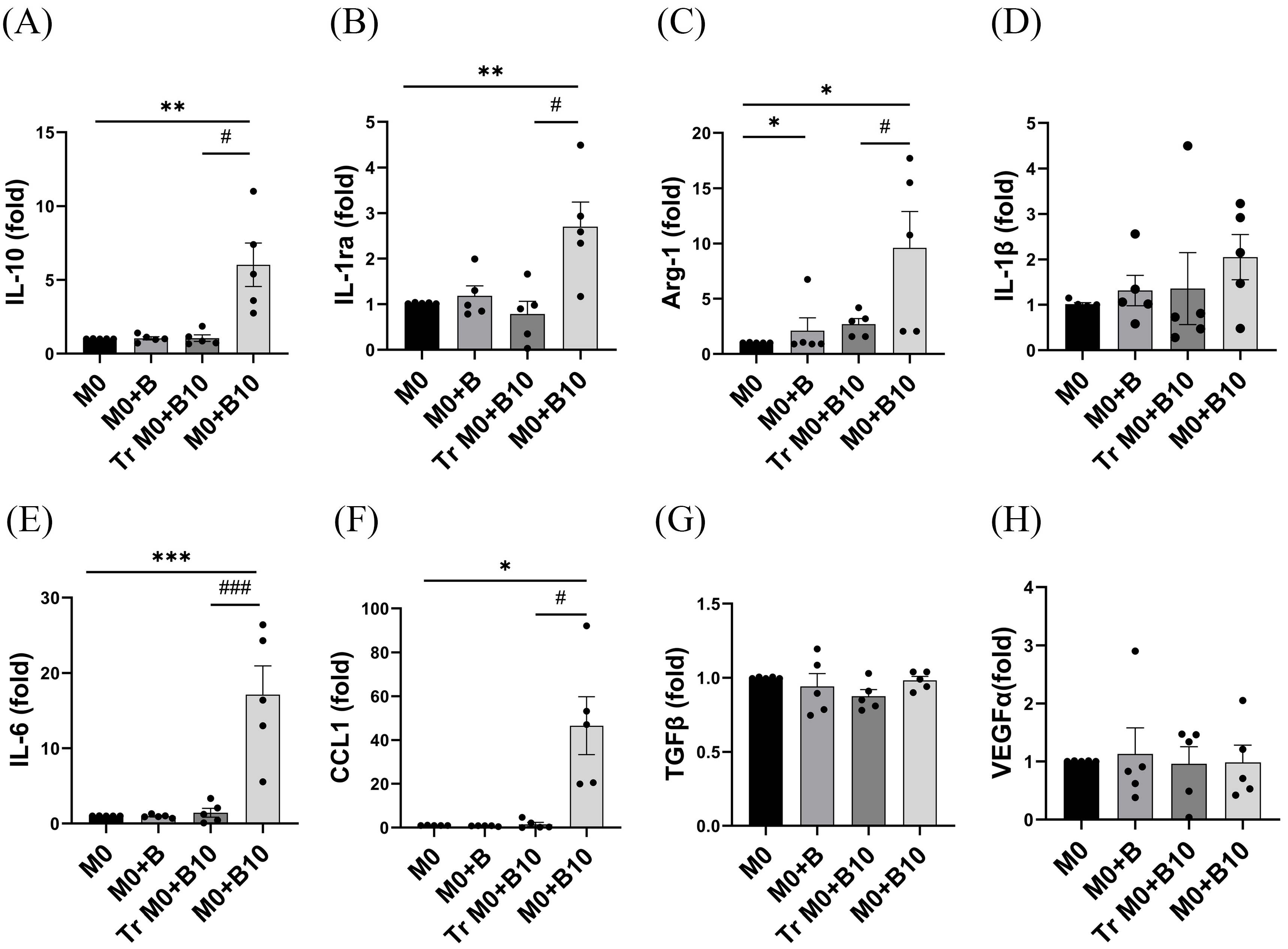
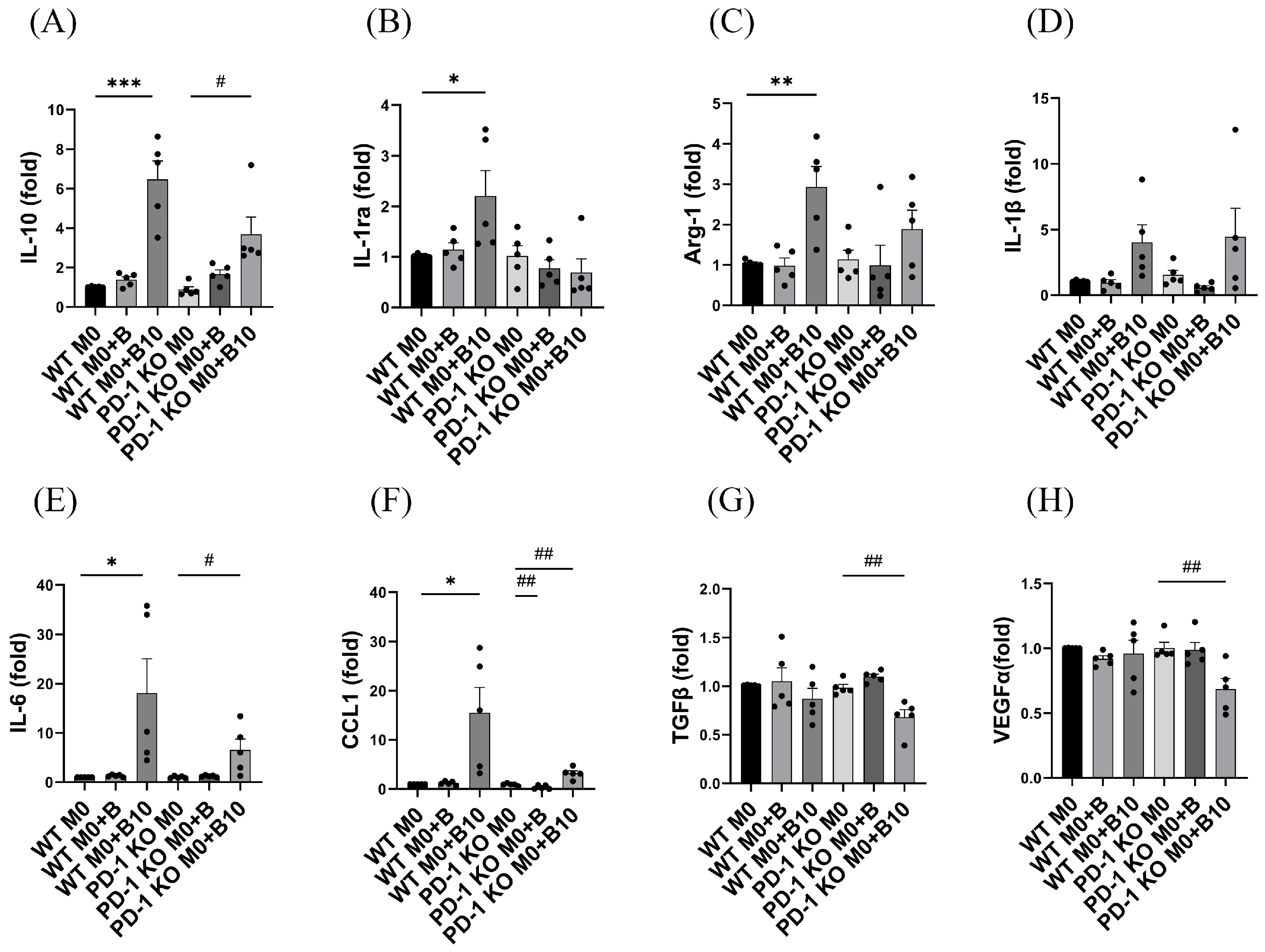
3.6. PD-1/PD-L1 Ligation Regulates the Release of Cytokines Related to Various M2 Type Macrophages
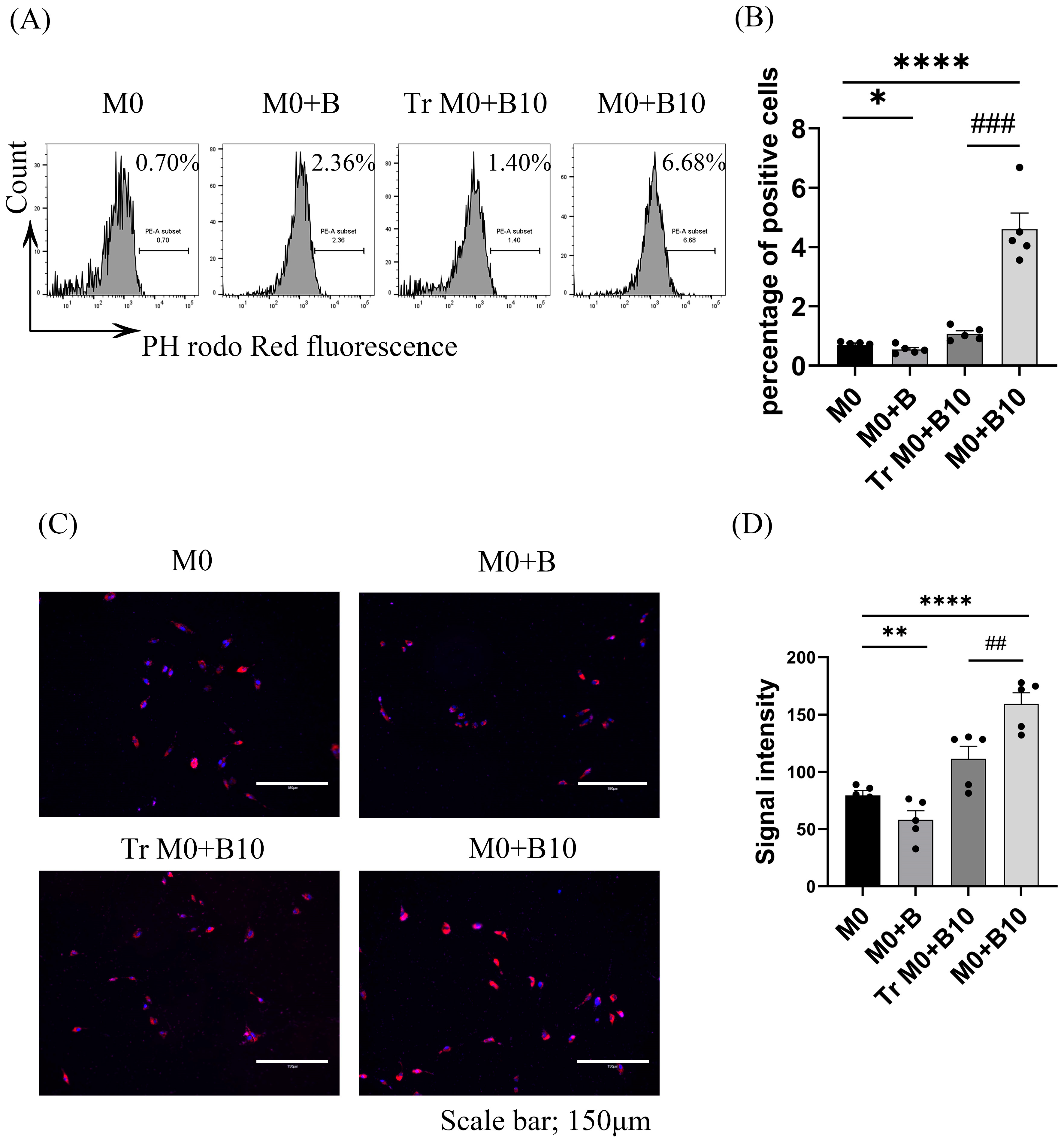
3.7. B-10 Cells Enhance Phagocytic Activity Through PD-L1/PD-1 Engagement
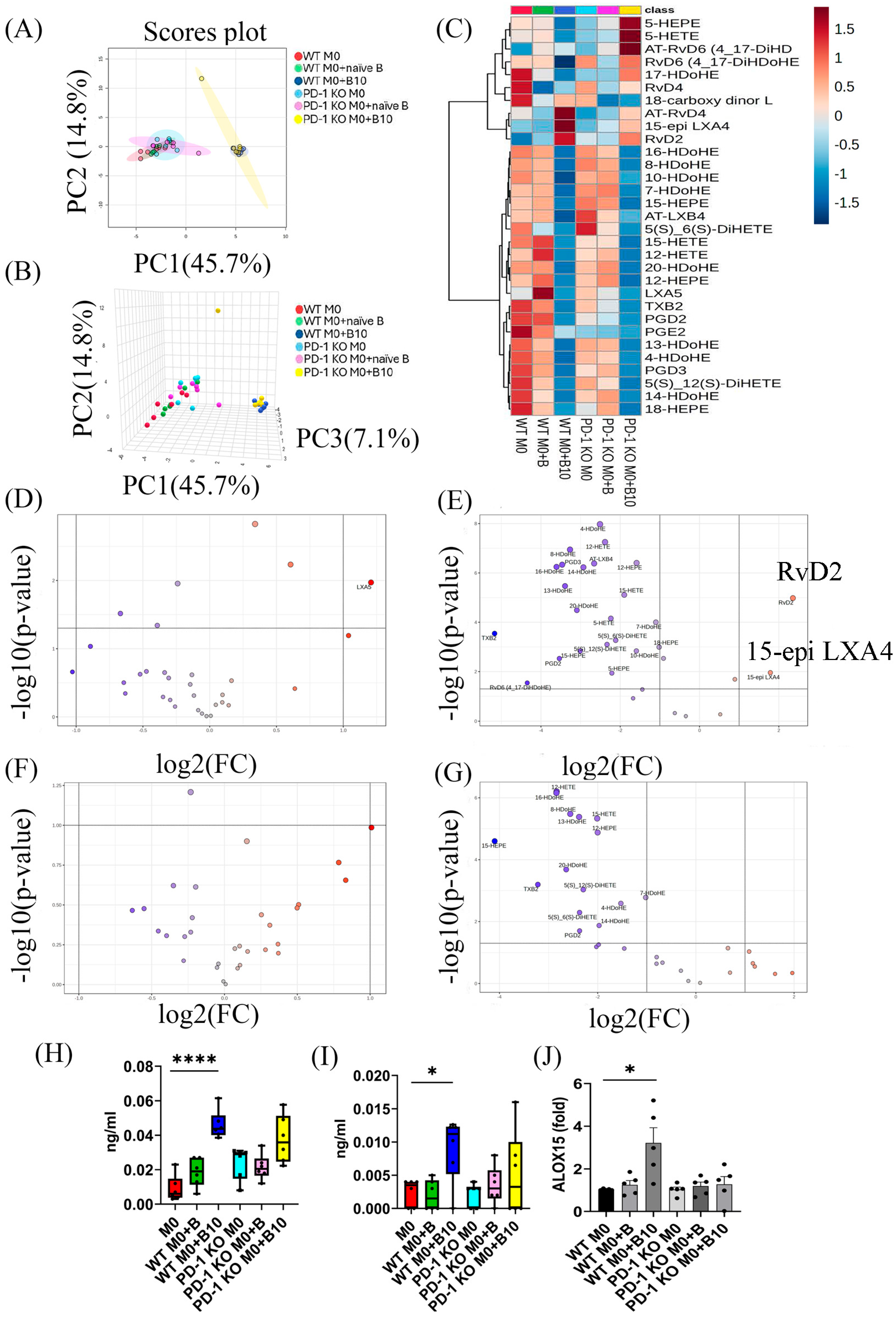
3.8. B-10 Cells Stimulate Specialized Pro-Resolving Mediator (SPM) Synthesis via PD-L1/PD-1 Interaction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mauri, C.; Bosma, A. Immune regulatory function of B cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, A.; Mizoguchi, E.; Takedatsu, H.; Blumberg, R.S.; Bhan, A.K. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 2002, 16, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Fillatreau, S.; Sweenie, C.H.; McGeachy, M.J.; Gray, D.; Anderton, S.M. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 2002, 3, 944–950. [Google Scholar] [CrossRef]
- Carter, N.A.; Vasconcellos, R.; Rosser, E.C.; Tulone, C.; Muñoz-Suano, A.; Kamanaka, M.; Ehrenstein, M.R.; Flavell, R.A.; Mauri, C. Mice lacking endogenous IL-10–producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J. Immunol. 2011, 186, 5569–5579. [Google Scholar] [CrossRef]
- Yoshizaki, A.; Miyagaki, T.; DiLillo, D.J.; Matsushita, T.; Horikawa, M.; Kountikov, E.I.; Spolski, R.; Poe, J.C.; Leonard, W.J.; Tedder, T.F. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature 2012, 491, 264–268. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Zou, X.-B.; Chai, Y.-F.; Yao, Y.-M. Macrophage polarization in inflammatory diseases. Int. J. Biol. Sci. 2014, 10, 520. [Google Scholar] [CrossRef]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of human macrophage polarization in inflammation during infectious diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [PubMed]
- Jordan, P.M.; Werz, O. Specialized pro-resolving mediators: Biosynthesis and biological role in bacterial infections. FEBS J. 2022, 289, 4212–4227. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Harwood, J.L. Polyunsaturated fatty acids: Conversion to lipid mediators, roles in inflammatory diseases and dietary sources. Int. J. Mol. Sci. 2023, 24, 8838. [Google Scholar] [CrossRef] [PubMed]
- Prescott, D.; McKay, D.M. Aspirin-triggered lipoxin enhances macrophage phagocytosis of bacteria while inhibiting inflammatory cytokine production. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 301, G487–G497. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.-H.; Yang, R.; Petasis, N.A.; Serhan, C.N. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; de la Rosa, X.; Libreros, S.; Serhan, C.N. Novel resolvin D2 receptor axis in infectious inflammation. J. Immunol. 2017, 198, 842–851. [Google Scholar] [CrossRef]
- Haas-Stapleton, E.J.; Lu, Y.; Hong, S.; Arita, M.; Favoreto, S.; Nigam, S.; Serhan, C.N.; Agabian, N. Candida albicans modulates host defense by biosynthesizing the pro-resolving mediator resolvin E1. PLoS ONE 2007, 2, e1316. [Google Scholar] [CrossRef]
- Decker, C.; Sadhu, S.; Fredman, G. Pro-resolving ligands orchestrate phagocytosis. Front. Immunol. 2021, 12, 660865. [Google Scholar] [CrossRef]
- Vannella, K.M.; Wynn, T.A. Mechanisms of organ injury and repair by macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, X.; Lin, J.; Hu, Y.; Zhao, Q.; Kawai, T.; Taubman, M.A.; Han, X. B10 cells alleviate periodontal bone loss in experimental periodontitis. Infect. Immun. 2017, 85, e00335-17. [Google Scholar] [CrossRef]
- Yu, P.; Hu, Y.; Liu, Z.; Kawai, T.; Taubman, M.A.; Li, W.; Han, X. Local induction of B cell interleukin-10 competency alleviates inflammation and bone loss in ligature-induced experimental periodontitis in mice. Infect. Immun. 2017, 85, e00645-16. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, Y.; Pan, K.; Li, H.; Shang, S.; Wang, Y.; Tang, G.; Han, X. In-vivo imaging revealed antigen-directed gingival B10 infiltration in experimental periodontitis. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2021, 1867, 165991. [Google Scholar] [CrossRef]
- Hasan, M.M.; Thompson-Snipes, L.; Klintmalm, G.; Demetris, A.J.; O’Leary, J.; Oh, S.; Joo, H. CD24hiCD38hi and CD24hiCD27+ human regulatory B cells display common and distinct functional characteristics. J. Immunol. 2019, 203, 2110–2120. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Latchman, Y.E.; Buhlmann, J.E.; Tomczak, M.F.; Horwitz, B.H.; Freeman, G.J.; Sharpe, A.H. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur. J. Immunol. 2003, 33, 2706–2716. [Google Scholar] [CrossRef] [PubMed]
- Azuma, T.; Yao, S.; Zhu, G.; Flies, A.S.; Flies, S.J.; Chen, L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood J. Am. Soc. Hematol. 2008, 111, 3635–3643. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef]
- Cao, G.; Xu, Q.; Huang, S.; Dai, D.; Wang, J.; Li, W.; Zhao, Y.; Lin, J.; Han, X. B10 cells regulate macrophage polarization to alleviate inflammation and bone loss in periodontitis. J. Periodontol. 2024, 96, 355–368. [Google Scholar] [CrossRef]
- Maddipati, K.R.; Zhou, S.-L. Stability and analysis of eicosanoids and docosanoids in tissue culture media. Prostaglandins Other Lipid Mediat. 2011, 94, 59–72. [Google Scholar] [CrossRef]
- Maddipati, K.R.; Romero, R.; Chaiworapongsa, T.; Zhou, S.-L.; Xu, Z.; Tarca, A.L.; Kusanovic, J.P.; Munoz, H.; Honn, K.V. Eicosanomic profiling reveals dominance of the epoxygenase pathway in human amniotic fluid at term in spontaneous labor. FASEB J. 2014, 28, 4835. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Wang, L.-X.; Zhang, S.-X.; Wu, H.-J.; Rong, X.-L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef]
- Iwata, Y.; Matsushita, T.; Horikawa, M.; DiLillo, D.J.; Yanaba, K.; Venturi, G.M.; Szabolcs, P.M.; Bernstein, S.H.; Magro, C.M.; Williams, A.D. Characterization of a rare IL-10–competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood J. Am. Soc. Hematol. 2011, 117, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Liang, M.; Xiong, L.; Su, N.; Gao, X.; Jiang, Z. PD-L1 induces macrophage polarization toward the M2 phenotype via Erk/Akt/mTOR. Exp. Cell Res. 2021, 402, 112575. [Google Scholar] [CrossRef]
- Ito, I.; Asai, A.; Suzuki, S.; Kobayashi, M.; Suzuki, F. M2b macrophage polarization accompanied with reduction of long noncoding RNA GAS5. Biochem. Biophys. Res. Commun. 2017, 493, 170–175. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, L.; Hu, X.; Ji, J.; Mor, G.; Liao, A. The role of the PD-1/PD-L1 axis in macrophage differentiation and function during pregnancy. Hum. Reprod. 2019, 34, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-H.; Aldo, P.; You, Y.; Ding, J.; Kaislasuo, J.; Petersen, J.F.; Lokkegaard, E.; Peng, G.; Paidas, M.J.; Simpson, S. Trophoblast-secreted soluble-PD-L1 modulates macrophage polarization and function. J. Leucoc. Biol. 2020, 108, 983–998. [Google Scholar] [CrossRef]
- Greenlee-Wacker, M.C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 2016, 273, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Khan, S. Specialized pro-resolving mediators enhance the clearance of dead cells. Immunol. Rev. 2023, 319, 151–157. [Google Scholar] [CrossRef]
- Spite, M.; Norling, L.V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N.A.; Flower, R.J.; Perretti, M.; Serhan, C.N. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 2009, 461, 1287–1291. [Google Scholar] [CrossRef]
- Mitchell, S.; Thomas, G.; Harvey, K.; Cottell, D.; Reville, K.; Berlasconi, G.; Petasis, N.A.; Erwig, L.; Rees, A.J.; Savill, J. Lipoxins, Aspirin-Triggered Epi-Lipoxins, Lipoxin Stable Analogues, and the Resolution of Inflammation: Stimulation of Macrophage Phagocytosis of Apoptotic Neutrophils: In Vivo. J. Am. Soc. Nephrol. 2002, 13, 2497–2507. [Google Scholar] [CrossRef]
- Kneidl, J.; Löffler, B.; Erat, M.C.; Kalinka, J.; Peters, G.; Roth, J.; Barczyk, K. Soluble CD163 promotes recognition, phagocytosis and killing of Staphylococcus aureus via binding of specific fibronectin peptides. Cell. Microbiol. 2012, 14, 914–936. [Google Scholar] [CrossRef] [PubMed]
- Jiao, K.; Zhang, J.; Zhang, M.; Wei, Y.; Wu, Y.; Qiu, Z.Y.; He, J.; Cao, Y.; Hu, J.; Zhu, H. The identification of CD163 expressing phagocytic chondrocytes in joint cartilage and its novel scavenger role in cartilage degradation. PLoS ONE 2013, 8, e53312. [Google Scholar] [CrossRef]
- Schulz, D.; Severin, Y.; Zanotelli, V.R.T.; Bodenmiller, B. In-depth characterization of monocyte-derived macrophages using a mass cytometry-based phagocytosis assay. Sci. Rep. 2019, 9, 1925. [Google Scholar] [CrossRef]
- Sezin, T.; Ferreirós, N.; Jennrich, M.; Ochirbold, K.; Seutter, M.; Attah, C.; Mousavi, S.; Zillikens, D.; Geisslinger, G.; Sadik, C.D. 12/15-Lipoxygenase choreographs the resolution of IgG-mediated skin inflammation. J. Autoimmun. 2020, 115, 102528. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Kuhn, H.; Heydeck, D. Structural and functional biology of arachidonic acid 15-lipoxygenase-1 (ALOX15). Gene 2015, 573, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Radmark, O. Formation of eicosanoids and other oxylipins in human macrophages. Biochem. Pharmacol. 2022, 204, 115210. [Google Scholar] [CrossRef]
- Li, W.; Wu, F.; Zhao, S.; Shi, P.; Wang, S.; Cui, D. Correlation between PD-1/PD-L1 expression and polarization in tumor-associated macrophages: A key player in tumor immunotherapy. Cytokine Growth Factor Rev. 2022, 67, 49–57. [Google Scholar] [CrossRef]
- van Roon, J.; Wijngaarden, S.; Lafeber, F.P.; Damen, C.; van de Winkel, J.; Bijlsma, J.W. Interleukin 10 treatment of patients with rheumatoid arthritis enhances Fc gamma receptor expression on monocytes and responsiveness to immune complex stimulation. J. Rheumatol. 2003, 30, 648–651. [Google Scholar]
- Blair, P.A.; Noreña, L.Y.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. CD19+ CD24hiCD38hi B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. immunity 2010, 32, 129–140. [Google Scholar] [CrossRef]
- Bystrom, J.; Evans, I.; Newson, J.; Stables, M.; Toor, I.; Van Rooijen, N.; Crawford, M.; Colville-Nash, P.; Farrow, S.; Gilroy, D.W. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood J. Am. Soc. Hematol. 2008, 112, 4117–4127. [Google Scholar] [CrossRef]
- Stables, M.J.; Shah, S.; Camon, E.B.; Lovering, R.C.; Newson, J.; Bystrom, J.; Farrow, S.; Gilroy, D.W. Transcriptomic analyses of murine resolution-phase macrophages. Blood J. Am. Soc. Hematol. 2011, 118, e192–e208. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.; Bradley, L.; Smith, A.; Foxwell, B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J. Immunol. 2004, 172, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Sarma, U.; Maiti, M.; Nair, A.; Bhadange, S.; Bansode, Y.; Srivastava, A.; Saha, B.; Mukherjee, D. Regulation of STAT3 signaling in IFNγ and IL10 pathways and in their cross-talk. Cytokine 2021, 148, 155665. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Wu, X.; Qin, H.; Yuan, Y.-C.; Schmolze, D.; Su, C.; Zain, J.; Moyal, L.; Hodak, E.; Sanchez, J.F. Reprogramming of PD-1+ M2-like tumor-associated macrophages with anti–PD-L1 and lenalidomide in cutaneous T cell lymphoma. JCI Insight 2023, 8, e163518. [Google Scholar] [CrossRef]
- He, X.; Xiao, J.; Li, Z.; Ye, M.; Lin, J.; Liu, Z.; Liang, Y.; Dai, H.; Jing, R.; Lin, F. Inhibition of PD-1 alters the SHP1/2-PI3K/Akt axis to decrease M1 polarization of alveolar macrophages in lung ischemia–reperfusion injury. Inflammation 2023, 46, 639–654. [Google Scholar] [CrossRef]
- Memida, T.; Abdolahinia, E.D.; Cao, G.; Ruiz, S.; Huang, S.; Shindo, S.; Nakamura, S.; Lin, J.; Kawai, T.; Han, X. B10 cells promote pro-resolving macrophage function through direct cell-cell contact and IL-10 secretion in Raw 264.7 cells. Int. Immunol. 2025. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Memida, T.; Cao, G.; Dalir Abdolahinia, E.; Ruiz, S.; Huang, S.; Hassantash, S.; Shindo, S.; Okamoto, M.; Yamashita, S.; Nakamura, S.; et al. B10 Promotes Polarization and Pro-Resolving Functions of Bone Marrow Derived Macrophages (BMDM) Through PD-1 Activation. Cells 2025, 14, 860. https://doi.org/10.3390/cells14120860
Memida T, Cao G, Dalir Abdolahinia E, Ruiz S, Huang S, Hassantash S, Shindo S, Okamoto M, Yamashita S, Nakamura S, et al. B10 Promotes Polarization and Pro-Resolving Functions of Bone Marrow Derived Macrophages (BMDM) Through PD-1 Activation. Cells. 2025; 14(12):860. https://doi.org/10.3390/cells14120860
Chicago/Turabian StyleMemida, Takumi, Guoqin Cao, Elaheh Dalir Abdolahinia, Sunniva Ruiz, Shengyuan Huang, Sahar Hassantash, Satoru Shindo, Motoki Okamoto, Shohei Yamashita, Shin Nakamura, and et al. 2025. "B10 Promotes Polarization and Pro-Resolving Functions of Bone Marrow Derived Macrophages (BMDM) Through PD-1 Activation" Cells 14, no. 12: 860. https://doi.org/10.3390/cells14120860
APA StyleMemida, T., Cao, G., Dalir Abdolahinia, E., Ruiz, S., Huang, S., Hassantash, S., Shindo, S., Okamoto, M., Yamashita, S., Nakamura, S., Suzuki, M., Kawai, T., & Han, X. (2025). B10 Promotes Polarization and Pro-Resolving Functions of Bone Marrow Derived Macrophages (BMDM) Through PD-1 Activation. Cells, 14(12), 860. https://doi.org/10.3390/cells14120860