1. Introduction
Currently, reprogramming technology for generating human-induced pluripotent stem cells (hiPSCs) has become widespread. These cells possess very similar characteristics to human embryonic stem cells (hESCs) and are widely used in research modeling of various human diseases, drug testing, and as a source of cells for regenerative medicine, for example, for autologous cell therapies. At the same time, our knowledge of the factors and signaling cascades that play an important role in the induction and maintenance of pluripotency is still very limited.
Previously, in searching for the new signaling molecules, which are important for hiPSCs generation, we used a high-throughput small interfering RNA (RNAi) screening assay during the initiation phase of reprogramming. We performed specific knockdown of 784 members of the different kinases and phosphatases from the Dharmacon library and revealed 6 members of the G-protein-coupled receptors (GPCRs) expected to be important for reprogramming [
1]. Adhesion GPR123 was identified by this screen as a potential effector for the reprogramming process and this was the rationale for the present study. Importantly, this is the first study which addresses in detail the role of GPR123 for hPSCs and in the reprogramming process during hiPSCs generation.
Adhesion GPCRs is a class of 33 human protein receptors, but despite their broad distribution and modern screening techniques, 17 of them are still without known ligands and most of these proteins are orphans [
2,
3,
4]. Adhesion GPCRs are known to be expressed in white blood cells, neurons, embryonic cells, reproductive tract cells, and various tumors. They are important in homeostasis, induction of PKA activity, c-AMP binding, activation of insulin signaling, and signaling of NOD-like receptors [
4].
Regrettably, the role of adhesion GPCRs is practically unknown or poorly studied for stem cells. At the same time, according to Nakamura and colleagues [
5], the
EDG5,
GPR20, and
GPR123 genes have a significantly higher expression level in hESCs compared with 100 types of somatic cells studied. Moreover, GPR125, which is a group III adhesion receptor, like GPR123, has been shown to be involved in maintaining pluripotency of stem cells and is a known germ line precursor marker [
6]. GPR123 together with GPR124 and GPR125 form a separate phylogenetic group among the adhesion GPCRs [
7]. It is important to note that, in its primary structure, GPR123 differs from the other members of the adhesion GPCRs. The functional specificity of these receptors is largely determined by the presence of a long extracellular N-terminal fragment (NTF) containing conserved protein domains, but such a domain has not been identified for GPR123 [
7]. The second important difference between GPR123 and the other adhesion family members is the absence of a GPCR proteolytic domain (GPS) that functions as an intracellular autocatalytic site [
8].
While the precise mechanism for signaling cascades via GPR123 in stem cells is not known, it is generally accepted that adhesion GPCRs use basic alpha subunits of G-proteins, such as Gαs, Gαi, Gαq, and Gα12/13 [
4,
6].
In the present study, to unravel the role of GPR123 for hPSCs and for hiPSCs generation we suppressed its expression by using RNAi. We demonstrated that the suppression of GPR123 in hPSCs leads to a loss of pluripotency, alteration in the colony morphology, an accumulation of cells at the G2 phase of the cell cycle, and an absence of the scratch closure in the wound assay, which is associated with decreased cell motility. Downregulation of GPR123 during the initiation stage of the reprogramming process leads to a decrease in the percentage of the “true” hiPSC colonies, a drop in E-cadherin expression, a decrease in the percentage of NANOG+ nuclei, a loss of GPR123-Gαi co-localization, and the absence of actin cytoskeleton remodeling. This leads to an absence of the alkaline-phosphatase-positive hiPSCs colonies on the 18th day of the reprogramming process. Together, these data indicate for the first time the essential role of GPR123 in pluripotency maintenance as well as pluripotency induction during the reprogramming process.
2. Materials and Methods
Cell culture and hiPSCs generation. H9 hESCs (WiCell Research Institute, Madison, MI, USA) and hiPSCs (line SB-NEO1) were cultured in a feeder-free condition on Matrigel-coated plates (Corning Matrigel Matrix, Life Sciences, hESC-qualified, High Wycombe, UK) with mTESR1 media (STEMCELL Technologies, Cambridge, UK). Human iPSC line (SB-NEO1) was generated from the reprogramming of neonatal fibroblasts using the Sendai-based CytoTune-iPS 2.0 Sendai reprogramming kit provided by Life Technologies (A16517, Invitrogen, Fisher Scientific U.K. Ltd.; Loughborough, UK) and described previously [
9,
10]. Human iPSCs were cultured in the same way as hESCs. The cells were analyzed 24 h, 48 h, 72 h, and 96 h after transfection.
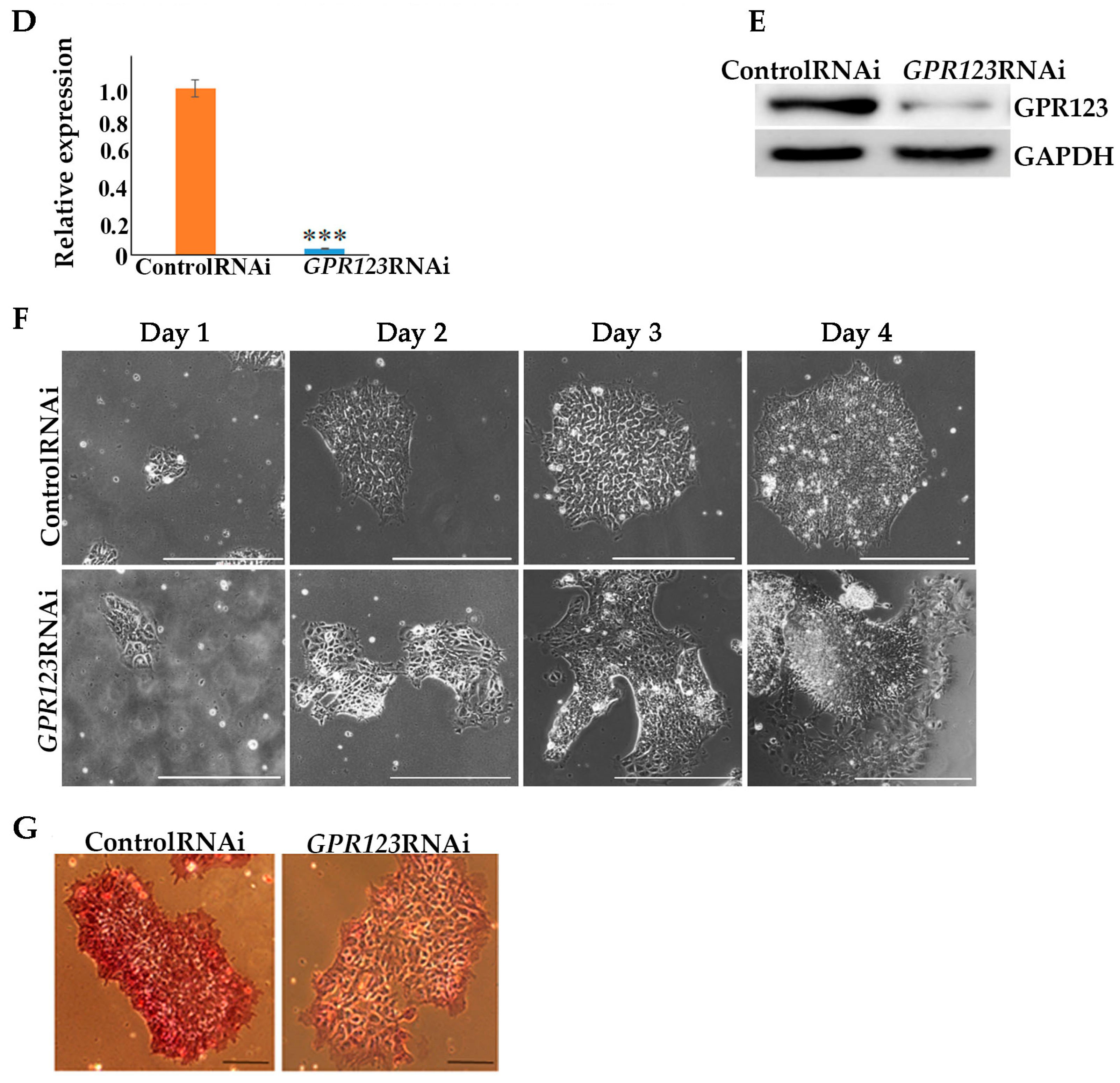
RNA Interference. SMARTpool: siGENOME small interfering RNA (siRNA) for GPR123 (ADGRA1) was purchased from Dharmacon (L-005539-02-0005), siRNA sequences are listed in parentheses (5′-CCACGAACAUCAGGAAUUA-3′, 5′-GGCACACGCUCCUGAAUUU-3′, 5′-GCAGAACGAGCACUCAUUC-3′, 5′-GCACACGGUCAUCCGGUUU-3′). The siRNA mixture at a final concentration of 10 nM was used for transfection with DharmaFECT1 Transfection reagent (Dharmacon, Cambridge, U.K., T-2001-01) according to manufacturer’s instructions with OPTI-MEM reduced serum Media (31985-062; Gibco, Dublin, Ireland) for the first 45 min of transfection. Then, an equal volume of the mTeSR1Medium was added to cells. Media was changed for mTeSR1 every day. As a control, ON-TARGETplus nontargeting control pool from Dharmacon (D-001810-10) was used.
Cell cycle analysis. hESCs and hiPSCs were collected using Accutase (Chemicon, Temecula, CA, USA). Cell cycle analysis was performed using the CycleTest Plus DNA reagent kit (BD Biosciences, Oxford, UK) using a FACS Canto (BD Biosciences) measuring FL2 area versus total counts. The data were analyzed using ModFit LT 4.1. (Verity Software House, Topsham, ME, USA) to generate percentages of cells in G1, S, and G2/M phases. At least 10,000 cells were analyzed in each experiment.
Immunocytochemistry and confocal microscopy. Briefly, hESCs and hiPSCs were cultured on Matrigel-covered glass slide flasks (SlideFlask, NUNC, Roskilde, Denmark) in the mTESR1 media. Cells were quickly washed with phosphate-buffered saline (PBS) prior to being fixed with 2% formaldehyde for 10 min and permeabilized with 0.1% Triton X-100 in PBS for 15 min at room temperature. Unspecific binding was blocked by the incubation of samples in PBS containing 5% normal goat serum for 40 min. Cells were incubated with primary antibodies overnight at 4 °C and with secondary antibodies: goat anti-rabbit-Alexa488 (A-11008, ThermoFisher Scientific, Eugene, OR, USA); goat anti-rabbit-Alexa594 (A-11012, ThermoFisher Scientific, Eugene, OR, USA); goat anti-mouse-Alexa488 (A28175, ThermoFisher Scientific, Bleiswijk, The Netherlands); and goat anti-mouse-Alexa594 (A-11032, ThermoFisher Scientific, Eugene, OR, USA) for 2 h in a dark at room temperature. Primary antibodies used in this study were: anti-GPR123 (PA5-39620, ThermoFisher Scientific, Waltham, MA, USA); anti-GNAI1 (MA5-12800, ThermoFisher Scientific, Bleiswijk, The Netherlands); Rhodamine phalloidin (R415, ThermoFisher Scientific, Eugene, OR, USA); and anti-Nanog (4893 s, Cell Signaling Technology, Danvers, MA, USA). The nuclei were counterstained with DAPI (4′,6-Diamidino-2-Phenylindole, Dihydrochloride; D1306, ThermoFisher Scientific, Eugene, OR, USA). Samples were covered with Vectashield Mounting Medium (Vector Laboratories Ltd., Peterborough, UK). The images were obtained using the Olympus FV3000 (Olympus, Nagano, Japan) microscope and the FluoView FV3000 software (Olympus, Nagano, Japan). At least 100 cells were analyzed for each technical replicate.
For an accurate comparison among immunofluorescence signals of GPR123 (FITC) between emerging hiPSC cells and fibroblasts during the 14th day of the reprogramming process, confocal microscopy images were captured using the same laser excitation and sample emission settings in all immunofluorescence preparations of each slide. In total, 25 fibroblast and 25 hiPSCs from three experimental repeats were analyzed according to Shihan et al. [
11]. Negative controls were performed avoiding the primary antibodies. Quantification of the GPR123 immunofluorescence intensity in both cell types was performed with ImageJ software (version 1.53c) [
12] and fluorescence values of CTCF (corrected total cell fluorescence) were expressed as arbitrary units/cells according to formula: CTCF = Integrated Density − (Area of selected cell × Mean fluorescence of background readings). The results are represented as the average ± the standard deviations of three independent experiments. We considered
p values < 0.05 to be statistically significant.
n = 25 cells for each type.
Transmission electron microscopy. For ultrastructural immunocytochemistry, the cells were fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4, for 1 h at 4 °C, and postfixed in 1% OsO4 for 1 h. Then, the cells were mechanically detached from the coverslips and centrifuged. The obtained pellets were dehydrated in graded alcohol solutions and embedded in Epon and Araldit. Prepared with a diamond knife on an LKB-ultratome (Stockholm, Sweden), ultrathin sections were placed on nickel grids, treated with 3% hydrogen peroxide for 20 min to loosen resin, and incubated in the first antibody solution–polyclonal anti-GPR123 (PA5-39620, ThermoFisher Scientific, Waltham, MA, USA) and monoclonal anti- anti-GNAI1 (MA5-12800, ThermoFisher Scientific, MA5-12800 Bleiswijk, The Netherlands) overnight in a moist chamber at 4 °C. After rinsing in PBS containing 0.1% fish gelatin and 0.05% Tween-20, the sections were incubated with secondary anti-mouse and anti-rabbit antibodies conjugated to 10 nm and 15 nm colloidal gold particles, respectively (Sigma, Burlington, ON, Canada). The sections were contrasted with uranyl acetate and lead citrate and examined with a Zeiss Libra 120 electron microscope (Carl Zeiss, Jena, Germany) at an accelerating voltage of 80 kV.
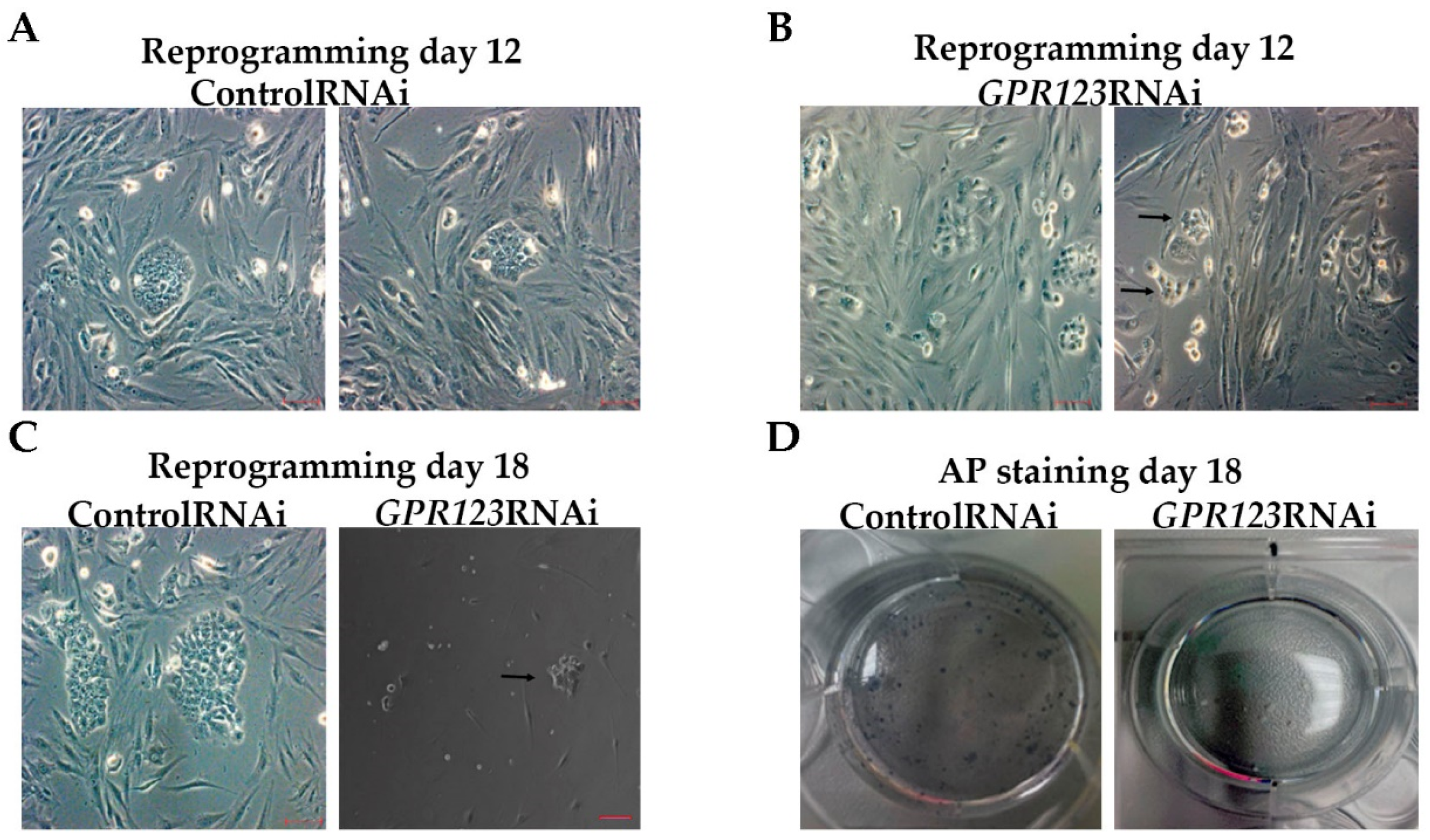
Alkaline-phosphatase staining. AP staining was carried out using the AP Detection kit according to manufacturer’s instructions (Chemicon, Temecula, CA, USA). The images were obtained using the imaging system EVOS FL Auto (ThermoFisher Scientific, Carlsbad, CA, USA) and the EVOS FL Auto 2 Software (ThermoFisher Scientific, Carlsbad, CA, USA).
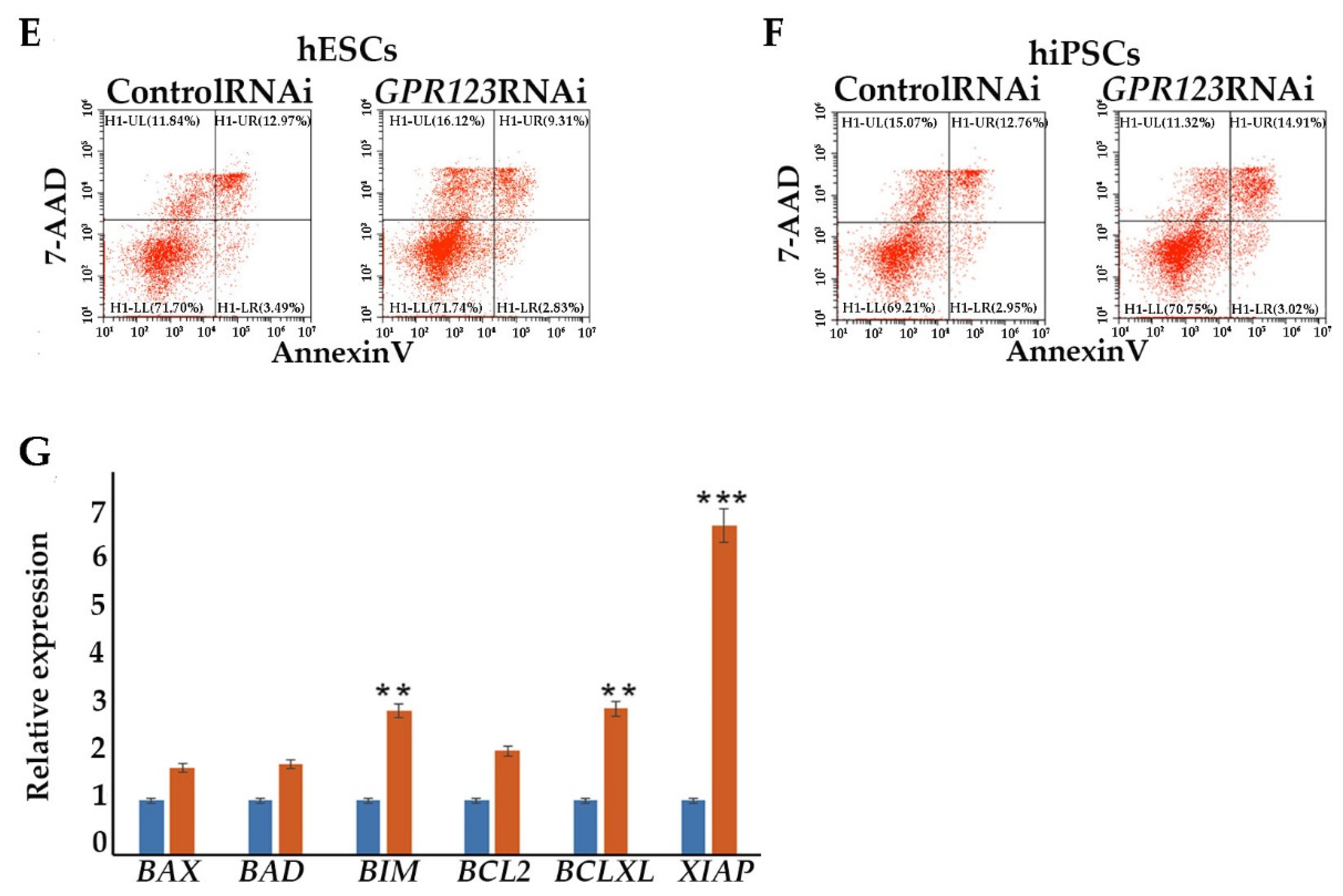
Flow cytometric analysis for assessing apoptosis. Apoptosis was addressed with the Annexin-V-PE apoptosis detection kit (#556547, BD Bioscience, Oxford, UK) as described previously in [
13]. At least 10,000 events were recorded for each sample.
Isolation of RNA and quantitative RT–PCR analysis. To analyze gene expression, total RNA was isolated with Aurum™ Total RNA Mini Kit (BioRad, Hercules, CA, USA) according to the manufacturer’s instructions. RNA was quantified in the NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE, USA). cDNA was obtained by reverse transcription of RNA using the RevertAid H Minus First Strand cDNA Synthesis Kit (ThermoFisher Scientific, Vilnius, Lithuania) according to the manufacturer’s instructions. For qRT-PCR, cDNA was amplified with specific primers and the sequences of the oligonucleotides used for the quantitative RT–PCR are shown in
Table S1. Regression curves were drawn for each sample and the relative amount was calculated from the threshold cycles with the CFX Manager software for the BioRad CFX-96 real-time system (Bio-Rad, Hercules, CA, USA) based on the manufacturer’s instructions. Relative expression levels of the target genes were normalized with the control gene
GAPDH or
RPL13A.Western immunoblotting. Protein extraction, Western blotting, and antibody/antigen complex detection were performed as published previously [
10,
14]. Densitometry analysis was performed using ImageLab software Version 6.0.0 (BioRad, Hercules, CA, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to normalize band intensities of proteins of interest. The details of the antibodies used in this work can be found in the
Table S2. The antibody to GAPDH was used after membrane stripping to confirm uniform protein loading.
In brief, cells in 6-well plates were washed with cold-phosphate-buffered saline and lysed in RIPA buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1% IGEPAL CA 630, 0.5% Na-DOC, and 0.1% SDS). Before the treatment of cells, 1mM PMSF and Roche protease inhibitors (1 tablet per 10 mL) were added to RIPA buffer. After 30 min on ice, the lysates were homogenized and centrifuged at 13,000 r.p.m. for 15 min. The total protein concentration was determined using Pierce™ BCA Protein Assay Kit (#23225, ThermoFisher Scientific, Rockford, IL, USA). Absorption at 595 nm was detected using a Thermo Labsystems Multiskan Ascent. Lysates were electrophoresed on a 8–12% SDS–polyacrylamide gel electrophoresis and electrophoretically transferred to a nitrocellulose membrane (Bio-Rad). Membranes were blocked in Tris-buffered saline with 5% milk and 0.1% Tween. The blots were probed overnight at 4C and the primary antibodies are listed in
Table S2. The next morning, the blots were washed and incubated for 2 h with horseradish-peroxidase-conjugated secondary antibodies: Goat Anti-Rabbit HRP (ab205718, Abcam, Cambridge, UK) and Goat Anti-Mouse HRP (ab205719, Abcam, Cambridge, UK). Antibody/antigen complexes were detected using ECL (Amersham Biosciences, Buckinghamshire, UK) and images were acquired using the ChemiDoc MP Imaging System (BioRad, Hercules, CA, USA) and ImageLab 6.0 (BioRad, Hercules, CA, USA) software.
Statistical analysis. All data are demonstrated as mean ± standard deviation (SD) of at least three biological replicates. The statistical tests were performed using GraphPad Prism (Version 7.0) software (GraphPad Software, Boston, MA, USA). Statistical significance between two groups (control RNAi and GPR123 RNAi) was analyzed using Student’s t-test. p < 0.05 was considered significant and is denoted as *, p < 0.01 as **, and p < 0.001 as ***.
4. Discussion
Stem cell technology is mainly dedicated to practical applications for regenerative medicine, disease modeling, drug screening, and understanding of human developmental biology. Currently, successful clinical trials with the use of stem cells are being carried out, as well as studies using stem cells in disease modeling related to the musculoskeletal system, heart, nervous system, immune system, etc. [
19,
20]. However, with such a widespread application of hiPSCs, our knowledge and understanding of the entire mechanism of the reprogramming process is still very limited. At the same time, successful clinical applications of hiPSCs will require overcoming serious downsides, one of which is incomplete reprogramming, which calls for a deeper understanding of the molecular machinery of the reprogramming process. Therefore, our new data highlighting the importance of the GPR123 and GPR123-Gαi signaling in hiPSCs generation could be the first step in this direction.
As mentioned here, GPR123 is an orphan receptor. Data on its function even in somatic cells are very limited [
4]. Further research is needed to discover specific agonists or antagonists for adhesion GPCRs, including GPR123. For example, recently, beclomethasone dipropionate was identified as a small molecular weight agonistic compound for GPR97 [
21,
22].
Many GPCRs have been shown to bind to more than one member of the G protein family. Adhesion GPCRs are no exception to this rule. For example, GPR126 and GPR133 interact with both Gαs and Gαi proteins [
23]. GPR64 interacts with Gαs and Gαq proteins [
24] and GPR56 binds to Gαq/11 and Gα12/13 [
25].
It is still largely a mystery how autoproteolysis and NTF removal occur. The signaling pathways associated with the above events may differ for different receptors and even for the same receptor depending on the cellular context. The structural features of the GPR123 molecule suggest that not all adhesion GPCRs rely on released NTF for their signaling [
21,
26]. This is supported by studies of lat-1 in C. elegans showing that separation of NTF and CTF is not necessary to achieve proper receptor function [
27]. For most adhesive GPCRs, their large size is determined by the NTF domain, comprising modular protein domains, such as cadherin, epidermal growth factor, immunoglobulin, and leucine-rich repeat domains. Some of these domains may mediate contacts with other cells or extracellular matrix-associated molecules. However, as noted earlier here, most of the adhesion GPCRs remain orphaned in terms of ligand binding [
4,
19]. In this context, more work is required to explain our findings on the co-localization of GPR123 and E-cadherin in hPSCs. Currently, there is no doubt that various GPCRs play an important role in the maintenance of stem cells and in reprogramming towards hiPSCs [
1,
5,
6,
18]. Moreover, serious changes in the gene expression of some GPCRs at different stages of stem cell differentiation once again emphasize their involvement in the maintenance of stemness [
28]. Our data showing that during the course of EBs differentiation the protein level of GPR123 demonstrated significant reduction are in good agreement with data about a significant drop at GPR123 during specification to ectoderm differentiation of the HUES64 hESC line. Throughout comprehensive transcriptional profiling of populations derived through directed differentiation of hESCs, Gifford and colleagues identified GPR123 among the most downregulated GPCRS, highlighting that a better understanding of the involvement of GPCRs in the specification events can lead to the development of the more effective differentiation strategies. Analysis of gene expression profiles performed by Choi and colleagues demonstrated that more than 106 GPCRs were over expressed in the PCSs or cancer stem cells, whereas the expression of the other 22 GPCRs was downregulated and 81 were differentially expressed during somatic reprogramming to iPSCs [
6]. However, since that time, very little has been done to shed light on the role and function of these molecules for pluripotency maintenance and other biological properties of hPSCs.
Here, we have shown that the adhesion family member GPCR, GPR123, is essential for pluripotency maintenance of hPSCs as well as for hiPSCs generation:
- (i).
By employing GPR123 RNAi from the 8th to the 10th day of the reprogramming process, we demonstrated significant downregulation in the number of hiPSCs colonies from the 12th to the 18th day of the reprogramming period, with complete absence of the AP+ colonies on the 18th day.
- (ii).
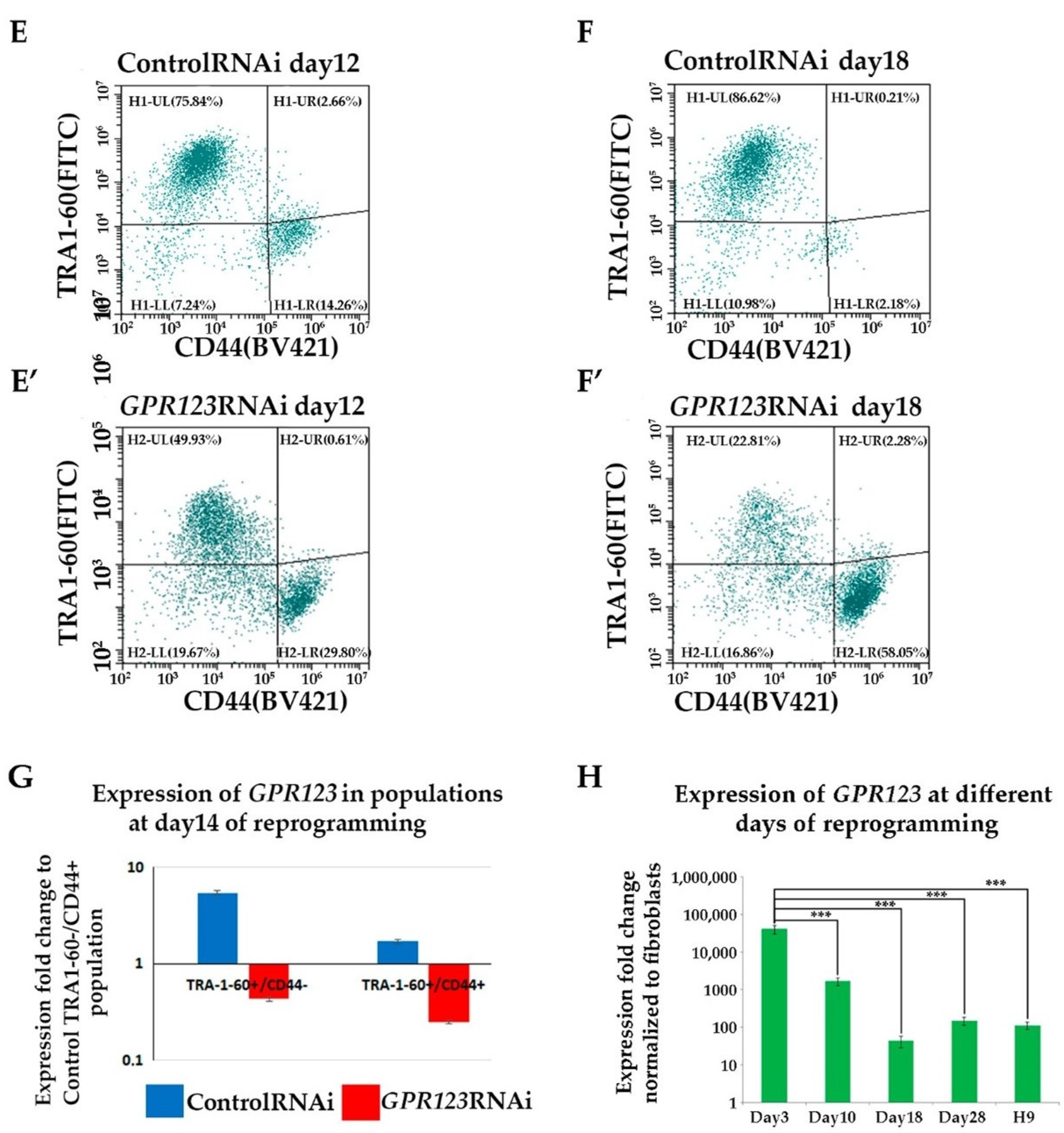
Flowcytometry cell populations analysis demonstrated significant decrease in the percentage of the “true” hiPSCs (TRA1-60+/CD44- population) by the 18th day of the reprogramming process under GPR123 RNAi.
- (iii).
We demonstrated significantly reduced expression of GPR123 in the emerging TRA1-60+/CD44- population and in the population of the partially reprogrammed cells (TRA+CD44+) from the GPR123 RNAi group on the 14th day of the reprogramming process.
Thus, our findings indicate that elevation of
GPR123 expression during the initial stages of the reprogramming process is an important and necessary requirement for induction and maintaining pluripotency during this process, corroborating previous data that the expression level of GPR123 in hESCs is much higher than in 100 somatic cell types tested by Nakamura and colleagues [
5].
We addressed the effect of GPR123 suppression on pluripotency maintenance as one of the most important characteristics of hPSCs and demonstrated an important role of GPR123 in pluripotency maintenance as:
- (iv).
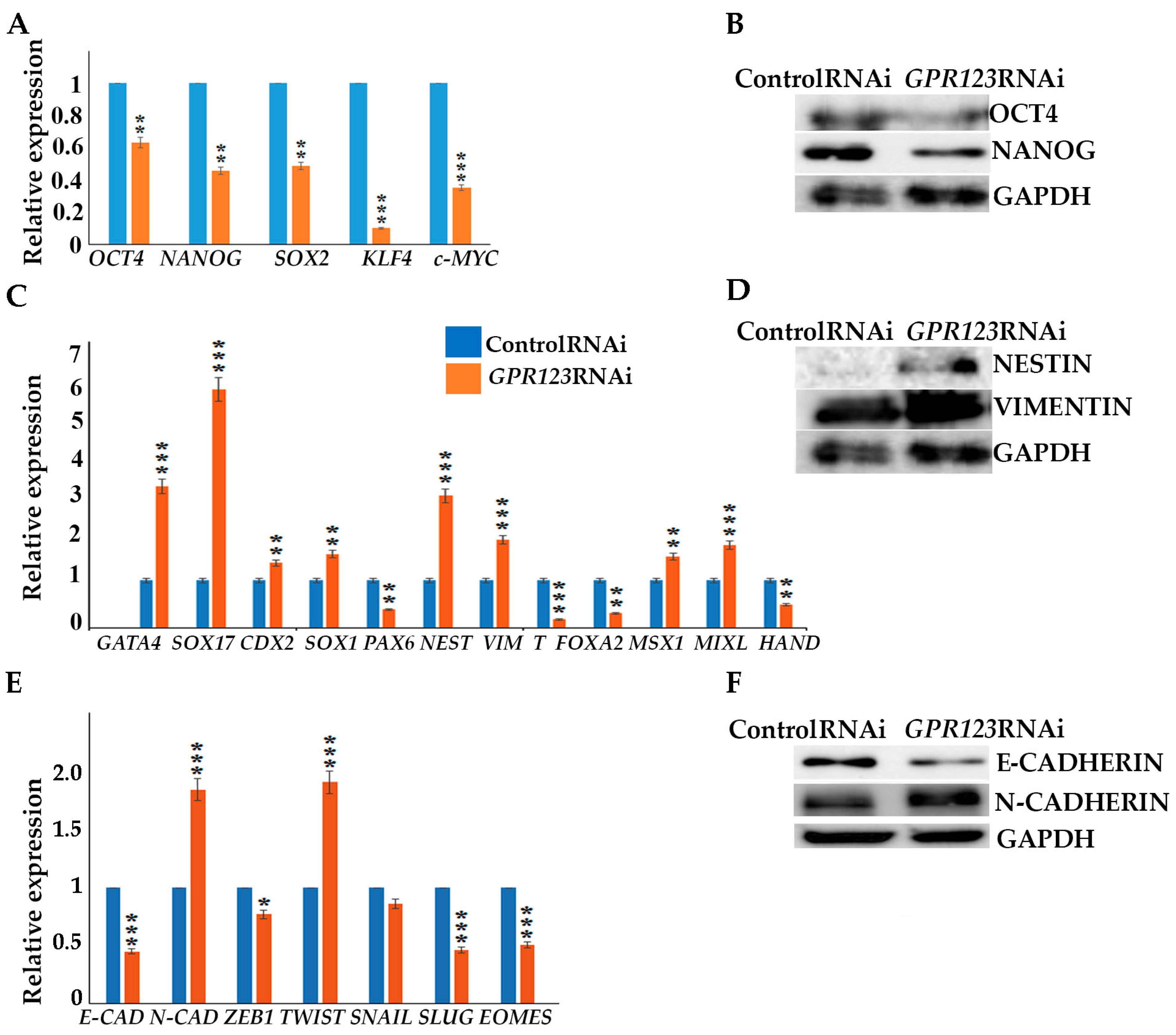
Suppression of GPR123 by RNAi leads to significant downregulation of OCT4, NANOG, SOX2, KLF4, and c-Myc pluripotency markers expression
- (v).
with simultaneous increase in expression of differentiation markers genes, namely GATA4, SOX17, NESTIN, Vimentin, MSX1 and MIXL.
- (vi).
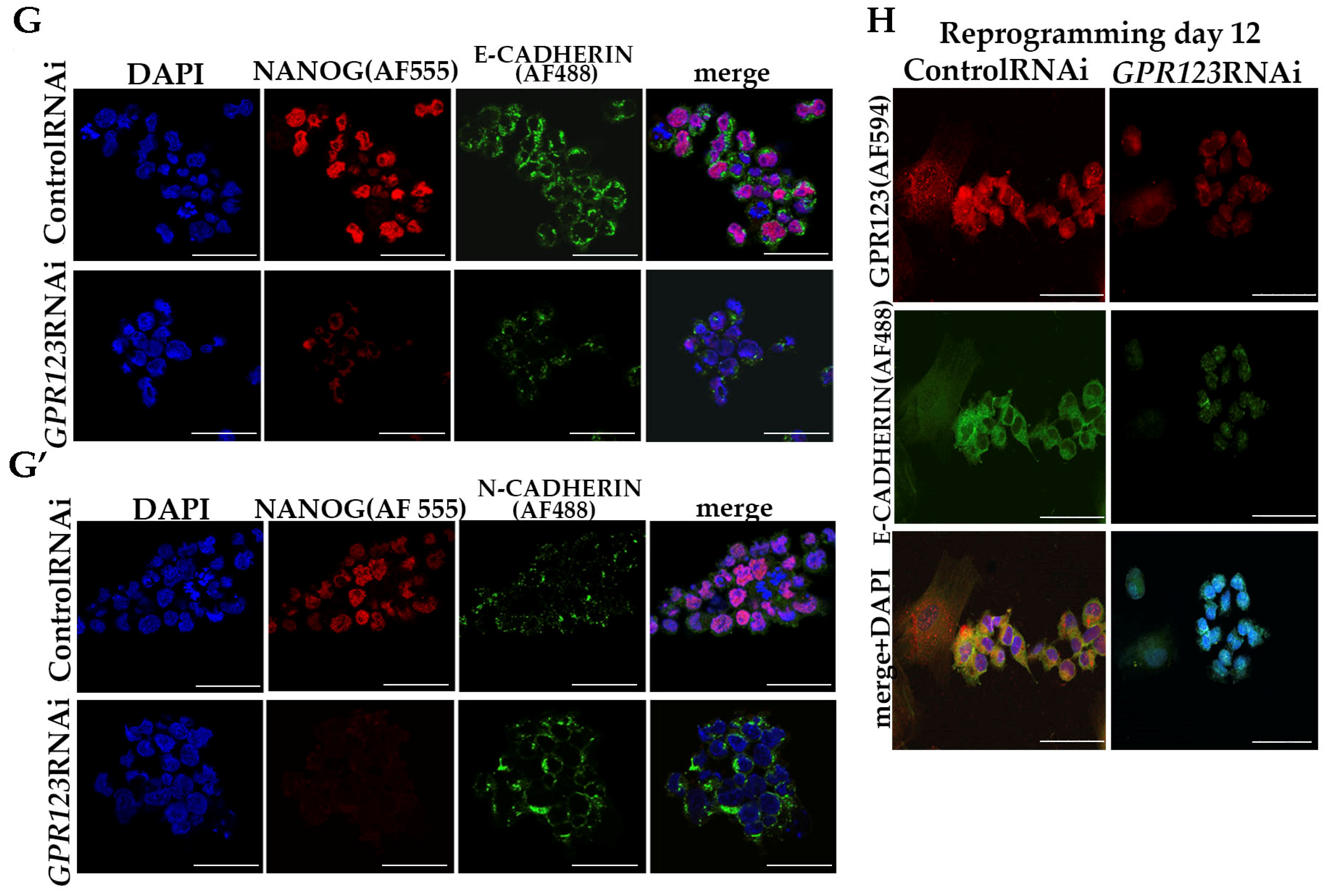
GPR123 RNAi causes a drop in E-cadherin expression with a significant rise in N-cadherin.
- (vii).
Confocal immunofluorescence observation revealed co-localization of E-cadherin and GPR123 at the surface of the control RNAi cells and the absence of such colocalization in the GPR123 RNAi population.
The close relationship between pluripotency and cell cycle regulation is well documented [
14,
16,
29,
30] and alterations in the expression of the cyclins and CDKs lead to abrogation of the hiPSCs [
1,
9,
10]. For this reason, we examine the cell cycle profile of the control and
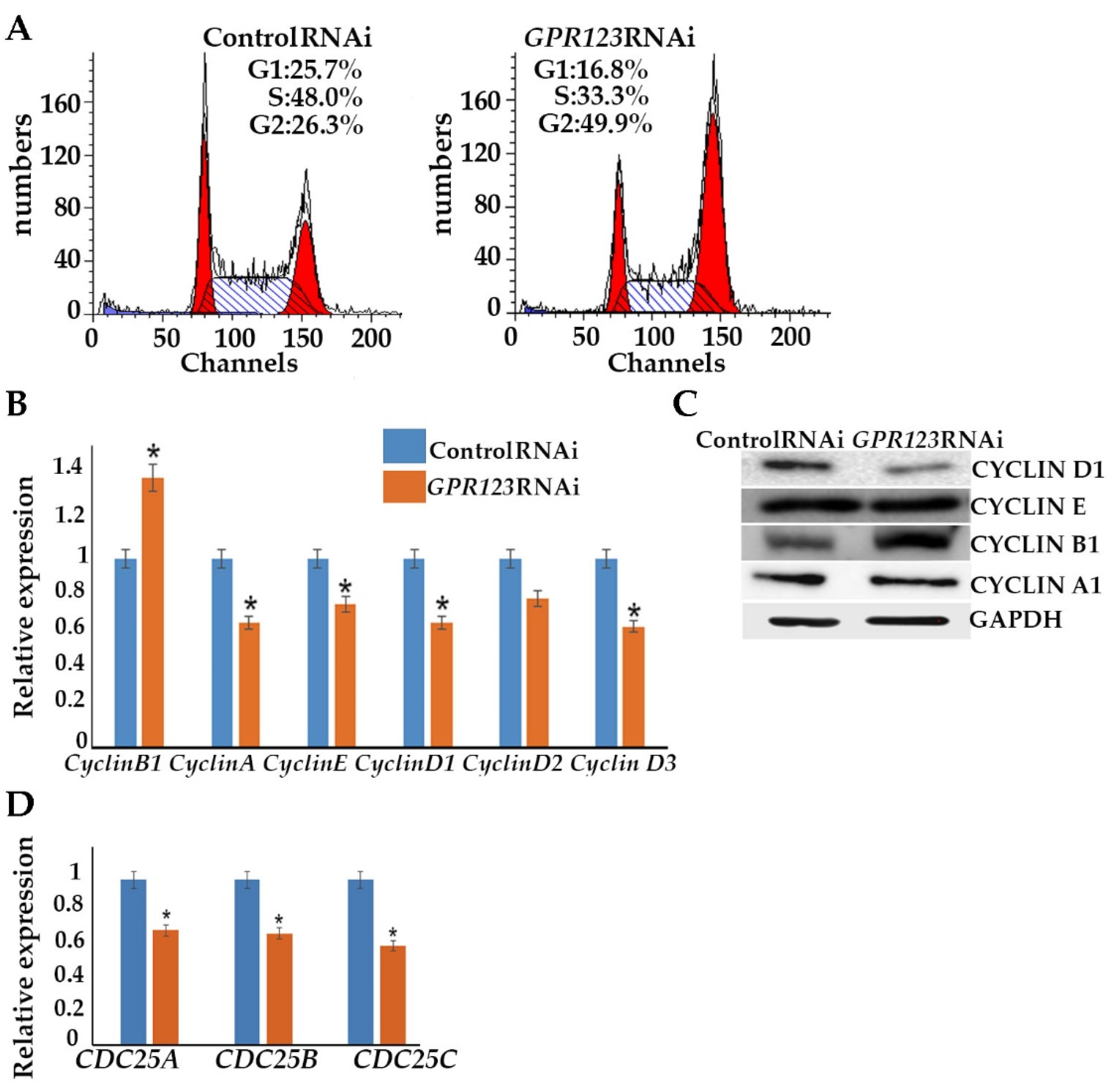
GPR123 RNAi hESCs and observed accumulation of the
GPR123 RNAi cells at the G2 phase of the cell cycle of the third day of transfection. This was further supported by the increased level of the cyclinB1 level. As was previously mentioned, one of the important prerequisites of successful hiPSCs generation is the acceleration of the cell cycle [
15]. Thus, accumulation of the
GPR123 RNAi cells at the G2 phase of the cell cycle might be one of the reasons for the absence of the hiPSCs colonies under
GPR123 RNAi. Therefore, we concluded that:
- (viii).
Expression of GPR123 is important for proper cell cycle regulation in hPSCs.
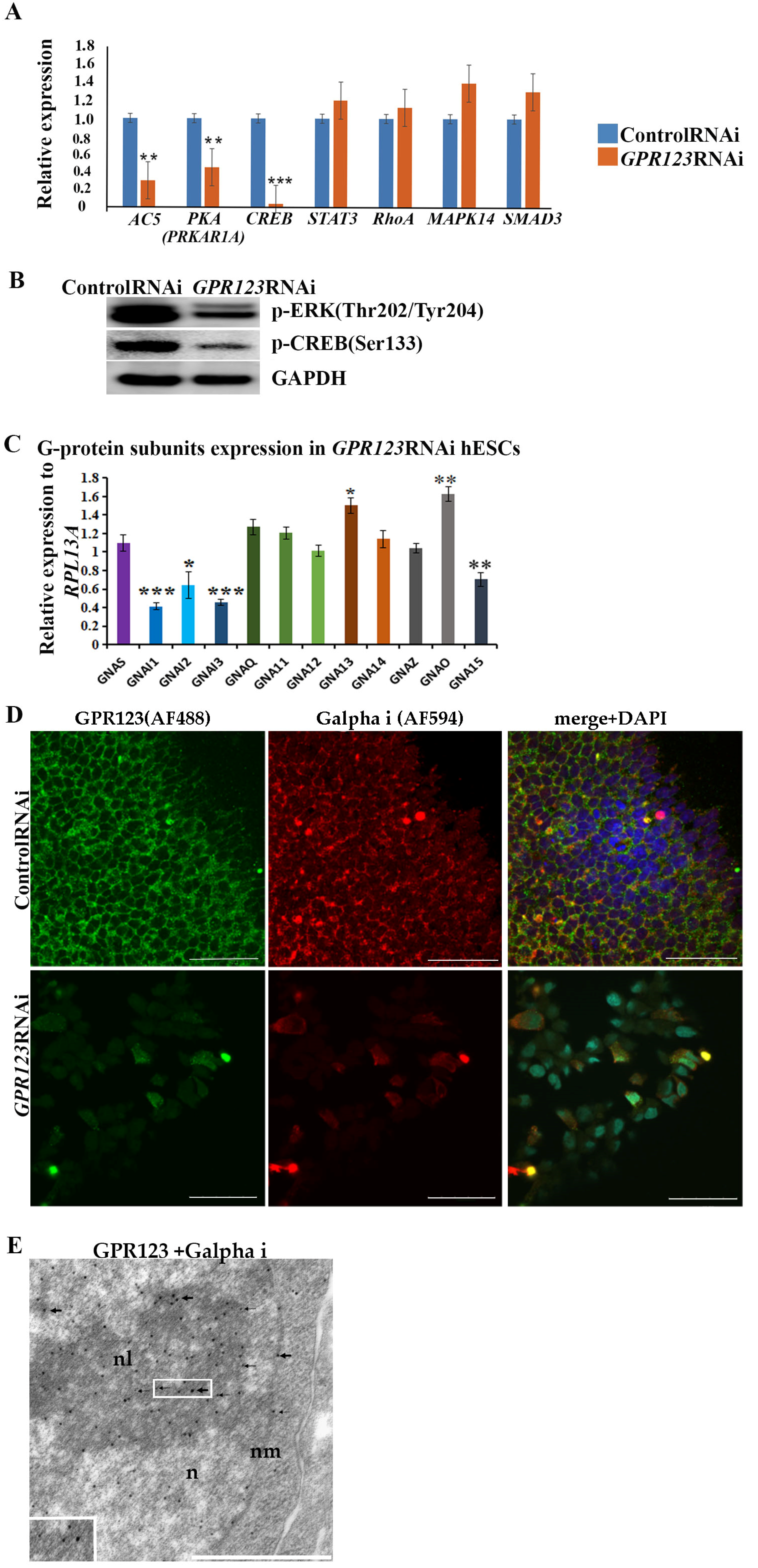
GPCRs are the largest family of cell surface receptors that modulate the activity of a variety of intracellular signals via G-protein signaling. G proteins are second messengers in intracellular signaling and consist of Gα, Gβ, and Gγ subunits. Gα subunits are subdivided into four subfamilies according to their structural and functional features: Gαs, Gαi/o, Gαq/11, and Gα12/13 [
31]. The G alpha (α) subunits partners are not known for GPR123, but it is expected that adhesion GPCRs can transduce signals via main G alpha subunits, including GαS and Gαi [
6,
21]. Previously, it was shown that hPSC colonies form and maintain characteristic pluripotent morphology and organization through Gαi function [
5]. Observed morphological changes in colonies under
GPR123 RNAi allow us to suggest that Gαi subunits may be involved in this process. To determine which G α subunits might be affected by
GPR123 RNAi in hPSCs, we analyzed the expression pattern of the main G α subunits by qRT-PCR and revealed that:
- (ix).
Expression levels of all three G alpha i subunits: GNAI1, GNAI2, and GNAI3 involved in regulation of cAMP and CREB pathways were significantly downregulated.
Corroborating the qRT-PCR data, confocal immunofluorescence analysis of the Gαi-staining pattern in the control and GPR123 RNAi hPSCs revealed a co-localization between GPR123 and the Gαi subunit in the control cells and loss of such co-localization between GPR123 and Gαi in GPR123 RNAi cells. Importantly, similar observation confirms loss of such co-localization on the 12th day of reprogramming in emerging hiPSC colonies.
Based on the above data, we concluded that in hPSCs the Gαi represents an important partner for GPR123 and the GPR123–Gαi axis is important for hPSC pluripotency maintenance and acquisition.
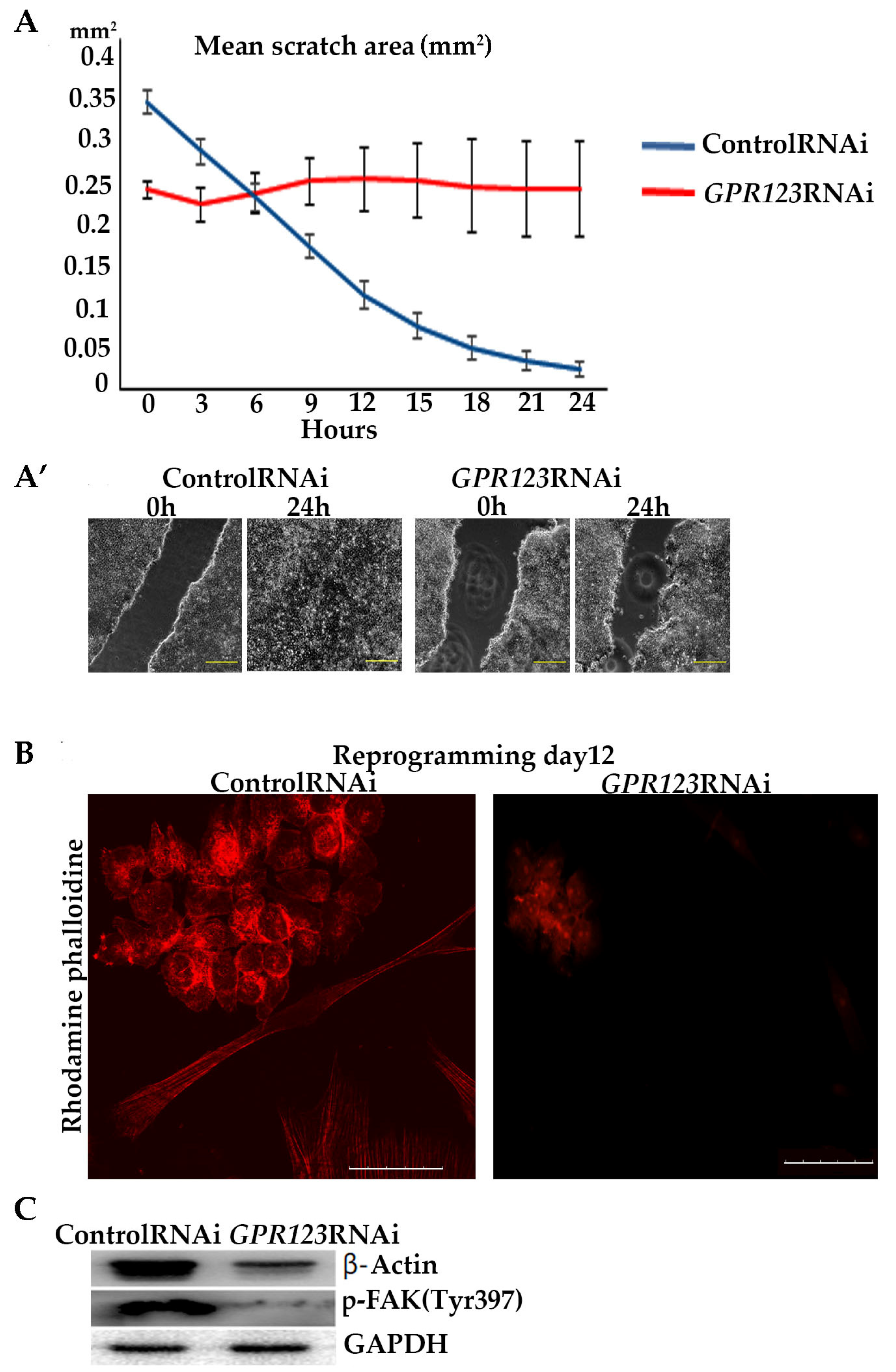
In addition to carrying out signaling cascades associated with the activation of cAMP and CREB, Gαi is also actively involved in cellular processes such as cell adhesion, cytoskeleton remodeling, actin nucleation and, accordingly, associated with cell mobility [
32,
33]. To reveal involvement of the GPR123–Gαi axis in cell motility, we performed scratch-overgrowth experiments in control RNAi and in
GPR123 RNAi hiPSCs. Our data demonstrated the absence of scratch closure in
GPR123 RNAi colonies in good agreement with the previous data of Nakamura and colleagues [
5]. In their work by experiments with the pertussis toxin, which suppresses Gαi signaling, these authors demonstrated the absence of overgrowth of the scratch in hPSCs. Having in mind a reduced expression of all three Gαi subunits in
GPR123 RNAi cells, we assumed that in our experimental settings, signaling cascades, which are important for cell motility and regulated by the GPR123–Gαi axis, are impaired. Thus, the GPR123–Gαi axis is important for cell motility in hPSCs.
Summarizing the obtained results, we can conclude that a high level of GPR123 expression is important for both: (1) the maintenance of pluripotency in hPSCs and (2) its acquisition during the reprogramming. All the above data are fundamentally new and deserve further attention.
Given the evidence discussed herein, the significance of the GPR123 in stem cell maintenance and somatic reprogramming to hiPSCs allows us to consider GPR123 as a new important hPSC marker, thus highlighting the need for further extensive research on its regulation. Our present and previous data [
1] demonstrate that GPCRs are a promising target for modulating the formation and organization of hPSC colonies and are important for understanding somatic cell reprogramming.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









