Abstract
Saccharomyces uvarum is one of the few fermentative species that can be used in winemaking, but its weak sulfite tolerance is the main reason for its further use. Previous studies have shown that the expression of the methionine synthase gene (MET4) is upregulated in FZF1 (a gene encoding a putative zinc finger protein, which is a positive regulator of the transcription of the cytosolic sulfotransferase gene SSU1) overexpression transformant strains, but its exact function is unknown. To gain insight into the function of the MET4 gene, in this study, a MET4 overexpression vector was constructed and transformed into S. uvarum strain A9. The MET4 transformants showed a 20 mM increase in sulfite tolerance compared to the starting strain. Ninety-two differential genes were found in the transcriptome of A9-MET4 compared to the A9 strain, of which 90 were upregulated, and two were downregulated. The results of RT-qPCR analyses confirmed that the expression of the HOMoserine requiring gene (HOM3) in the sulfate assimilation pathway and some fermentation-stress-related genes were upregulated in the transformants. The overexpression of the MET4 gene resulted in a significant increase in sulfite tolerance, the upregulation of fermentation-stress-related gene expression, and significant changes in the transcriptome profile of the S. uvarum strain.
1. Introduction
The quality of wine depends, to a certain extent, on the species or strain of yeast used in the fermentation process [1]. In addition to the common brewing yeasts, Saccharomyces uvarum is often used in winemaking because of its ability to ferment at low temperatures and its ability to produce specific aromas [2]. Sulfites are known to be a widely used preservative that is toxic to many microorganisms, and can also give wines a specific flavor during fermentation in the presence of yeast [3]. There are no compounds that can completely replace this additive because of its multifunctional properties, for example, the inhibition of the growth and enzymatic activity of the other microorganisms during winemaking and preservation [3,4]. However, sulfites not only destroy the cell structure, but also bind to some enzymes or metabolites, hindering the normal metabolic activities of the cells and seriously affecting the fermentation efficiency of the winemaking yeast in the later stages of fermentation [5]. Therefore, sulfite resistance in winemaking yeast is considered to be a vital trait in winemaking.
An overexpression analysis, transcriptome analysis, and quantitative reverse transcription-polymerase chain reaction (RT-qPCR) analysis are effective methods for gene function analysis and have been used in many applications in the study of gene function in S. cerevisiae [6,7] and S. uvarum [8,9]. In sulfite tolerance studies, the sulfotransferase gene (SSU1) was first identified as an important gene in the regulation of sulfite tolerance traits, and its overexpression significantly enhanced sulfite tolerance in S. cerevisiae [10]. S. cerevisiae cells have different mechanisms to deal with the stress of sulfite production, including an increased production of bound acetaldehyde, regulation of the sulfite uptake pathway, and efflux of sulfite through a plasma membrane pump encoded by the SSU1 gene [11]. Another important gene for sulfite resistance is the five zinc fingers protein encoding gene (FZF1), a positive regulator of SSU1 gene transcription. The FZF1 gene encodes a 5-finger transcription factor that plays a vital role in sulfite resistance in S. cerevisiae. The protein encoded by FZF1 in S. cerevisiae contains five C2H2-type zinc finger structural domains, whereas in S. uvarum, only four of the proteins encoded by the FZF1 gene are present [12,13]. Sulfite resistances of many S. cerevisiae strains are conferred by changes in FZF1 expression and changes in protein structure.
MET4p, encoded by the methionine synthase gene (MET4), is a transcriptional activator belonging to the family of leucine zip proteins [12]. MET4p can stimulate the positive transactivator of MET gene transcription in the methionine biosynthetic pathway [14], participating in the transcriptional activation of Met28 [15] and Met30 [16], and resisting sulfur stress by assembling the MET4–Met28–MET31 [17] and MET4–Met28–Met32 [18] complexes, inducing glutathione synthesis [18], and so on. Thus, MET4 can play a vital regulatory role in the relative stability of Saccharomyces cerevisiae in response to sulfur stress.
There are just two reports on the mechanism of sulfite tolerance in S. uvarum [8,9]. Our previous study found that the sulfite tolerance-related gene FZF1 regulates the expression of MET4 and HAL4, but not SSU1 [9]. Meanwhile, elevating the expression of the SSU1 gene could also enhance the sulfite tolerance of S. uvarum [8]. To gain insight into the function of the MET4 gene, and validate the hypothesis that the MET4 gene can regulate sulfite tolerance in S. uvarum, the MET4 gene function in S. uvarum was mined by the construction of MET4 overexpression strains, sulfite tolerance phenotype screening, polymerase chain reaction (PCR) analysis, fermentation weight loss analysis, transcriptome analysis, and RT-qPCR analysis.
2. Materials and Methods
Saccharomycesuvarum A9, pCAMBIA1301, and E. coli DH5α were stored at the Key Laboratory of Southwest Biodiversity Conservation, National Forest and Grassland Administration, Southwest Forestry University (Kunming, China). MET4 was synthesized and constructed in Pgem-T Easy vector by Baiqi Biotechnology Co. (Wuhan, China) The sequence of the MET4 gene is from the S. uvarum strain A9 [9] (access number: OL804291). Primers were synthesized by Shanghai Biotechnology Co., Ltd., Shanghai, China, and molecular reagents or kits were purchased from Shanghai Biotechnology Co.
2.1. Construction of the MET4 Gene Expression Vector
The small fragment of Pgem-T-MET4, cleaved by NcoI and BglII and purified according to [19], was ligated to the pCAMBIA1301 vector, which was also cleaved by these two enzymes. pCAMBIA1301-MET4 was transferred into E. coli DH5α receptor cells by electrotransformation and then coated on LB (1% yeast extract, 1% tryptone, 2% agar, and 0.5% NaCl) plates containing 10 mg/mL Hygromycin (HYG) and incubated overnight at 37 °C. Larger transformed colonies were picked into 3 mL LB liquid medium (1% yeast extract, 1% tryptone, and 0.5% NaCl) containing 10 mg/mL HYG and incubated overnight at 37 °C. Cells were harvested, and plasmids were extracted and purified. The expression vector was then verified by Polymerase Chain Reaction (PCR) and sent to Shanghai Biotechnology for sequencing to test the success of the vector construction.
2.2. Genetic Transformation
After the preparation of S. uvarum receptor cells, the cells and recombinant plasmid pCAMBIA1301-MET4 were mixed at a volume ratio of 10:1 in an electroporation cup and electroporated for 5 ms at 1500 V using an Eppendorf electroporator (Eppendorf, Hamburg, Germany). Eight hundred μL of YEP medium (1% yeast extract, 1% peptone, and 0.5% NaCl, pH 7.5) was added and incubated for 1 h at 28 °C. After incubation for 2 h in a 200 rpm shaker at 28 °C, the larger transformants were selected by incubating overnight on YPD (1% yeast extract, 2% peptone, 2% agar, and 2% glucose) plates containing 30 mg/L HYG and 30 mM sulfite.
2.3. Sulfite Tolerant Trait Typing
The strains were inoculated and grew on fresh YPD medium containing 5, 10, 20, 40, and 60 mM sodium sulfite and 80 mM succinate at pH 3.5. After 24 h, 48 h, 72 h, 96 h, 120 h, 144 h, and 168 h, the sulfite tolerance levels were recorded, according to colony growth measurements.
2.4. PCR Analysis
Deoxyribonucleic acid (DNA) samples were prepared according to the method of Nardi [20]. Ten candidate colonies were randomly selected from MET4 transformants for PCR analysis. The PCR reaction mix (25 μL) consisted of 0.2 μL of 5 U/μL Taqase, 0.5 μL of 10 mM dNTP, 1 μL of 10 μM primer, 2 μL of DNA template, 2.5 μL of 10× PCR buffer (including mg2+), and 17.8 μL of dH2O. The primers used for PCR were HYG-F: 5′-TGCTGCTCCATACAAGCCAA-3′ and HYG-R: 5′-ACCGCAAGGAATCGGTCAAT-3. The PCR reactions were performed according to the following procedure: 95 °C pre-denaturation for 5 min; 35 cycles of denaturation at 95 °C for 30 s, annealing at 56 °C for 30 s, and extension at 72 °C for 60 s, with a final extension at 72 °C for 8 min.
2.5. Fermentation Weight Loss Analysis
The transgenic strain A9-MET4 was fermented in 15 g of grape juice (grapes from Aziying, Panlong District, Kunming, China) containing 20 mM sodium sulfite and 80 mM succinic acid at pH 3.5, together with the starting strain A9 and EC1118. The Petri dishes containing the strains were initially weighed and their weight recorded as W0, weighed every 1 day for a total of 7 measurements as W1–7, and the weight loss every 1 day as W = Wn − Wn − 1. Based on these data, graphs were made in excel.
2.6. Ribonucleic Acid (RNA) Extraction and cDNA Synthesis
The yeast strains were collected by incubation in liquid YPD for 24 h, followed by treatment in a medium containing 20 mM sulfite for 10 min. Yeast cells were ground in liquid nitrogen and RNA was extracted using the QIAGEN kit (Qiagen China, Shanghai, China). RNA samples were then reverse transcribed into cDNA using the Reverse Transcription Kit (Takara, Dalian, China). These samples would be used for further RT-qPCR analysis and transcriptome analysis.
2.7. RT-qPCR Analysis
RT-qPCR analysis was performed using an ABI 7500 fluorescent qPCR instrument (Thermo Fisher Scientific, Carlsbad, CA, USA) according to the method described by Liu [9]. The primers are shown in Table 1. Actin-1 (ACT1) was used as the reference gene (in Table 1). Three replicates were set for each gene. Gene expression levels were calculated according to the method of Liu [8].

Table 1.
Primers used for RT-qPCR and PCR analysis.
2.8. Transcriptome Analysis
RNA-Seq was performed by Nextomics Biosciences Co. Ltd. (Wuhan, China) using an Illumina HiSeqTM (Illumina, San Diego, CA, USA). RNA-Seq data were submitted to the National Center for Biotechnology Information (NCBI) (Bethesda, MD, USA) repository SRA (Accession no. PRJNA786265). Transcriptome assembly, sequence alignment, gene orthology determination, and gene set enrichment analysis were performed according to the method described by Bian [21]. Differential gene expression heat maps were plotted using Log2 (Fragments Per Kilobase of Exon Model per Million Mapped Fragments (FPKM) + 1) values with the R 3.0.2 software.
2.9. Data Analysis
Histograms were calculated and plotted using GraphPad Prism Software (v9.0), and one-way ANOVAs were performed by macros, with p-values and standard errors calculated using GraphPad Prism Software.
3. Results
3.1. Detection of Transgenic Strains
Transformants grown on sulfite-containing media were analyzed by PCR, and the results showed that all ten transformants picked were positive. Three of the larger transformants grown in sulfite-tolerant medium were picked for RT-qPCR analysis, which showed that their relative expression was 10.11-fold higher than the mean value of the starting strain (see Figure 1), with the difference reaching significance (p-value < 0.01).
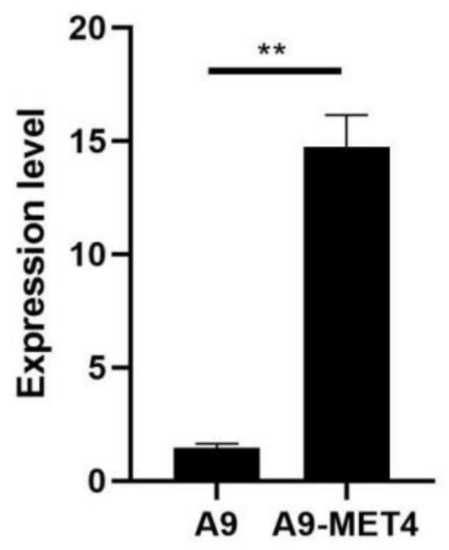
Figure 1.
The expression level of the MET4 gene in the S. uvarum starting strain A9 and its transformants. Enhanced expression or depression of the MET4 gene was assessed using the 2−ΔΔCT method to determine relative gene expression from RT-qPCR data with ACT1 as a housekeeping gene. Values were means ± standard error (SE) of 2−ΔΔCT (n = 3). ** p < 0.01.
Comparison with transformants transfected with the SSU1 gene and the FZF1 gene indicates that the transfection of this gene can enhance sulfite tolerance in grape juice yeast, but to a lesser extent than the overexpression transformants of the two genes mentioned above (See Table 2) [8,9].
Table 2.
The genotype and sulfite resistance ability of MET4 transgenic strain and its starting stain.
During the first three days of fermentation, the weight loss of the MET4 transformants was significantly higher than that of the starting strain. As the fermentation progressed, the weight loss of the MET4 transformants was still higher than that of the starting strain, but the value of the difference decreased (see Figure 2). It indicates that the MET4 transformants were significantly more capable of fermenting in grape juice containing 20 mM sodium sulfite than the starting strain in the early stages of fermentation. As time progressed, the weight loss between the MET4 transformants and the starting strain fermentate gradually drew closer as the sugars were gradually consumed, but the weight loss of the MET4 transformants fermentate was still higher than that of the starting strain. It indicates that the fermentability of MET4 transformants in sulfite-containing grape juice was also higher than that of the starting strain in the later stages of fermentation.
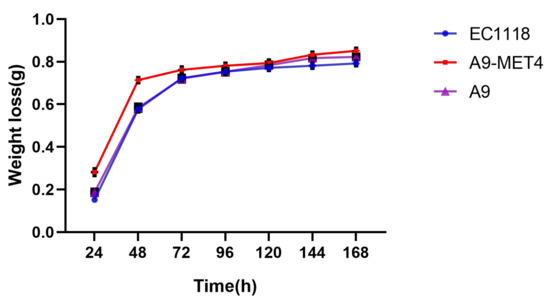
Figure 2.
Weight loss curve of the transformant. Three replicates were set for each strain, each point in the figure was the mean value of three values. Values were means ± SE.
3.2. Transcriptome Differential Gene Analysis
Transcriptome sequencing yielded over 250 million high-quality reads, with over 4 million high-quality gene data obtained per replicate. Only 1.1% of FPKM values were between 0–1 in A9, and 2.5% of FPKM values were between 0–1 in A9-MET4. In the A9 strain, 86.1% of FPKM values were above 10, and 75.8% of FPKM values were above 10 in the A9-MET4 strain (Table 3). These results demonstrate the high quality of the reads obtained.
Table 3.
Differential expression analysis of different stains.
The transcriptome profiles of A9-MET4 and the starting strain A9 were compared, and a total of 92 differential genes were obtained, with 90 upregulated genes and two downregulated ones. These differential genes were annotated to the three major functional classes, such as molecular function, cellular component, and biological process (Figure 3). The differentially expressed cellular-component-related genes are mainly involved in protein-containing complexes, cellular anatomical entities, and intracellular proteins. Molecular functional genes include genes related to binding, catalytic activity, transcriptional activity, transport activity, and protein folding chaperones, while biological-process-related genes are related to cellular processes, metabolic processes, bioregulation, and response to stimuli. The expression of genes related to fermentation-stress response, protein folding, and transcriptional regulation of RNA polymerase II was upregulated (see Table 4), e.g., HOM3, NRG1, and VID24. The methylenetetrahydrofolate dehydrogenase gene MIS1, which is related to NADPH synthesis, is downregulated.
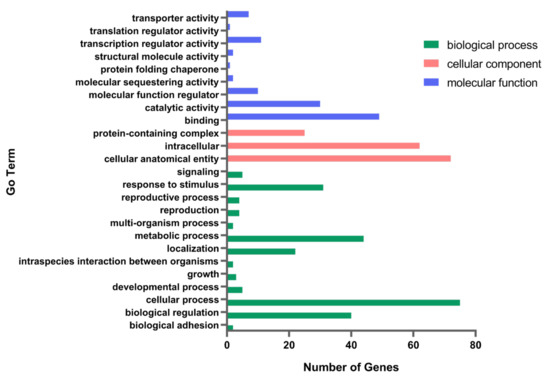
Figure 3.
Pathway enrichment of differentially expressed genes between A9-MET4 and the starting strain A9 with Gene Ontology (GO) interpretation.
Table 4.
The gene expression levels of A9-MET4 compared to the starting strain A9.
An expression heat map analysis was performed for genes involved in the stress response (Figure 4). The eight most differentially expressed genes compared to the starting strain A9 were HOM3, HSP30, NRG1, VID24, APJ1, YAP6, Hsp104, and FES1. The differential expression of these genes may be associated with increased sulfite tolerance.
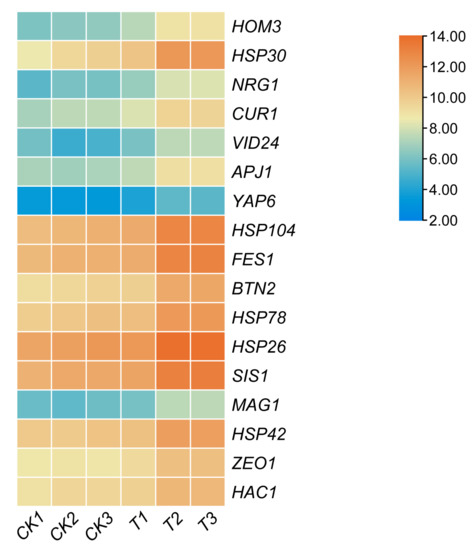
Figure 4.
Heat map of the expression levels of differentially expressed, stress-related genes. Color represents expression change. CK1, CK2, and CK3, control; T1, T2, and T3, A9-MET4, treated in a sulfite-containing medium for 10 min.
To identify the major pathways affecting sulfite tolerance in S. uvarum, differential expression genes (DEGs) were analyzed for enrichment in the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway website [22]. Among these, protein processing in the endoplasmic reticulum was the most significantly enriched pathway for differential genes, with a p-value = 0.002 < 0.05 (see Figure 5). Most upregulated genes were enriched in the endoplasmic reticulum-associated degradation (ERAD) response, e.g., Hsp104, HSP78, etc.; the molecular chaperone binding protein (BiP) was also enriched with an uregulated gene, and BIP induces the ERAD response [23]. Isolation of misfolded proteins into insoluble aggregates under proteotoxic stress conditions is a way for cells to attempt to maintain function [24], and the ERAD response can remove misfolded proteins in response to stress [25]. This pathway suggests that MET4 overexpression in S. uvarum transformants enhance their sulfite adaptation by activating the ERAD response.
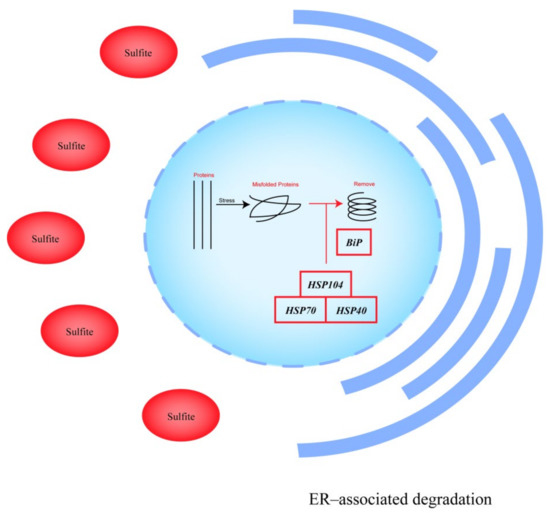
Figure 5.
Map of protein processing pathways in the endoplasmic reticulum of A9-MET4 in a sulfite environment. The main upregulated genes in the figure are: (a) HSP104, which regulates the expression of the nucleotide-exchange factor (NEF), with an expression level Log2 value of 1.50; (b) KAR2, which regulates the expression of BiP (Binding immunoglobulin protein), with a Log2 value of 1.29; (c) SSA4, which regulates the expression of the HSP70 family, with a Log2 value of 1.43; (d) APJ1, which regulates the expression of the HSP40 family, with a Log2 value of 1.61. HSP104 combined with HSP70 and HSP40 activates denatured protein refolding under stress conditions [26]. The figure is adapted from the KEGG pathway.
In the cysteine and methionine metabolic pathways, the gene encoding aspartate kinase (HOM3, EC: 2.7.2.4) is differentially expressed in a sulfite environment. Aspartate kinase is the enzyme of the first step in methionine biosynthesis [27]. In this study, HOM3 was the most differentially expressed gene, which may indicate that overexpression of MET4 in S. uvarum regulates methionine synthesis by increasing HOM3 expression, thereby ultimately enhancing its sulfite adaptation.
3.3. RT-qPCR Validation
The results of the RT-qPCR analysis confirmed that the ten genes selected in the transcriptome analysis were indeed upregulated in the MET4 transformants compared to the starting strain, although the correlation coefficient (R2 = 0.0659) between these two sets of data was small. Both the transformants and the starting strain were treated with the same method and for the same duration, but the expression of genes involved in sulfite depletion (e.g., HOM3) and stress-related genes (e.g., Hsp104) were significantly upregulated in the transformants (see Figure 6). It suggests that the overexpression of the MET4 gene promotes the expression of genes involved in sulfite depletion and stress-related genes, which in turn leads to enhanced sulfite tolerance in S. uvarum.
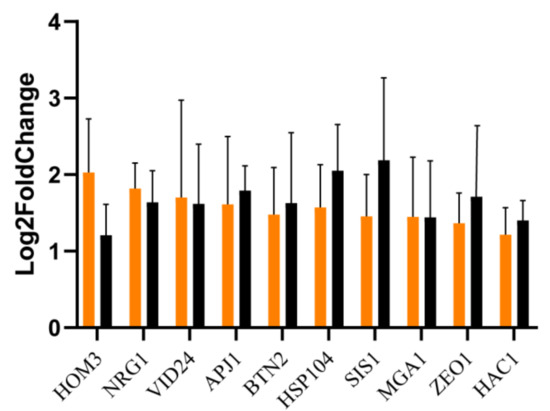
Figure 6.
Comparison of transcriptome and RT-qPCR analysis results. Orange, transcriptome analysis; Black, RT-qPCR analysis. The standard deviation values of the treatments (error bars) are treated with log10 because they are too large after dividing by the control values; the rest are log2 values. Each gene was replicated three times.
4. Discussion
In winemaking, the sulfite assimilation pathway in S. cerevisiae has been revealed to be controlled by five transcriptional regulators, including three DNA-binding proteins (Met31p, Met32p, and Cbf1p), an activator (MET4p), a cofactor (Met28p), and a combination of ubiquitin ligase subunits (Met30p). Of these, MET4p is the only transcriptional activator in the sulfite assimilation pathway [28]. In a strain of S. uvarum isolated from wine that exhibits high sulfite tolerance, the sulfite tolerance is regulated through the FZF1 gene, not SSU1 [9]. In a previous study, we compared the transcriptional profiles of FZF1-overexpressing and FZF1-silenced S. uvarum transformants and found that the FZF1 gene regulates S. uvarum sulfite tolerance by modulating MET4 gene expression levels [9]. The different expression levels of MET4 may be one of the important reasons for the different sulfite tolerance observed in the MET4 transformants and the starting strain. The MET4 transformants were sulfite tolerant up to 40 mM, which was 20 mM higher than the 20 mM of the starting strain A9, but 60 mM and 40 mM lower than the transformants of the FZF1 and SSU1 genes, respectively, suggesting that the MET4 gene could play a smaller role in sulfite tolerance in S. uvarum strains than the FZF1 and SSU1 genes.
After a comparison in the SGD database (Data updated on 1 February 2022) [29], we found that the genes in the expression profile of the MET4 overexpression S. uvarum transformants did not have any overlapping parts with the S. cerevisiae MET4 reciprocal gene [30,31], indicating that the MET4 gene of the S. uvarum A9 strain is functionally distinct from the MET4 gene of S. cerevisiae. Both the MET4 gene of S. uvarum and S. cerevisiae contain a basic leucine zip structural domain (bZip) at the end, but about 29.57% of the DNA sequence of S. uvarum MET4 is different from that of S. cerevisiae MET4 (NM_001182941.3, S. cerevisiae S288C) and the encoded amino acids differ by about 33.24%; these differences may be the main reason for the change in gene function of MET4.
Fermentation of grape juice in the presence of sulfite exposes the yeast to a cascade of stresses including osmotic pressure, hypoxia, nitrogen depletion, and increased ethanol concentration. Fermentation-stress-response genes exhibit sustained and significantly induced expression in response to stress conditions during fermentation. In this study, fermentation-stress-response and heat-stress-protein-encoding genes accounted for approximately 40% of the differentially upregulated expressed genes in the expression profile of the MET4 overexpression S. uvarum transformants. The significant upregulation of these genes facilitated the sustained fermentation of S. uvarum in a sulfite environment. In this study, RNA-polymerase-II-related genes were upregulated, and the methylenetetrahydrofolate dehydrogenase gene MIS1, associated with NADPH synthesis, was downregulated in the MET4 overexpression S. uvarum transformants compared to the starting strain. The reason for this might be due to the positive regulatory function of the MET4 gene on RNA polymerase II on transcription, and the toxic effect of sulfite on enzymes associated with NADPH production or utilization [32,33].
In the sulfate assimilation pathway, sulfite is reduced by sulfite reductase and eventually synthesized into sulfur-containing compounds. It has been shown that HOM3 is involved in regulating the synthesis of the sulfur-containing compound methionine [34], and can consume a portion of the sulfite. In the present study, the overexpression of the MET4 gene in S. uvarum upregulated the expression of the HOM3 gene. We suggested that upregulating the expression of the HOM3 gene depleted more of the sulfite in the medium, causing the concentration of sulfite in the environment to decrease, which in turn allowed the transformants to survive or ferment in succession with higher levels of sulfite, i.e., to be more tolerant of sulfite. Therefore, the upregulation of the expression of HOM3 might be one of the key reasons for the increased sulfite tolerance in MET4 S. uvarum transformants. Some populations are allergic to sulfite, and common symptoms in sulfite-allergic people after exposure to sulfite include nasal congestion, headache, breathing difficulties, nausea, dizziness, and abdominal pain. The MET4 S. uvarum transformants obtained in this study could deplete the sulfite in its fermentation environment, which in turn would further reduce the adverse effects of its fermentation products on sulfite-allergic people.
Mechanisms of sulfite tolerance in S. cerevisiae have been uncovered, including (A) the mediation of sulfite efflux via SSU1p and FZF1p, (B) the synthesis of non-toxic compounds with acetaldehyde, and (C) the reduction of sulfite in vivo [11]. Of these, sulfate reduction is mainly via the reduction to sulfur-containing compounds by sulfite reductase in the sulfate assimilation pathway [10]. We suggested that in this study, MET4 overexpression transformants may also deplete the sulfite in the culture environment through sulfate assimilation, thereby reducing the sulfite concentration in the environment (i.e., medium) and achieving higher sulfite concentration tolerance. Interestingly, we found that in S. uvarum, the upregulation of SSU1 gene expression was accompanied by increased expression of the HOM3 gene [8], but that the upregulation of FZF1 expression was not accompanied by a corresponding increase in HOM3 gene expression [9].
It has been suggested that the Hsp104 gene is associated with ethanol tolerance [24]. It has been further shown that other members of the HSP gene family are also upregulated in other adverse environments [35]. The expression of Hsp104 is elevated in FZF1 overexpressing S. uvarum strains [9] and upregulated in MET4 overexpressing S. uvarum strains. The elevated expression of the Hsp104 gene in the present study suggested that this gene may be associated with the ability of transformants to tolerate higher concentrations of sulfite.
5. Conclusions
- (1)
- We suggest that a mechanism of sulfite tolerance exists in S. uvarum, i.e., tolerance to higher sulfite concentrations is achieved by depleting the sulfite in the culture environment. S. uvarum may be able to tolerate higher concentrations of sulfite by continuously growing and dividing, and by upregulating the expression of genes, such as HOM3, which consume part of the sulfite in the culture medium or fermentation broth.
- (2)
- S. uvarum may be able to tolerate higher concentrations of sulfite by increasing the expression of MET4, and thus the stress-related genes. The hypothesis that the ‘MET4 gene can regulate sulfite tolerance in S. uvarum’ is proved.
- (3)
- Overexpression of the MET4 gene resulted in a significant increase in sulfite tolerance, an upregulation of fermentation-stress-related gene expression, and a significant change in the transcriptome profile of the strain. The transformants could broaden the application of S. uvarum in the winemaking industry.
Author Contributions
H.Z. conceived and designed the experiments. Z.Z., X.L. and Z.W. performed the experiments. W.Z. and X.L. analyzed the data. Z.W. and Z.Z. wrote the paper. T.Y. and Z.Z. contributed analysis tools. X.L. edited the paper. All authors have read and agreed to the published version of the manuscript.
Funding
The study was supported by grants from the National Natural Science Foundation of China (32160556, 31760450) and the Joint Project of Agricultural Basic Research in Yunnan Province (2018FG001-038). The funders had no role in the design of the study and collection, analysis, and interpretation of data, or in writing the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
The experiment materials do not include human beings or animals. Hence, ethics approval and consent to participate are not applicable.
Data Availability Statement
All data generated or analyzed during this study are included in this published article. RNA-Seq data were presented at the Genome Sequence Archive of the Beijing Institute of Genomics (BIG) Data Center (accession number CRA001986).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, H.Y.; Lee, S.A.; Bradbury, J.E.; Warren, R.N.; Sheth, H.; Hooks, D.O.; Richards, K.D.; Gardner, R.C. Yeasts isolated from New Zealand vineyards and wineries. Aust. J. Grape Wine Res. 2010, 16, 491–496. [Google Scholar] [CrossRef]
- Zhang, H.; Richards, K.D.; Wilson, S.; Lee, S.A.; Sheehan, H.; Roncoroni, M.; Gardner, R.C. Genetic characterization of strains of Saccharomyces uvarum from New Zealand wineries. Food Microbiol. 2015, 46, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Nadai, C.; Treu, L.; Campanaro, S.; Giacomini, A.; Corich, V. Different mechanisms of resistance modulate sulfite tolerance in wine yeasts. Appl. Microbiol. Biotechnol. 2015, 100, 797–813. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y. Formation and characteristics of wine bouquet produced by wine yeasts. Microbiology 2007, 34, 757–760. (In Chinese) [Google Scholar]
- Liu, X.Z.; Zhang, Z.M.; Zhang, H.Y. Cross breeding and hybrid identification of sulphite-tolerant hybrids of Saccharomyces uvarum. S. Afr. J. Enol. Vitic. 2017, 38, 125–131. [Google Scholar] [CrossRef][Green Version]
- Vargas-Maya, N.I.; González-Hernández, G.A.; Padilla-Guerrero, I.E.; Torres-Guzmán, J.C. Overexpression of smORF YNR034W-A/EGO4 in Saccharomyces cerevisiae increases the fermentative efficiency of Agave tequilana Weber must. J. Ind. Microbiol. Biotechnol. 2017, 44, 63–74. [Google Scholar] [CrossRef]
- Bergman, A.; Vitay, D.; Hellgren, J.; Chen, Y.; Nielsen, J.; Siewers, V. Effects of overexpression of STB5 in Saccharomyces cerevisiae on fatty acid biosynthesis, physiology and transcriptome. FEMS Yeast Res. 2019, 19, foz027. [Google Scholar] [CrossRef]
- Liu, X.Z.; Sang, M.; Zhang, X.A.; Zhang, T.K.; Zhang, H.Y.; He, X.; Li, S.X.; Sun, X.D.; Zhang, Z.M. Enhancing expression of SSU1 genes in Saccharomyces uvarum leads to an increase in sulfite tolerance and a transcriptome profile change. FEMS Yeast Res. 2017, 17, fox023. [Google Scholar] [CrossRef]
- Liu, X.; Liu, X.; Zhang, Z.; Sang, M.; Sun, X.D.; He, C.Z.; Xin, P.Y.; Zhang, H.Y. Functional analysis of the FZF1 genes of Saccharomyces uvarum. Front. Microbiol. 2018, 9, 96. [Google Scholar] [CrossRef]
- Hansen, J.; Johannesen, P.F. Cysteine is essential for transcriptional regulation of the sulfur assimilation genes in Saccharomyces cerevisiae. Mol. Gen. Genet. 2000, 263, 535–542. [Google Scholar] [CrossRef]
- Thomas, D.; Surdinkerjan, Y. Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1997, 61, 503–532. [Google Scholar] [PubMed]
- Kuras, L.; Thomas, D. Functional analysis of MET4, a yeast transcriptional activator responsive to S-adenosylmethionine. Mol. Cell. Biol. 1995, 15, 208. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhang, X.A.; Liu, X.Z.; Li, S.X.; Zhang, H.Y. Cloning and sequence analysis of the FZF1 gene concerning sulfur tolerance from Saccharomyces bayanus. Sci. Technol. Food Ind. 2015, 36, 166–172. (In Chinese) [Google Scholar]
- Thomas, D.; Jacquemin, I.; Surdin-Kerjan, Y. MET4, a leucine zipper protein, and centromere-binding factor 1 are both required for transcriptional activation of sulfur metabolism in Saccharomyces cerevisiae. Mol. Cell. Biol. 1992, 12, 1719–1727. [Google Scholar] [PubMed]
- Kuras, L.; Cherest, H.; Surdin-Kerjan, Y.; Thomas, D. A heteromeric complex containing the centromere binding factor 1 and two basic leucine zipper factors, MET4 and Met28, mediates the transcription activation of yeast sulfur metabolism. EMBO J. 1996, 15, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Su, N.Y.; Ouni, I.; Papagiannis, C.V.; Kaiser, P. A dominant suppressor mutation of the Met30 cell cycle defect suggests regulation of the Saccharomyces cerevisiae MET4-Cbf1 transcription complex by Met32. J. Biol. Chem. 2008, 283, 11615–11624. [Google Scholar] [CrossRef] [PubMed]
- Kuras, L.; Barbey, R.; Thomas, D. Assembly of a bZIP-bHLH transcription activation complex: Formation of the yeast Cbf1-MET4-Met28 complex is regulated through Met28 stimulation of Cbf1 DNA binding. EMBO J. 1997, 116, 2441–2451. [Google Scholar] [CrossRef] [PubMed]
- Blaiseau, P.L.; Thomas, D. Multiple transcriptional activation complexes tether the yeast activator MET4 to DNA. EMBO J. 2014, 17, 6327–6336. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, New York: Cold Spring Harbor Laboratory Press. BioScience 1989, 33, 721–722. [Google Scholar]
- Nardi, T.; Corich, V.; Giacomini, A.; Blondin, B. A sulphite-inducible form of the sulphite efflux gene SSU1 in a Saccharomyces cerevisiae wine yeast. Microbiology 2010, 156, 1686–1696. [Google Scholar] [CrossRef]
- Bian, W.; Liu, X.; Zhang, Z.; Zhang, H. Transcriptome analysis of diploid and triploid Populus tomentosa. Peer J. 2020, 8, e10204. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Wakasa, Y.; Yasuda, H.; Oono, Y.; Kawakatsu, T.; Hirose, S.; Takahashi, H.; Hayashi, S.; Yang, L.; Takaiwa, F. Expression of ER quality control-related genes in response to changes in BiP1 levels in developing rice endosperm. Plant J. Cell Mol. Biol. 2015, 65, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Sathyanarayanan, U.; Musa, M.; Dib, P.B.; Raimundo, N.; Milosevic, I.; Krisko, A. ATP hydrolysis by yeast Hsp104 determines protein aggregate dissolution and size in vivo. Nat. Commun. 2020, 11, 5226. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Hu, X.; Guo, Y.; Wang, L.; Ji, J.; Li, J.; Zhang, Z.R. A technique for delineating the unfolding requirements for substrate entry into retrotranslocons during endoplasmic reticulum-associated degradation. J. Biol. Chem. 2019, 294, 20084–20096. [Google Scholar] [CrossRef]
- Glover, J.R.; Lindquist, S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell 1998, 94, 73. [Google Scholar] [CrossRef]
- Rafalski, J.A.; Falco, S.C. Structure of the yeast HOM3 gene which encodes aspartokinase. J. Biol. Chem. 1988, 263, 2146–2151. [Google Scholar] [CrossRef]
- Lee, T.A.; Jorgensen, P.; Bognar, A.L.; Peyraud, C.; Thomas, D.; Tyers, M. Dissection of combinatorial control by the MET4 transcriptional complex. Mol. Biol. Cell 2010, 21, 456. [Google Scholar] [CrossRef]
- Park, H.; Bakalinsky, A.T. SSU1 mediates sulphite efflux in Saccharomyces cerevisiae. Yeast 2000, 16, 881–888. [Google Scholar] [CrossRef]
- Thorsen, M.; Lagniel, G.; Kristiansson, E.; Junot, C.; Nerman, O.; Labarre, J.; Tamas, M.J. Quantitative transcriptome, proteome, and sulfur metabolite profiling of the Saccharomyces cerevisiae response to arsenite. Physiol. Genom. 2007, 30, 35–43. [Google Scholar] [CrossRef]
- Youn, J.Y.; Friesen, H.; Nguyen Ba, A.N.; Liang, W.; Messier, V.; Cox, M.J.; Moses, A.M.; Andrews, B. Functional analysis of kinases and transcription factors in Saccharomyces cerevisiae using an integrated overexpression library. G3 Genes|Genomes|Genetics 2017, 7, 911–921. [Google Scholar] [CrossRef]
- Kobayashi, K.; Yoshimoto, A. Studies on yeast sulfite reductase. IV. Structure and steady-state kinetics. Biochim. Biophys. Acta 1982, 705, 348–356. [Google Scholar] [CrossRef]
- McIsaac, R.S.; Petti, A.A.; Bussemaker, H.J.; Bussemaker, D. Perturbation-based analysis and modeling of combinatorial regulation in the yeast sulfur assimilation pathway. Mol. Biol. Cell 2012, 23, 2993–3007. [Google Scholar] [CrossRef] [PubMed]
- Arévalo-Rodríguez, M.; Calderón, I.L.; Holmberg, S. Mutations that cause threonine sensitivity identify catalytic and regulatory regions of the aspartate kinase of Saccharomyces cerevisiae. Yeast 2010, 15, 1331–1345. [Google Scholar] [CrossRef]
- Chen, J.; Gao, T.; Wan, S.; Zhang, Y.H.; Yang, J.K.; Yu, Y.B.; Wang, W.D. Genome-wide identification, classification and expression analysis of the HSP gene superfamily in tea plant (Camellia sinensis). Int. J. Mol. Sci. 2018, 19, 2633. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).