
Does Disruption of Optic Atrophy-1 (OPA1) Contribute to Cell Death in HL-1 Cardiomyocytes Subjected to Lethal Ischemia-Reperfusion Injury?

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
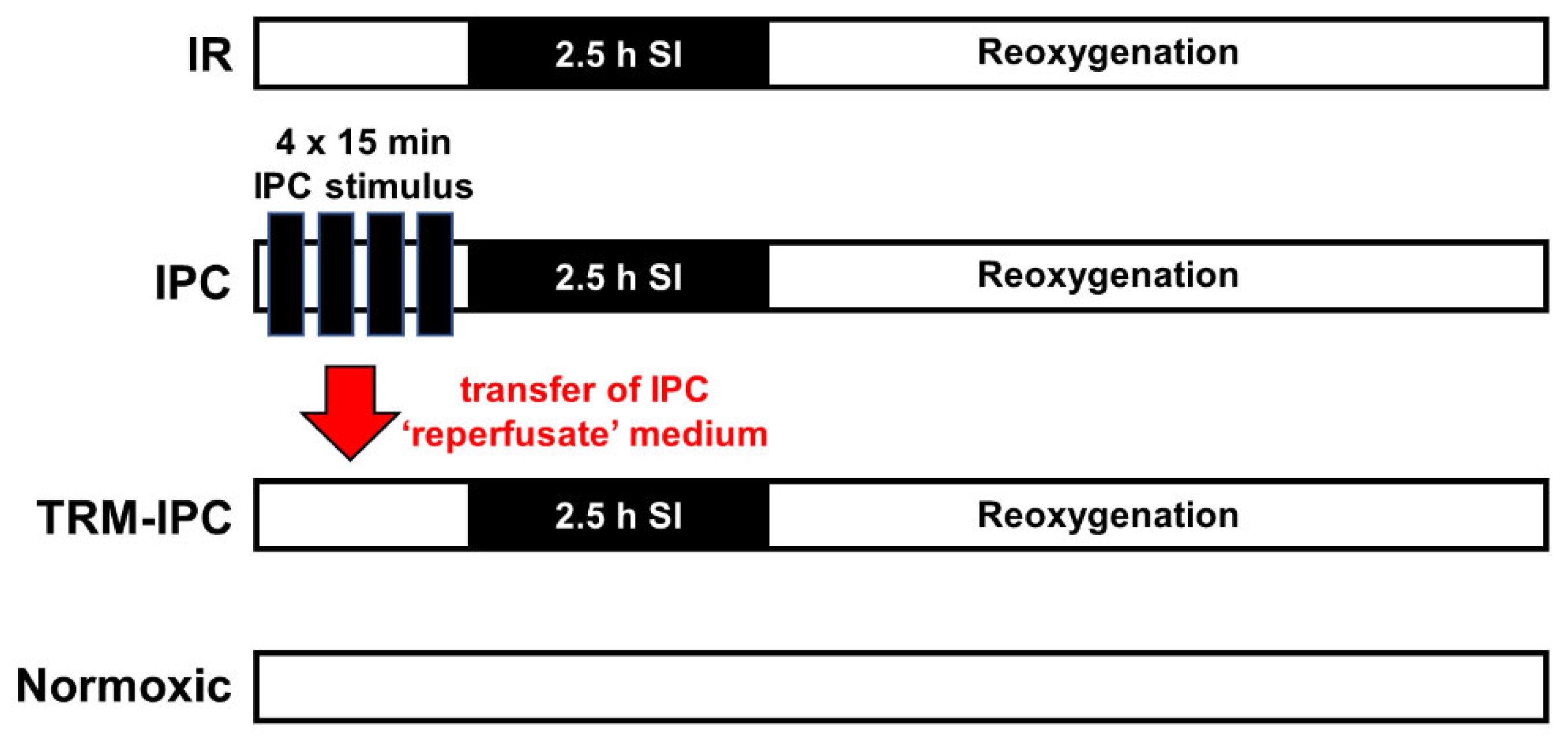
2.1. Protocol 1: Ischemic Conditioning Attenuates Cardiomyocyte Death in the HL-1 Cell Model

2.2. Protocol 2
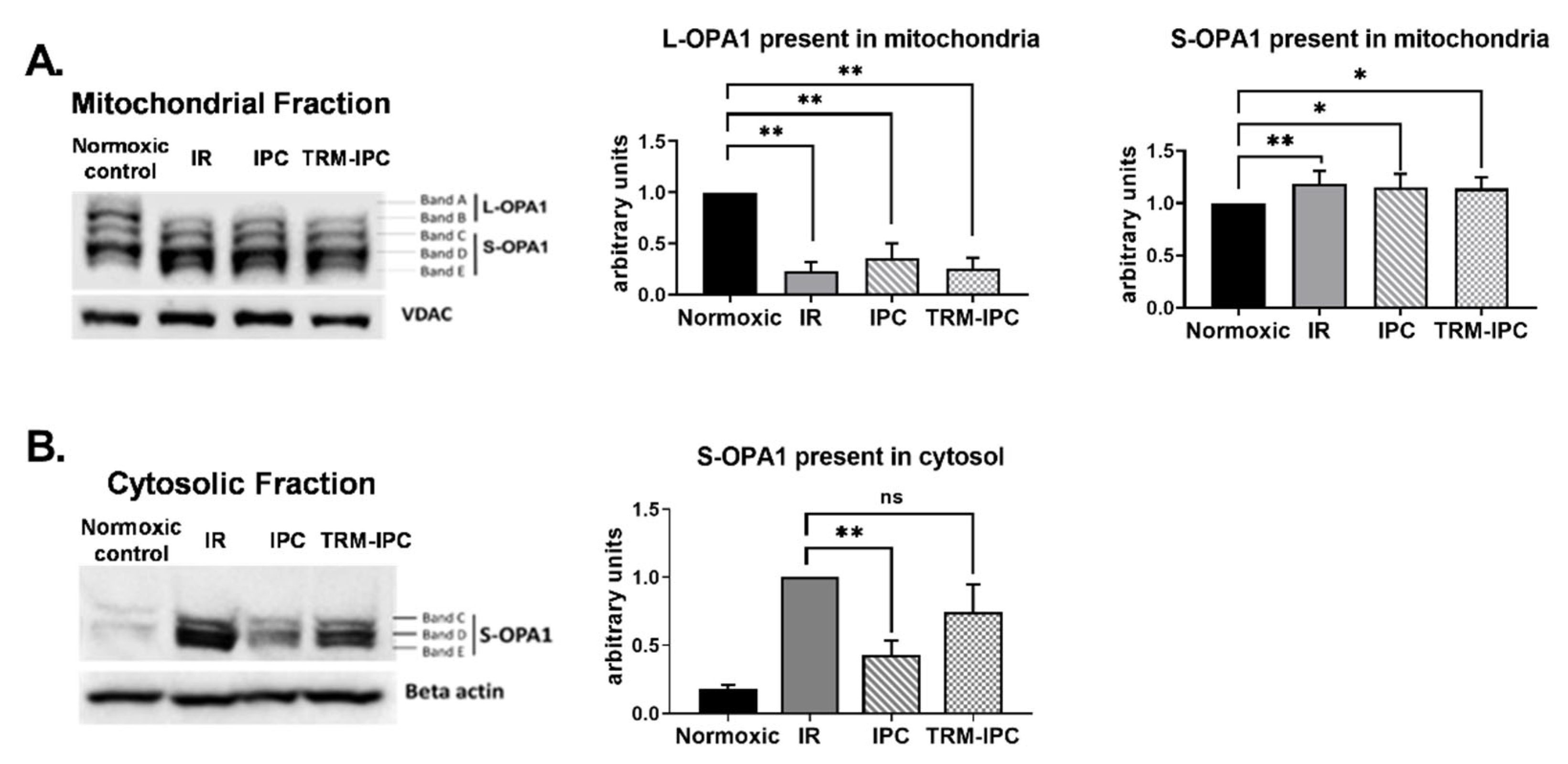
2.2.1. Protocol 2A: Effect of Simulated Ischemia-Reperfusion Injury on the Integrity and Subcellular Localization of OPA1
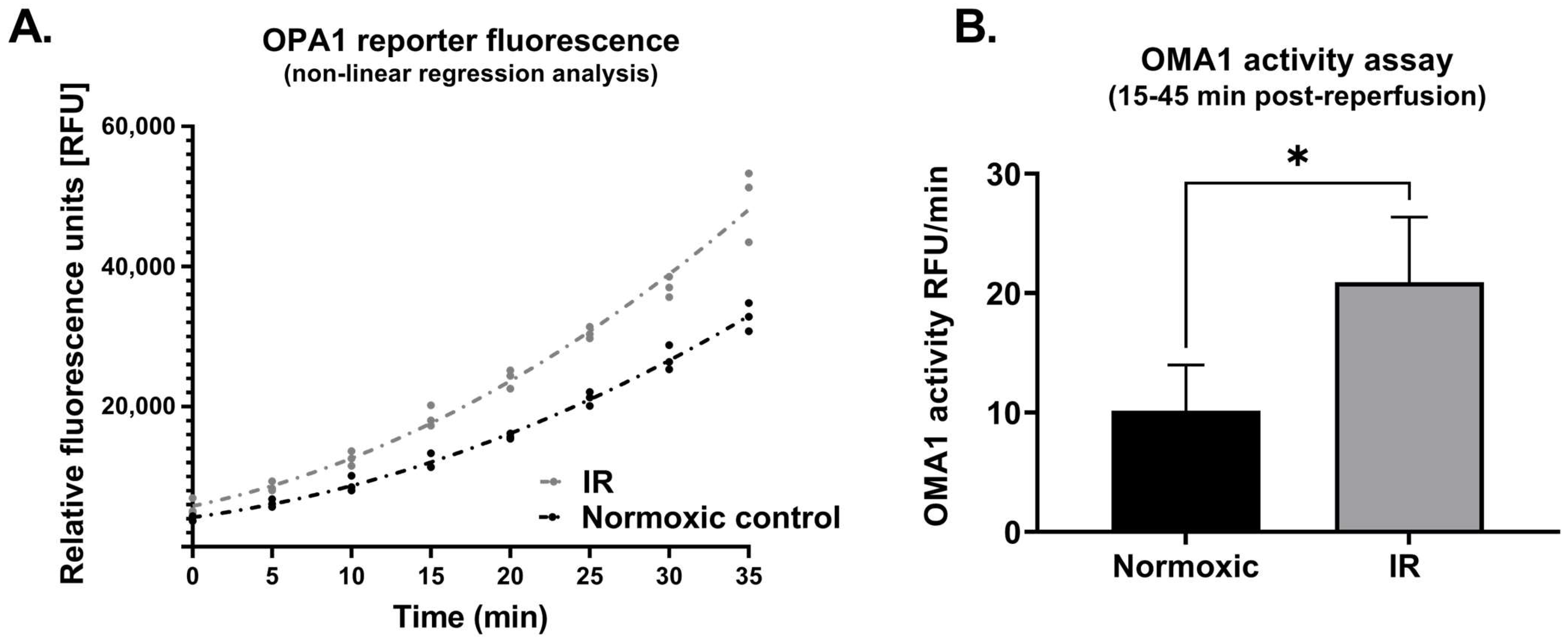
2.2.2. Protocol 2B: Effect of Simulated Ischemia-Reperfusion on OMA1 Activity
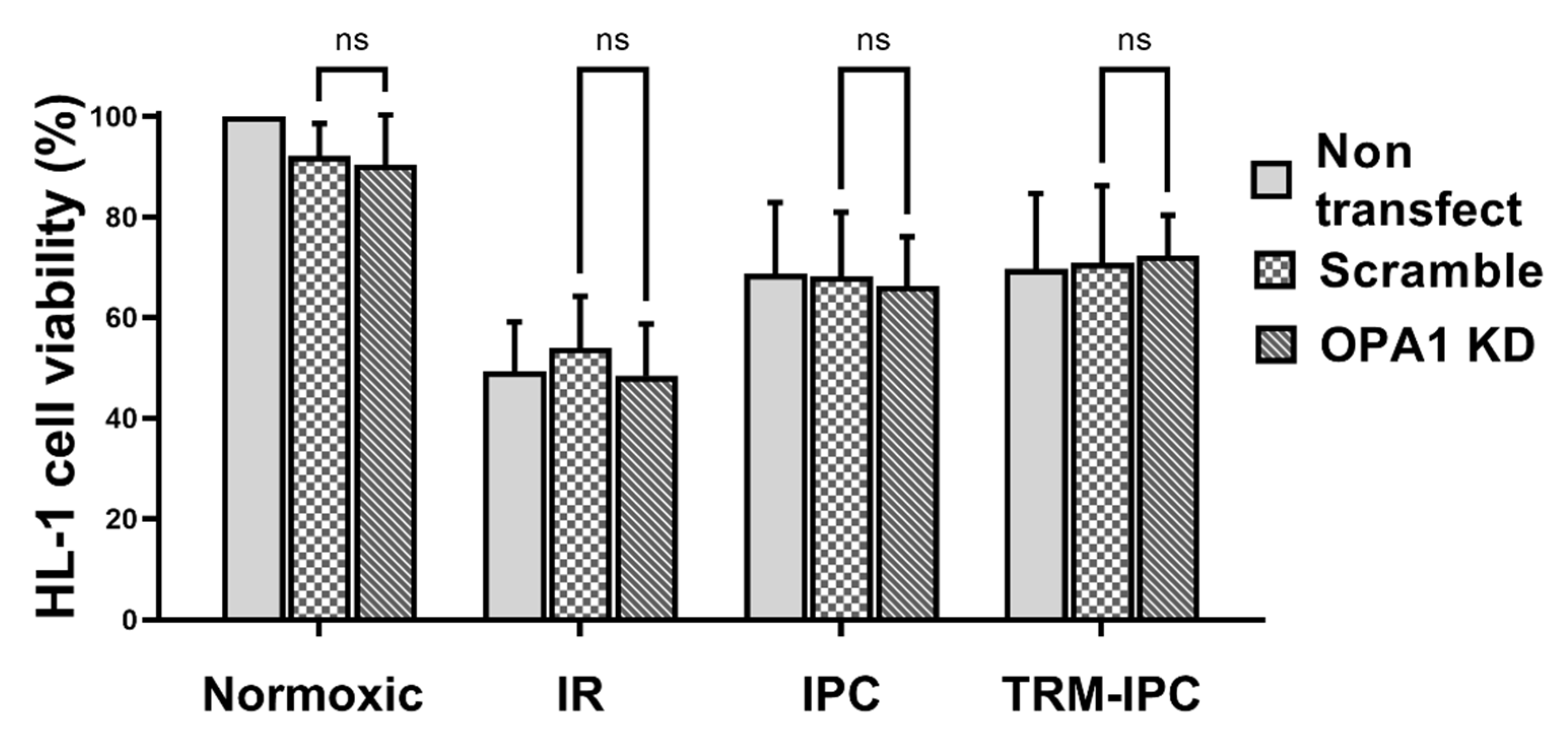
2.3. Protocol 3: Effect of OPA1 Knockdown on Cardiomyocyte Fate
2.4. Statistical Analysis
3. Results
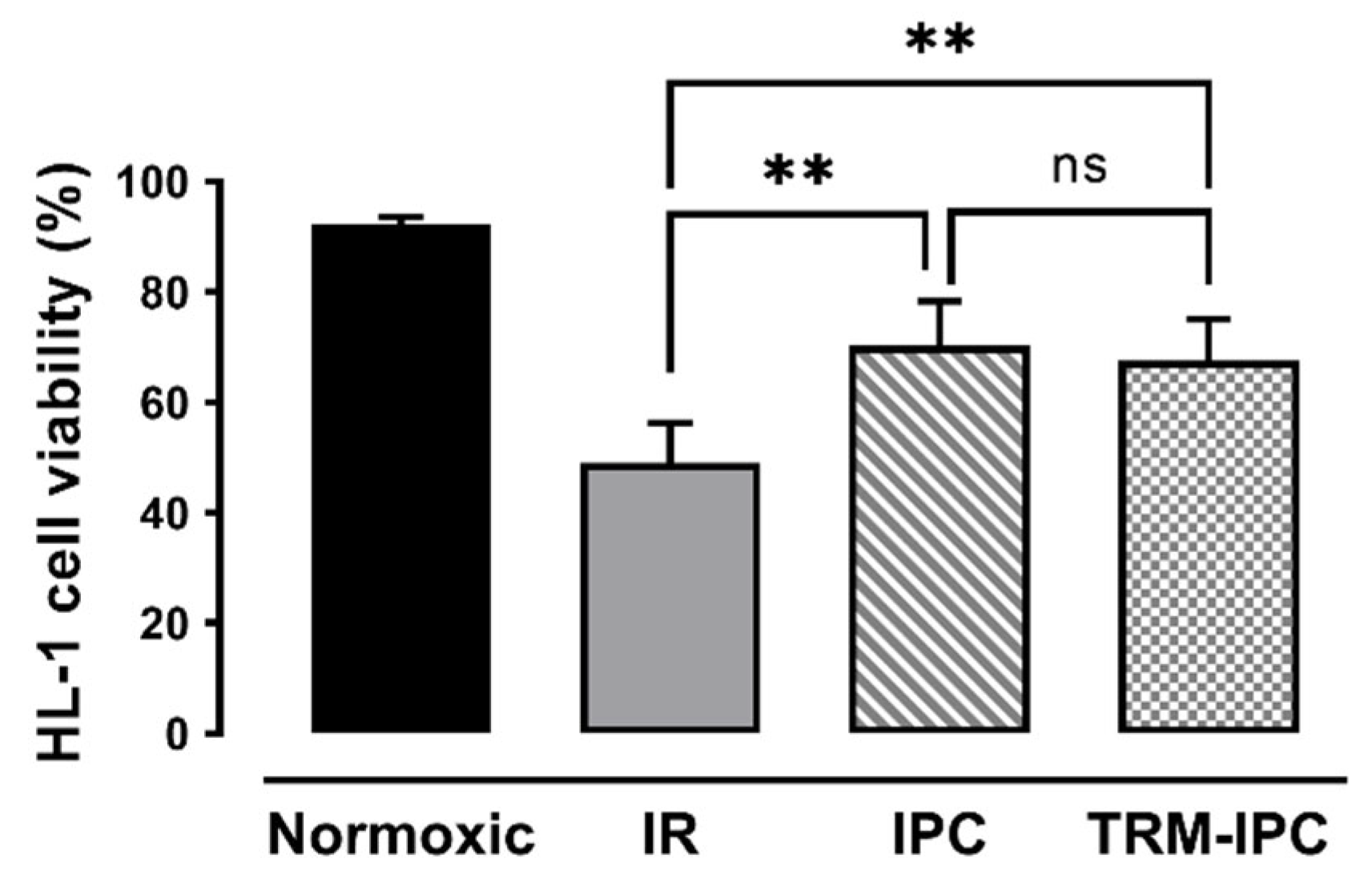
3.1. Protocol 1
3.2. Protocol 2

3.3. Protocol 3
4. Discussion
4.1. Ischemic Conditioning in Cell Culture Models
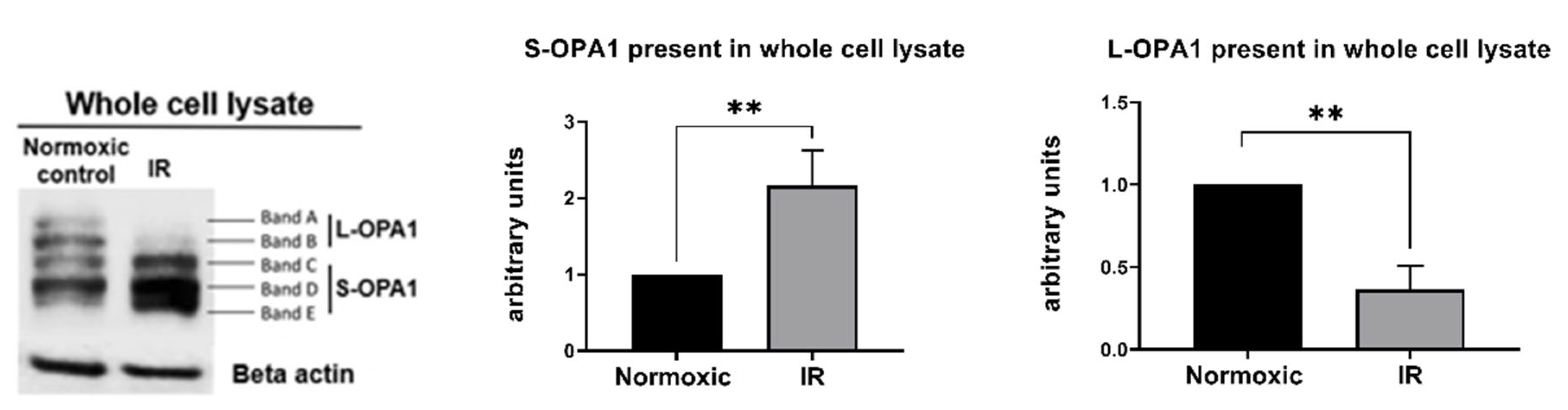
4.2. Total OPA1 Content and Lethal Ischemia-Reperfusion Injury
4.3. Ischemia-Reperfusion and OPA Processing
4.4. OPA1 Content, OPA1 Processing, and Cardioprotection
4.5. Summary, Limitations and Future Directions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tullio, F.; Angotti, C.; Perrelli, M.G.; Penna, C.; Pagliaro, P. Redox balance and cardioprotection. Basic Res. Cardiol. 2013, 108, 392. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Maddock, H.L.; Baxter, G.F.; Yellon, D.M. Inhibiting mitochondrial permeability transition pore opening: A new paradigm for myocardial preconditioning? Cardiovasc. Res. 2002, 55, 534–543. [Google Scholar] [CrossRef]
- Halestrap, A.P. Calcium, mitochondria and reperfusion injury: A pore way to die. Biochem. Soc. Trans. 2006, 34, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Canton, M.; Menabo, R.; Dodoni, G.; Bernardi, P. Mitochondria and reperfusion injury. The role of permeability transition. Basic Res. Cardiol. 2003, 98, 235–241. [Google Scholar] [CrossRef]
- Archer, S.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010, 88, 16–29. [Google Scholar] [CrossRef]
- Ong, S.B.; Hall, A.R.; Hausenloy, D.J. Mitochondrial dynamics in cardiovascular health and disease. Antioxid. Redox Signal. 2013, 19, 400–414. [Google Scholar] [CrossRef]
- Ong, S.-B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef]
- Disatnik, M.H.; Ferreira, J.C.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.; Qi, X.; Mochly-Rosen, D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Undyala, V.V.R.; Przyklenk, K. Inhibition of mitochondrial fission as a molecular target for cardioprotection: Critical importance of the timing of treatment. Basic Res. Cardiol. 2016, 111, 59. [Google Scholar] [CrossRef] [PubMed]
- Wendt, L.; Vider, J.; Hoe, L.E.S.; Du Toit, E.; Peart, J.N.; Headrick, J.P. Complex Effects of Putative DRP-1 Inhibitors on Stress Responses in Mouse Heart and Rat Cardiomyoblasts. J. Pharmacol. Exp. Ther. 2020, 372, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Kwek, X.Y.; Katwadi, K.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Ismail, N.I.; Lin, Y.H.; Yap, E.P.; Lim, S.Y.; Ja, K.; et al. Targeting Mitochondrial Fission Using Mdivi-1 in A Clinically Relevant Large Animal Model of Acute Myocardial Infarction: A Pilot Study. Int. J. Mol. Sci. 2019, 20, 3972. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; de Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Garcia-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Ruperez, F.J.; Barbas, C.; Ibanez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef]
- Rodriguez-Graciani, K.M.; Chapa-Dubocq, X.R.; MacMillan-Crow, L.A.; Javadov, S. Association between L-OPA1 Cleavage and Cardiac Dysfunction during Ischemia-Reperfusion Injury in Rats. Cell. Physiol. Biochem. 2020, 54, 1101–1114. [Google Scholar] [CrossRef]
- Jones, E.; Gaytan, N.; Garcia, I.; Herrera, A.; Ramos, M.; Agarwala, D.; Rana, M.; Innis-Whitehouse, W.; Schuenzel, E.; Gilkerson, R. A threshold of transmembrane potential is required for mitochondrial dynamic balance mediated by DRP1 and OMA1. Cell. Mol. Life Sci. 2017, 74, 1347–1363. [Google Scholar] [CrossRef]
- Zhang, K.; Li, H.; Song, Z. Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep. 2014, 15, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Griparic, L.; Kanazawa, T.; van der Bliek, A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabo, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef]
- Chen, L.; Liu, T.; Tran, A.; Lu, X.; Tomilov, A.A.; Davies, V.; Cortopassi, G.; Chiamvimonvat, N.; Bers, D.M.; Votruba, M.; et al. OPA1 mutation and late-onset cardiomyopathy: Mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 2012, 1, e003012. [Google Scholar] [CrossRef]
- Le Page, S.; Niro, M.; Fauconnier, J.; Cellier, L.; Tamareille, S.; Gharib, A.; Chevrollier, A.; Loufrani, L.; Grenier, C.; Kamel, R.; et al. Increase in Cardiac Ischemia-Reperfusion Injuries in Opa1+/- Mouse Model. PLoS ONE 2016, 11, e0164066. [Google Scholar] [CrossRef]
- Cellier, L.; Tamareille, S.; Kalakech, H.; Guillou, S.; Lenaers, G.; Prunier, F.; Mirebeau-Prunier, D. Remote ischemic conditioning influences mitochondrial dynamics. Shock 2016, 45, 192–197. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef]
- Przyklenk, K.; Bauer, B.; Ovize, M.; Kloner, R.A.; Whittaker, P. Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1993, 87, 893–899. [Google Scholar] [CrossRef]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Wider, J.; Undyala, V.V.R.; Whittaker, P.; Woods, J.; Chen, X.; Przyklenk, K. Remote ischemic preconditioning fails to reduce infarct size in the Zucker fatty rat model of type-2 diabetes: Role of defective humoral communication. Basic Res. Cardiol. 2018, 113, 16. [Google Scholar] [CrossRef] [PubMed]
- Brewer, J.H.; Allgeier, D.L. Safe Self-contained Carbon Dioxide-Hydrogen Anaerobic System. Appl. Microbiol. 1966, 14, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Brewer, J.H.; Allgeier, D.L. Disposable Hydrogen Generator. Science 1965, 147, 1033–1034. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 2006, 281, 29776–29787. [Google Scholar] [CrossRef]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B.1–A3.B.3. [Google Scholar] [CrossRef]
- Gerlier, D.; Thomasset, N. Use of MTT colorimetric assay to measure cell activation. J. Immunol. Methods 1986, 94, 57–63. [Google Scholar] [CrossRef]
- Gomez, L.A.; Alekseev, A.E.; Aleksandrova, L.A.; Brady, P.A.; Terzic, A. Use of the MTT assay in adult ventricular cardiomyocytes to assess viability: Effects of adenosine and potassium on cellular survival. J. Mol. Cell. Cardiol. 1997, 29, 1255–1266. [Google Scholar] [CrossRef]
- Berridge, M.V.; Tan, A.S. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): Subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 1993, 303, 474–482. [Google Scholar] [CrossRef]
- Cung, T.T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guerin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Tobacyk, J.; Parajuli, N.; Shrum, S.; Crow, J.P.; MacMillan-Crow, L.A. The first direct activity assay for the mitochondrial protease OMA1. Mitochondrion 2019, 46, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Nan, J.; Nan, C.; Ye, J.; Qian, L.; Geng, Y.; Xing, D.; Rahman, M.S.U.; Huang, M. EGCG protects cardiomyocytes against hypoxia-reperfusion injury through inhibition of OMA1 activation. J. Cell Sci. 2019, 132, jcs220871. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Hu, Y.; Quiros, P.M.; Wei, Q.; Lopez-Otin, C.; Dong, Z. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am. J. Physiol. Renal Physiol. 2014, 306, F1318–F1326. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef]
- Bohovych, I.; Fernandez, M.R.; Rahn, J.J.; Stackley, K.D.; Bestman, J.E.; Anandhan, A.; Franco, R.; Claypool, S.M.; Lewis, R.E.; Chan, S.S.; et al. Metalloprotease OMA1 Fine-tunes Mitochondrial Bioenergetic Function and Respiratory Supercomplex Stability. Sci. Rep. 2015, 5, 13989. [Google Scholar] [CrossRef]
- Pan, Y.X.; Ren, A.J.; Zheng, J.; Rong, W.F.; Chen, H.; Yan, X.H.; Wu, C.; Yuan, W.J.; Lin, L. Delayed cytoprotection induced by hypoxic preconditioning in cultured neonatal rat cardiomyocytes: Role of GRP78. Life Sci. 2007, 81, 1042–1049. [Google Scholar] [CrossRef]
- Maslov, L.N.; Lishmanov, I.B.; Kolar, F.; Portnichenko, A.G.; Podoksenov, I.K.; Khaliulin, I.G.; Wang, H.; Pei, J.M. Hypoxic preconditioning is a phenomenon increasing tolerance of cardiomyocytes to hypoxia-reoxygenation. Ross. Fiziol. Zhurnal Im. IM Sechenova 2010, 96, 1170–1189. [Google Scholar] [CrossRef]
- Xu, R.; Sun, Y.; Chen, Z.; Yao, Y.; Ma, G. Hypoxic Preconditioning Inhibits Hypoxia-induced Apoptosis of Cardiac Progenitor Cells via the PI3K/Akt-DNMT1-p53 Pathway. Sci. Rep. 2016, 6, 30922. [Google Scholar] [CrossRef]
- Yan, F.; Yao, Y.; Chen, L.; Li, Y.; Sheng, Z.; Ma, G. Hypoxic preconditioning improves survival of cardiac progenitor cells: Role of stromal cell derived factor-1alpha-CXCR4 axis. PLoS ONE 2012, 7, e37948. [Google Scholar] [CrossRef]
- Cokkinos, A.D.; Tzeis, S.; Moraitis, P.; Pantos, C.; Carageorgiou, H.; Panousopoulos, D.; Varonos, D.D.; Cokkinos, D.V. Loss of cardioprotection induced by ischemic preconditioning after an initial ischemic period in isolated rat hearts. Exp. Clin. Cardiol. 2003, 8, 5–9. [Google Scholar]
- Shimizu, M.; Tropak, M.; Diaz, R.J.; Suto, F.; Surendra, H.; Kuzmin, E.; Li, J.; Gross, G.; Wilson, G.J.; Callahan, J.; et al. Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: Evidence suggesting cross-species protection. Clin. Sci. 2009, 117, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T.H.; Raghunayakula, S.; Kumar, R. Release of mitochondrial Opa1 following oxidative stress in HT22 cells. Mol. Cell. Neurosci. 2015, 64, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.K.; Lindsey, J.D.; Angert, M.; Patel, A.; Weinreb, R.N. Glutamate receptor activation triggers OPA1 release and induces apoptotic cell death in ischemic rat retina. Mol. Vis. 2008, 14, 2629–2638. [Google Scholar] [PubMed]
- Arnoult, D.; Grodet, A.; Lee, Y.J.; Estaquier, J.; Blackstone, C. Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J. Biol. Chem. 2005, 280, 35742–35750. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Botker, H.E. Why did remote ischaemic conditioning not improve clinical outcomes in acute myocardial infarction in the CONDI-2/ERIC-PPCI trial? Cardiovasc. Res. 2019, 115, e161–e163. [Google Scholar] [CrossRef] [PubMed]
- Trankle, C.; Thurber, C.J.; Toldo, S.; Abbate, A. Mitochondrial Membrane Permeability Inhibitors in Acute Myocardial Infarction: Still Awaiting Translation. JACC Basic Transl. Sci. 2016, 1, 524–535. [Google Scholar] [CrossRef]
- Ibanez, B.; Heusch, G.; Ovize, M.; Van de Werf, F. Evolving therapies for myocardial ischemia/reperfusion injury. J. Am. Coll. Cardiol. 2015, 65, 1454–1471. [Google Scholar] [CrossRef]
- Heusch, G.; Gersh, B.J. ERICCA and RIPHeart: Two nails in the coffin for cardioprotection by remote ischemic conditioning? Probably not! Eur. Heart J. 2016, 37, 200–202. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Candilio, L.; Evans, R.; Ariti, C.; Jenkins, D.P.; Kolvekar, S.; Knight, R.; Kunst, G.; Laing, C.; Nicholas, J.; et al. Remote Ischemic Preconditioning and Outcomes of Cardiac Surgery. N. Engl. J. Med. 2015, 373, 1408–1417. [Google Scholar] [CrossRef]
- Meybohm, P.; Bein, B.; Brosteanu, O.; Cremer, J.; Gruenewald, M.; Stoppe, C.; Coburn, M.; Schaelte, G.; Boning, A.; Niemann, B.; et al. A Multicenter Trial of Remote Ischemic Preconditioning for Heart Surgery. N. Engl. J. Med. 2015, 373, 1397–1407. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Kharbanda, R.K.; Moller, U.K.; Ramlall, M.; Aaroe, J.; Butler, R.; Bulluck, H.; Clayton, T.; Dana, A.; Dodd, M.; et al. Effect of remote ischaemic conditioning on clinical outcomes in patients with acute myocardial infarction (CONDI-2/ERIC-PPCI): A single-blind randomised controlled trial. Lancet 2019, 394, 1415–1424. [Google Scholar] [CrossRef]
- Cohen, M.V.; Downey, J.M. AMISTAD trials: Possible reasons for lack of success. J. Am. Coll. Cardiol. 2006, 47, 1236, author reply 1236–1237. [Google Scholar] [CrossRef] [PubMed]
- Chen-Scarabelli, C.; Scarabelli, T.M. Cyclosporine A Prior to Primary PCI in STEMI Patients: The Coup de Grace to Post-Conditioning? J. Am. Coll. Cardiol. 2016, 67, 375–378. [Google Scholar] [CrossRef]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Costi, A. A heart mitochondrial Ca2(+)-dependent pore of possible relevance to re-perfusion-induced injury. Evidence that ADP facilitates pore interconversion between the closed and open states. Biochem. J. 1990, 266, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Ellinger, H.; Costi, A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 1988, 255, 357–360. [Google Scholar]
- Griffiths, E.J.; Halestrap, A.P. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 1995, 307 Pt 1, 93–98. [Google Scholar] [CrossRef]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef]
- Ong, S.B.; Hall, A.R.; Dongworth, R.K.; Kalkhoran, S.; Pyakurel, A.; Scorrano, L.; Hausenloy, D.J. Akt protects the heart against ischaemia-reperfusion injury by modulating mitochondrial morphology. Thromb. Haemost. 2015, 113, 513–521. [Google Scholar] [CrossRef]
- Otera, H.; Miyata, N.; Kuge, O.; Mihara, K. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J. Cell Biol. 2016, 212, 531–544. [Google Scholar] [CrossRef]
- Jiang, X.; Jiang, H.; Shen, Z.; Wang, X. Activation of mitochondrial protease OMA1 by Bax and Bak promotes cytochrome c release during apoptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 14782–14787. [Google Scholar] [CrossRef] [PubMed]
- Germain, M.; Mathai, J.P.; McBride, H.M.; Shore, G.C. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005, 24, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Mopert, K.; Hajek, P.; Frank, S.; Chen, C.; Kaufmann, J.; Santel, A. Loss of Drp1 function alters OPA1 processing and changes mitochondrial membrane organization. Exp. Cell Res. 2009, 315, 2165–2180. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Bukowski, M.J.; Wider, J.M.; Reynolds, C.A.; Calo, L.; Lepore, B.; Tousignant, R.; Jones, M.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics following global cerebral ischemia. Mol. Cell. Neurosci. 2016, 76, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Lecour, S.; Andreadou, I.; Botker, H.E.; Davidson, S.M.; Heusch, G.; Ruiz-Meana, M.; Schulz, R.; Zuurbier, C.J.; Ferdinandy, P.; Hausenloy, D.J.; et al. IMproving Preclinical Assessment of Cardioprotective Therapies (IMPACT) criteria: Guidelines of the EU-CARDIOPROTECTION COST Action. Basic Res. Cardiol. 2021, 116, 52. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulek, A.R.; Undyala, V.V.R.; Anzell, A.R.; Raghunayakula, S.; MacMillan-Crow, L.A.; Sanderson, T.H.; Przyklenk, K. Does Disruption of Optic Atrophy-1 (OPA1) Contribute to Cell Death in HL-1 Cardiomyocytes Subjected to Lethal Ischemia-Reperfusion Injury? Cells 2022, 11, 3083. https://doi.org/10.3390/cells11193083
Kulek AR, Undyala VVR, Anzell AR, Raghunayakula S, MacMillan-Crow LA, Sanderson TH, Przyklenk K. Does Disruption of Optic Atrophy-1 (OPA1) Contribute to Cell Death in HL-1 Cardiomyocytes Subjected to Lethal Ischemia-Reperfusion Injury? Cells. 2022; 11(19):3083. https://doi.org/10.3390/cells11193083
Chicago/Turabian StyleKulek, Andrew R., Vishnu V. R. Undyala, Anthony R. Anzell, Sarita Raghunayakula, Lee Ann MacMillan-Crow, Thomas H. Sanderson, and Karin Przyklenk. 2022. "Does Disruption of Optic Atrophy-1 (OPA1) Contribute to Cell Death in HL-1 Cardiomyocytes Subjected to Lethal Ischemia-Reperfusion Injury?" Cells 11, no. 19: 3083. https://doi.org/10.3390/cells11193083
APA StyleKulek, A. R., Undyala, V. V. R., Anzell, A. R., Raghunayakula, S., MacMillan-Crow, L. A., Sanderson, T. H., & Przyklenk, K. (2022). Does Disruption of Optic Atrophy-1 (OPA1) Contribute to Cell Death in HL-1 Cardiomyocytes Subjected to Lethal Ischemia-Reperfusion Injury? Cells, 11(19), 3083. https://doi.org/10.3390/cells11193083